Synthesis of thermo-responsive glycopolymersviacopper catalysed azide–alkyne ‘click’ chemistry for inhibition of ricin: the effect of spacer between polymer backbone and galactose†
Jatin
Kumar
ab,
Lyndal
McDowall
ac,
Gaojian
Chen
d and
Martina H.
Stenzel
*a
aCentre of Advanced Macromolecular Design (CAMD), School of Chemical Engineering, The University of New South Wales, New South Wales 2052, Australia. E-mail: M.Stenzel@unsw.edu.au
bCooperative Research Centre for Polymers, 8 Redwood Drive, Notting Hill, Victoria 3168, Australia
cHuman Protection and Performance Division, Defence Science and Technology Organisation, 506 Lorimer Street, Fishermans Bend, Victoria 3207, Australia
dCentre for Soft Condensed Matter Physics and Interdisciplinary Research, Soochow University, Suzhou, 215006, China
First published on 8th June 2011
Abstract
Block copolymers poly(diethylene glycol methyl ether methacrylate)-block-poly(2-hydroxy ethyl methacrylate) (PDEGMA-b-PHEMA) and poly(diethylene glycol methyl ether methacrylate)-block-poly(ethylene glycol methyl ether methacrylate) PDEGMA-b-PPEGMA as well as homopolymers of PPEGMA and PHEMA were generated via Reversible Addition–Fragmentation chain Transfer (RAFT)polymerisation. The terminal hydroxyl moieties on the HEMA and PEGMA blocks were alkyne functionalised and conjugated to azide functionalised galactoseviaCuAAC leading to the glycopolymers PDEGMA-b-P(HEMA-Gal) and PDEGMA-b-P(PEGMA-Gal). Both dynamic light scattering (DLS) and transmission electron microscopy (TEM) prove the thermo-responsive characteristics of the polymers, due to the PDEGMA blocks, with the formation of polymeric micelles above a temperature of 40 °C. These glycopolymers were tested for their binding efficiency to ricin, a dangerous plant toxin reportedly weaponised by terrorist organisations. The binding experiments establish an order of ricin inhibition of P(HEMA-Gal) < P(PEGMA-Gal) < PDEGMA-b-P(HEMA-Gal) ≤ PDEGMA-b-P(PEGMA-Gal) showing that micelles are advantageous, but also that the introduction of a flexible spacer between polymer and galactose can enhance binding to a certain extent.
Introduction
Carbohydrates hold great significance to life due to their role in important biological functions such as immune response and cell recognition.1–3In recent times, the scientific community has shown that synthetic glycopolymers are a class of polymers that have high potential for biomedical applications due to their ability to interact with carbohydrate moieties and lectins in a manner similar to natural glycoproteins.3 The key to successful carbohydrate–lectin interaction lies in multivalent interactions, which result in specificity and high affinity. This effect, possible by having multiple saccharide functionalities on polymers, is known as the ‘cluster glycoside effect’.4
Ricin is a well-known plant-derived toxin that has reportedly been weaponised for use in terrorism.5 It can be easily isolated from castor beans and there are presently no known countermeasures for it.5,6 Binding to galactose can block its cytotoxic action,2 although bonds with single galactose moieties are generally weak and ineffective. As with all carbohydrate–lectin interactions the multivalency effect is crucial for biologically relevant binding.7,8 This suggests a potential application for galactose based glycopolymers.
There are generally two approaches in synthesizing glycopolymers, one of which is polymerizing a monomer that holds saccharide functionality. Various polymerisation techniques can be employed to generate glycopolymers from these monomers including living free radical techniques such as RAFT.9 The major drawback of this approach, however, is that it has been found that a number of these sugar monomers have the tendency to self-polymerise.10
The alternative technique involves post-functionalising a polymer with a saccharide.11 Post-functionalisation allows for greater control and variability of the glycopolymer as different saccharides can be conjugated to the same polymeric architecture. The post-functionalisation route has the added advantage of not requiring exotic or new monomer species, which could in turn also affect the control in RAFT polymerisations resulting in long inhibition periods or non-living characteristics.
Fernández-García and coworkers have reported conjugating amino-saccharides to polymer containing activated esters forming amide linkages.12,13 A technique that has been more widely used is the copper catalysed azide–alkyne Huisgen 1,3-dipolar cycloaddition (CuAAC) reaction. This is due to the flexibility and versatility of the reaction—possible under mild conditions in both aqueous and organic medium.14CuAAC conjugation techniques are widely used for various different functionalisations and have consistently reported high yields and orthogonality.15 Haddleton and co-workers have successfully reported a number of applications for the use of reactions to post-functionalise linear polymers with sugars and showed effective binding of the generated glycopolymers to lectins.16–18
Similar to CuAAC are other ‘click’ chemistry techniques such as the thiol–alkene conjugation. This involves the reaction of a thiol and an alkene, by either a radical approach or alternatively viaMichael Addition.19 Chen et al. synthesized glucose based glycopolymer micelles via this approach in a copolymer with a thermoresponsive poly(diethylene glycol methyl ether methacrylate) (PDEGMA) block. The studies found that there was an enhancement of glycopolymer interaction with the Concanavalin A lectin when micelles were formed.20 The increased binding to lectins upon micelle formation was also observed with mannose based thermoresponsive glycopolymers21 as well as galactose based micelles.22,23
Binding efficacy is optimised when the distance between saccharide units is close to or the same as the spacing between binding sites in a lectin. As a result, polymer rigidity as well as the distance of the sugar moiety to the polymer backbone may have an effect on protein binding.24 A more flexible polymer, or a polymer with pendant sugars linked with a spacer away from the backbone, would therefore be able to conform to the distance between binding sites. In the case of a rigid polymer, the only way to achieve optimum binding would be if the spaces between the sugars on the polymer matched the binding sites on the lectin exactly.25
In this study, we synthesize a series of thermoresponsive block copolymers containing PDEGMA and either poly 2-hydroxy ethyl methacrylate (PHEMA) or poly(polyethylene glycol methacrylate) (PPEGMA). The PHEMA and PPEGMA blocks are functionalised with terminal alkynes and then glycosylated with an azide-galactose. The purpose of the use of two different monomers for this study is to investigate how the introduction of a flexible poly(ethylene glycol) PEG spacer affects binding to ricin. (Scheme 1)
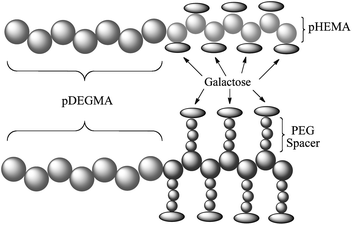 | ||
| Scheme 1 Overview of the different polymeric designs. | ||
Experimental procedures
Materials
The RAFT agent cumyl dithiobenzoate (CDB),264-oxo-4-(prop-2-ynyloxy)butanoic anhydride27 and azide-galactose28 were prepared according to the procedure described in previous publications. Diethylene glycol methyl ether methacrylate (DEGMA), 2-hydroxy ethyl methacrylate (HEMA) and polyethylene glycol methacrylate of Mn = 360 (PEGMA360) were all purchased from Aldrich and destabilized by passing them over a column of basic alumina. 2,2-Azobisisobutyronitrile (AIBN), (Fluka 98%) was recrystallised twice from methanol. All other chemicals were used as supplied by manufacturers unless otherwise stated.Syntheses
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHN3); 5.60 (n2H, d, anomeric glucose proton); 8.20 (1n2, COOCH2CCHN3). n1 and n2 are the degree of polymerisation of DEGMA and HEMA respectively. For PDEGMA-b-PPEGMA: 1H NMR (300 MHz, DMSO-d6, δ, ppm): 1.50–2.10 (5n1H + 5n3H, CH3CCH2 (backbone)); 2.70 (4n3H, COOCH2CH2COOCH2CCH); 3.30 (3n1H, CH3OCH2CH2); 3.40–4.30 (6n1H, CH3OCH2CH2OCH2CH2; 4n3(m − 1), COOCH2CH2O(CH2CH2O)m−1OC; 2n1H + 2n3H, OCH2CH2COO; 11n2, galactose); 5.20 (4n2, COOCH2CCHN3); 5.60 (n2H, d, anomeric glucose proton); 8.20 (1n2, COOCH2CCHN3). n1 and n3 are the degree of polymerisation of DEGMA and PEGMA360 respectively. m is the degree of polymerisation of ethylene glycol on PEGMA360.
CHN3); 5.60 (n2H, d, anomeric glucose proton); 8.20 (1n2, COOCH2CCHN3). n1 and n2 are the degree of polymerisation of DEGMA and HEMA respectively. For PDEGMA-b-PPEGMA: 1H NMR (300 MHz, DMSO-d6, δ, ppm): 1.50–2.10 (5n1H + 5n3H, CH3CCH2 (backbone)); 2.70 (4n3H, COOCH2CH2COOCH2CCH); 3.30 (3n1H, CH3OCH2CH2); 3.40–4.30 (6n1H, CH3OCH2CH2OCH2CH2; 4n3(m − 1), COOCH2CH2O(CH2CH2O)m−1OC; 2n1H + 2n3H, OCH2CH2COO; 11n2, galactose); 5.20 (4n2, COOCH2CCHN3); 5.60 (n2H, d, anomeric glucose proton); 8.20 (1n2, COOCH2CCHN3). n1 and n3 are the degree of polymerisation of DEGMA and PEGMA360 respectively. m is the degree of polymerisation of ethylene glycol on PEGMA360.
Analysis
Inhibition of ricin was also tested at 40 °C. For these assays the plate was incubated with blocking buffer in a Ratek orbital mixer incubator at 40 °C with gentle shaking for 1 h. The ricin and polymer/galactose mixtures were also incubated at 40 °C with gentle shaking for 1 h before being transferred to the plate at 40 °C and subsequently incubated for 1 h on the plate. The rest of the assay was performed at room temperature.
Results and discussion
The synthesis of different PDEGMA-b-PHEMA and PDEGMA-b-PPEGMA was achieved by RAFT polymerization using cumyl dithiobenzoate (CDB) as the RAFT agent and AIBN as a thermal initiator (Scheme 2). PDEGMA was generated first, but since both blocks are based on methacrylates the reverse approach would be possible. Synthesis of PDEGMA macroRAFT agent was carried out in bulk so as to keep the reaction time as low as possible and avoid unnecessary side reactions, thus maximising the percentage of RAFT capped chains. The Mn of the carefully purified polymer was obtained viaSEC and 1H NMR and was found to be 8200 g mol−1 and 7900 g mol−1 respectively. | ||
| Scheme 2 Outline of synthesis route of block copolymers. (a) The synthesis of PDEGMA macroRAFT agent; (b) the synthesis of PDEGMA-b-PHEMA and (c) of PDEGMA-b-PPEGMA. | ||
In a subsequent step, PDEGMA was used as the macroRAFT and was then chain extended with either HEMA or PEGMA360 using now DMAc as solvent to reduce viscosity. The same-sized PDEGMA was used for all copolymerizations to generate a constant block size of the stimuli-responsive block while the glycopolymer block was varied in chain length. Samples were then drawn at different time intervals and the block copolymers were analyzed viaGPC as well as 1H NMR.
Previous studies30 involving the polymerization of PEGMA360 recommended to keep the conversion of the reaction below 20% as there is evidence of gelation. It is suggested that the monomer as supplied by the manufacturer contains small amounts of poly(ethylene glycol) dimethacrylate, which could polymerise and cross-link the propagating chains.
The SEC traces of the copolymer show a narrow distribution with no shoulders at higher molecular weights or lower molecular weights tailings and a low PDI. Table 1 summarises the different homo and block (co)polymers prepared with their respective PDIs (Table 1, column: Mn (PDI) unfunct).
| No | DEGMA units | HEMA units | PEGMA360 units | M n Theo unfunct | M n (PDI) unfunct | M n (PDI) alkyne | M n (PDI) glyco |
|---|---|---|---|---|---|---|---|
a Commercial polymer purchased from Sigma-Aldrich. Mv ≈ 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000. 000.
|
|||||||
| 0 | 43 | — | — | 7900 | 8200 (1.25) | — | — |
| 1 | — | — | 24 | 8600 | 9400 (1.10) | 11300 (1.23) |
34800 (1.52) |
| 2 | — | 90 | — | 12000 |
11800 (1.18) |
24600 (1.22) |
35900(1.23) |
| 3a | — | 154 | — | 20000 |
17200 (1.80) |
34000 (1.88) |
45000 (1.84) |
| 4 | 43 | — | 20 | 15000 |
15700 (1.11) |
16900 (1.16) |
58400 (1.82) |
| 5 | 43 | 16 | 13800 |
14200 (1.17) |
16000 (1.23) |
53400 (1.75) |
|
| 6 | 43 | 20 | — | 10400 |
11000 (1.13) |
12500 (1.15) |
13700 (1.14) |
| 7 | 43 | 79 | — | 18000 |
18400 (1.16) |
23600 (1.21) |
26900 (1.28) |
| 8 | 43 | 68 | — | 16500 |
17000 (1.16) |
21800 (1.20) |
24100 (1.35) |
PEGMA360 and HEMA homopolymers were also generated with CDB as the RAFT agent. The purpose of this is to serve as a basis of comparison between non-thermoresponsive homopolymers and the thermoresponsive DEGMA containing copolymers.
The terminal hydroxyl functionalities of the (co)polymers were then converted to alkynes with 4-oxo-4-(prop-2-ynyloxy)butanoic anhydride in DMF in the presence of DMAP and TEA. The product of this functionalisation was precipitated in cold ether 3 times to ensure that all reactants are removed from the polymer. Full alkyne functionalisation was verified via1H NMR in DMSO-d6 by the appearance of peaks at 2.60 and 4.69 ppm, with integrals that verify the correct ratios of the associated protons. The NMR also serves to identify any excess 4-oxo-4-(prop-2-ynyloxy)butanoic anhydride that still might be present (Fig. 1).
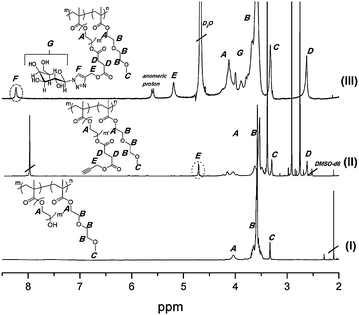 | ||
| Fig. 1 1H-NMR (from bottom to top): (I) pre-functionalised block copolymer (d-DMSO); (II) alkyne functionalisation of terminal hydroxyls on the PPDEGMA block (d-DMSO); (III) after ‘Clicking’ of azide galactose on polymer (D2O). | ||
Alkyne functionalized polymer was then reacted with azide functionalized galactosevia a copper catalysed azide–alkyne cycloaddition using copper(II) sulfate and sodium ascorbate. The reaction was left to run over 72 hours to ensure that all alkynes were reacted with the azide containing galactose molecules since their close proximity to one another could be sterically hindered. Scheme 3 depicts the various stages of functionalisation of the (co)polymers. The product of the ‘click’ reaction was dialysed against water for 2 days with water changes every 6 hours to remove excess galactose, copper sulfate, sodium ascorbate and solvent. However, traces of the catalyst were often still present and had to be removed using pentaerythritol tetrakis(3-mercaptopropionate) as a strong ligand for copper ions. Copper has been known to ligate to the triazole rings formed in azide–alkyne coupling reactions.15,31 To ensure that most of the copper was removed from the gly(co)polymer product, pentaerythritol tetrakis(3-mercaptopropionate) was added dropwise to an aqueous solution of the product. The mercaptan forms a white precipitate as it preferentially ligates to the copper. The mixture was filtered, the excess mercaptan separated, and then freeze-dried before any further analyses were carried out. The product was analysed via1H NMR. The disappearance of the peak at 2.5 ppm corresponding to the CH from the alkyne together with the appearance of the peak at 8.28 ppm due to the presence of a triazole ring suggests that all alkyne had reacted as illustrated in Fig. 1. In addition the methylene group adjacent to the ester functionality (E in Fig. 1) shifts from 4.7 ppm to 5.2 ppm. The signals 8.28, 5.60 and 5.20 ppm are with an intensity ratio of 1:1:2 in good agreement with the expected value. The Mns of the various glycopolymers generated at each stage of functionality are also reported in Table 1.
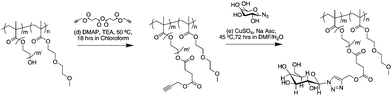 | ||
| Scheme 3 Postfunctionalization of the terminal hydroxyl moieties on the polymers. For the PHEMA based (co)polymers, m′ = 1. | ||
Fig. 2 and 3 show the evolution of SEC traces at each step of polymerisation and functionalisation for PDEGMA-b-PPEGMA and PDEGMA-b-PHEMA copolymers respectively.
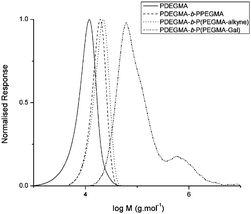 | ||
| Fig. 2 SEC traces for the stages of functionalisation for polymer 4. | ||
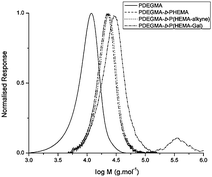 | ||
| Fig. 3 SEC traces for the stages of functionalisation for polymer 7. | ||
There is a noticeable high molecular weight hump in the SEC of all the glycosylated polymers. Lau et al.32 report artifacts similar to this where they found that triazole rings can have a synergistic effect on the interchain hydrogen bonding to the point of gelation. A phenomenon such as this one reasonably explains the high molecular weight humps seen in the SECs, especially due to the close proximity of the galactose moieties to the triazole rings on the polymers. To exclude the possibility of the formation of disulfide formation, which could be formed from the RAFT endgroup after hydrolysis, the SEC experiments were carried out in the presence of a small amount of DTT to reduce the disulfide to thiol. Changes in the intensity of the high molecular weight shoulder were however negligible. Another reason could be the occurrence of some transesterification reactions taking place under the conditions of the click reaction.
The glycopolymers (Table 3) were tested for the thermoresponsive characteristics by measuring their average size at various temperatures by dynamic light scattering (DLS). The polymers were dissolved in de-ionised water at a concentration of 1 mg mL−1. These polymers were left in water for 96 hours at room temperature to ensure complete dissolution since dissolution could be hindered by inter- and intra-molecular hydrogen bonding. The polymer solution was filtered using a 0.45 μm filter and placed in a 1 cm disposable plastic cuvette for DLS analysis.
It is expected that the polymers dissolve in water unimolecularly at 20 °C, while above the LCST of PDEGMA, micelle formation occurs resulting in aggregates with a PDEGMA core and aglycopolymer shell. The number-average and volume-average hydrodynamic diameter at 20 °C and 40 °C are displayed in Table 2. The strong discrepancy between both values at 20 °C is synonymous with a broad and multi-modal size distribution indicating the formation of unexpected aggregates. The particle size recorded for all polymers at a temperature of 20 °C is much larger than the average size of linear polymers in solution, which are often merely a few nanometres. Strong aggregation caused by the presence of carbohydrates, PEG chains and triazole (see above) may hamper the full dissolution of the theoretically fully water-soluble polymer. Aggregation of polysaccharides and glycopolymers has been observed earlier and investigated in detail.33,34 Once at 40 °C, above the LCST of PDEGMA of 29 °C, all the measured distributions have similar mean-values indicative for a better defined product with a smaller particle size distribution. A notable increase in the recorded number-mean particle diameter and count rate confirms that these polymers do indeed hold some thermoresponsive characteristic. The drop in mean volume and intensity diameters suggests that at higher temperatures, the hydrogen bonds break and allow the polymers to self assemble into micelles.
| No | Polymer | Number mean/nm | Volume mean/nm | Count rate | |||
|---|---|---|---|---|---|---|---|
| 20 °C | 40 °C | 20 °C | 40 °C | 20 °C | 40 °C | ||
| 4 | PDEGMA43-b-P(PEGMA-Gal)20 | 50 | 136 | 357 | 174 | 180 | 373 |
| 5 | PDEGMA43-b-P(PEGMA-Gal)16 | 92 | 145 | 526 | 198 | 175 | 338 |
| 6 | PDEGMA43-b-P(HEMA-Gal)20 | 84 | 114 | 558 | 116 | 98 | 252 |
| 7 | PDEGMA43-b-P(HEMA-Gal)79 | 60 | 119 | 199 | 140 | 110 | 261 |
| 8 | PDEGMA43-b-P(HEMA-Gal)68 | 81 | 158 | 414 | 198 | 161 | 418 |
Images of the polymer samples were also obtained through TEM. The samples were prepared at a room temperature of 20 °C as well as at a temperature of 40 °C and stained with phosphor tungstic acid. The TEM images at 20 °C do not have any characteristically significant features, however the images at 40 °C show particles which are not present in the samples prepared at 20 °C (Fig. 4). The shape of the particles obtained in the TEM samples prepared at 40 °C appears to be round which further suggests that there is a formation of micelles although the formation of large compound micelles could be possible considering the measured large sizes. The particle size through TEM appears to be comparable with the sizes reported through DLS (Table 3).
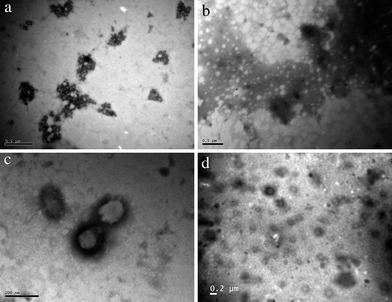 | ||
| Fig. 4 (Clockwise from top left) Polymer 7 prepared on the copper grid at 20 °C (a); polymer 8 prepared on the copper grid at 20 °C (b); polymer 8 prepared on the copper grid at 40 °C (c); polymer 7 prepared on the copper grid at 40 °C (d). All samples negatively stained with phosphor tungstic acid. | ||
Ricin binding assays
Galactose is a highly bioactive carbohydrate, which plays a major role in many recognition events including specific binding to plant and animal lectins including ricin.9Ricin, a highly toxic lectin isolated from castor bean (Ricinus communis), is a hetero lectin consisting of two parts, ricin A and ricin B chains. The B chain has two or three galactose binding sites35,36 and therefore a multivalent galactose system may provide a means of neutralisation. The focus of interest in this study is how the polymers prepared may increase binding to ricin compared to free galactose thus displaying a multivalency effect. The polymers were tested by asialofetuin competition assay in comparison to galactose at room temperature and at 40 °C. The concentrations were standardised as galactose equivalents. Only the IC50 value—the concentration of galactose that inhibits 50% of ricin—is listed in Table 4. It should be noted here that the concentrations are only approximate since solubility issues in the media led to uncertainties.| No | Sample/polymer | Approximate IC50 ratios | |
|---|---|---|---|
|
|
|
||
| Galactose | 0.75 | 1 | |
| 1 | P(PEGMA-Gal)24 | — | >0.4 |
| 2 | P(HEMA-Gal)90 | 0.2 | 1.2 |
| 3 | P(HEMA-Gal)154 | — | >0.75 (likely actual ratio of 1, tracks along with galactose) |
| 4 | PDEGMA43-b-P(PEGMA-Gal)20 | — | >0.175 |
| 5 | PDEGMA43-b-P(PEGMA-Gal)16 | <0.5, >0.19 | <0.3, >0.125 |
| 6 | PDEGMA43-b-P(HEMA-Gal)20 | — | >0.125 |
| 7 | PDEGMA43-b-P(HEMA-Gal)79 | 0.2 | 1 |
| 8 | PDEGMA43-b-P(HEMA-Gal)68 | — | >0.3 |
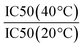

All polymers were tested at 40 °C and three polymer samples were tested additionally at 20 °C and their performance were compared to galactose. Increase in temperature meant a decrease of IC50 for all the four samples tested (Table 4, middle column). While galactose alone acted only slightly better with increasing temperature, the polymers led to noticeable better binding at higher temperature. This includes the tested homo glycopolymer (2) for which thermoresponsive self-assembly is not possible. It is not known how the buffer system used in these assays may affect the self-assembly of the polymers. An explanation for the increase in binding for the three polymers can be found in the changes in aggregate formation with temperature. It is possible that at room temperature the polymer is too closely associated with itself to participate fully in ricin binding. The DLS studies, which indicate strong aggregation and also the difficulties in solubilising these polymers may be an indication of tight inter- and intra-molecular binding. However on increase of temperature these associations would lessen, potentially freeing the galactose moieties for ricin binding. Block copolymers in addition will form more ordered structures at higher temperature with a high surface concentration of galactose. The increased binding here is in good agreement with earlier studies using thermo-responsive block copolymer, which show that upon micelle formation binding is more efficient.21,23 However, the absolute inhibition of ricin was only conclusively better for sample 5 (PDEGMA43-b-P(PEGMA-Gal)16) compared to monovalent Galactose. For polymers 2 and 7 the increase at higher temperature only brings them into line with what is seen for monovalent galactose, whereas polymer 5 is better than galactose at room (IC50 is half that of galactose) and elevated temperature.
Comparison of the performance of all the polymers with monovalent galactose at 40 °C shows that P(HEMA-Gal) has the weakest binding barely showing any improvement to galactose. Introduction of the flexible PEG spacer is indeed beneficial and the amount necessary to inhibit 50% of ricin is reduced by more than half. This would not be entirely unexpected due to the added flexibility this polymer has in its side-chains with the PEG spacer. Mammen and coworkers24 discuss the importance of matching the distance between two saccharides on a polymer to binding sites on a lectin to obtain good binding. It is evident from these data that having a PEG spacer in a polymer could indeed give the polymer the flexibility it requires to achieve good protein binding. Even better inhibition was observed by the micellar system (with exception of sample 7, whose bad performance cannot currently be explained). Upon micelle formation, the glycopolymers take on a more brush-like structure; the shorter the hydrophilic block, the more stretched are the glycopolymers, and thus the more brush-like they become. Comparing the glycopolymer block length with the values displayed in Table 4, last column, there seems to be a trend between the potential correlation between block length and performance with shorter block (therefore more brush-like structures) exhibiting better inhibition. In addition, a slight improvement may also be observed when a PEG spacer is employed, although this may be within errors.
These initial experiments establish an order of ricin inhibition of P(HEMA-Gal) < P(PEGMA-Gal) < PDEGMA43-b-P(HEMA-Gal) ≤ PDEGMA43-b-P(PEGMA-Gal) with better binding observed in shorter glycopolymer blocks.
The IC50 values reported here show only a modest enhancement in binding between galactose and galactose polymers. The IC50 is proportional to the inhibitor dissociation constant by the following equation: Ki = IC50/(1 + R/Kr) where R is ricin conc. and Kr is the ricin–asialofetuin dissociation constant.37 The ratio of IC50 (multivalent polymer)/IC50(galactose) is equivalent to Ki(polymer)/Ki(galactose) and is a measure of enhancement (as a reduction in dissociation constant). However Lundquist and Toone4 caution against making such interpretations of these types of assays and relying on Scatchard plots (where this equation is derived from) is problematic for covalently tethered ligands. More than just the kinetics affects the IC50 determined in an assay.38 This type of assay is useful as a guide only and to quantify any kinetic or thermodynamic enhancements due to strict multivalency effects techniques such as SPR and ITC should be used. However kinetic data may not account for all mechanisms of inhibition in real biological systems such as clustering or steric hinderance.
Conclusion
Block copolymers of DEGMA and HEMA as well as DEGMA and PEGMA were synthesized by RAFT polymerisation. The terminal hydroxyl moieties on the HEMA and PEGMA blocks were alkyne functionalised with an alkyne anhydride and then later conjugated to azide functionalised galactoseviaCuAAC in mild conditions. DLS studies show that raising a solution of the glycopolymers to 40 °C has an effect on its particle size. Below this temperature, the particle size is generally found to be larger, however this phenomenon can be attributed to inter-molecular hydrogen bonding. TEM images taken of the polymers prove that micelles form above the LCST, while those samples prepared below the LCST had no significant characteristics. This confirms that the glycopolymers do exhibit thermoresponsive characteristics and form micelles at temperatures above 40 °C.The purpose of these glycopolymers was to investigate the effect of the distance of galactose from the polymer backbone on lectin/protein and glycopolymer interaction. Studies were carried out to assess the binding efficiency of the glycopolymers to the plant toxin, ricin. PDEGMA-b-PPEGMA glycopolymers showed the highest degree of binding with the polymer with the shorter glycopolymeric chain having a better inhibition of ricin as compared to monovalent galactose. We have established an order of ricin inhibition of P(HEMA-Gal) < P(PEGMA-Gal) < PDEGMA43-b-P(HEMA-Gal) ≤ PDEGMA43-b-P(PEGMA-Gal) with better binding observed in shorter glycopolymer blocks and P(HEMA-Gal) not showing any significant improvement in ricin inhibition over monovalent galactose.
Acknowledgements
The authors would like to thank the Australian Research Council (ARC) for financial support. The authors would like to acknowledge the Mark Wainwright analytical centre at UNSW for help in NMR analysis and microscopy analysis.Notes and references
- Y. Miura, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 5031–5036 CrossRef CAS.
- J. M. Lord, L. M. Roberts and J. D. Robertus, FASEB J., 1994, 8, 201–208 CAS.
- S. G. Spain and N. R. Cameron, Polym. Chem., 2011, 2, 60–68 RSC.
- J. J. Lundquist and E. J. Toone, Chem. Rev. (Washington, DC, U. S.), 2002, 102, 555–578 CrossRef CAS.
- J. Audi, M. Belson, M. Patel, J. Schier and J. Osterloh, JAMA, J. Am. Med. Assoc., 2005, 294, 2342–2351 CrossRef CAS.
- S. Lundberg, L. Melin, C. Nilsson and P. von Schoenberg, Ricin. Threat, effects and protection, User Report FOI-R–1261–SE, FOI-Swedish Defence Research Agency, Umea, 2004 Search PubMed.
- L. L. Kiessling and N. L. Pohl, Chem. Biol., 1996, 3, 71–77 CrossRef.
- V. Ladmiral, E. Melia and D. M. Haddleton, Eur. Polym. J., 2004, 40, 431–449 CrossRef CAS.
- S. R. S. Ting, G. Chen and M. H. Stenzel, Polym. Chem., 2010, 1, 1392–1412 RSC.
- S. G. Spain, M. I. Gibson and N. R. Cameron, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2059–2072 CrossRef CAS.
- M. A. Gauthier, M. I. Gibson and H.-A. Klok, Angew. Chem., Int. Ed., 2009, 48, 48–58 CrossRef CAS.
- G. Martínez, M. Fernández-García and M. Sánchez-Chaves, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 18–27 CrossRef CAS.
- M. L. Cerrada, M. Sánchez-Chaves, C. Ruiz and M. Fernández-Garcia, Biomacromolecules, 2009, 10, 1828–1837 CrossRef CAS.
- R. A. Evans, Aust. J. Chem., 2007, 60, 384–395 CrossRef CAS.
- W. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2007, 28, 15–54 CrossRef CAS.
- V. Ladmiral, G. Mantovani, G. J. Clarkson, S. Cauet, J. L. Irwin and D. M. Haddleton, J. Am. Chem. Soc., 2006, 128, 4823–4830 CrossRef CAS.
- J. Geng, G. Mantovani, L. Tao, J. Nicolas, G. Chen, R. Wallis, D. A. Mitchell, B. R. G. Johnson, S. D. Evans and D. M. Haddleton, J. Am. Chem. Soc., 2007, 129, 15156–15163 CrossRef CAS.
- L. Nurmi, J. Lindqvist, R. Randev, J. Syrett and D. M. Haddleton, Chem. Commun., 2009, 2727–2729 RSC.
- A. B. Lowe, Polym. Chem., 2010, 1, 17–36 RSC.
- G. Chen, S. Amajjahe and M. H. Stenzel, Chem. Commun., 2009, 1198–1200 RSC.
- M. Hetzer, G. Chen, C. Barner-Kowollik and M. H. Stenzel, Macromol. Biosci., 2010, 10, 119–126 CrossRef CAS.
- S. R. S. Ting, E. H. Min, P. Escalé, M. Save, L. Billon and M. H. Stenzel, Macromolecules, 2009, 42, 9422–9434 CrossRef CAS.
- S. R. S. Ting, E. H. Min, P. B. Zetterlund and M. H. Stenzel, Macromolecules, 2010, 43, 5211–5221 CrossRef CAS.
- M. Mammen, S. K. Choi and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 2754–2794 CrossRef.
- T. Hasegawa, S. Kondoh, K. Matsuura and K. Kobayashi, Macromolecules, 1999, 32, 6595–6603 CrossRef CAS.
- L. Albertin, M. Stenzel, C. Barner-Kowollik, L. J. R. Foster and T. P. Davis, Macromolecules, 2004, 37, 7530–7537 CrossRef CAS.
- P. Antoni, Y. Hed, A. Nordberg, D. Nyström, H. von Holst, A. Hult and M. Malkoch, Angew. Chem., Int. Ed., 2009, 48, 2126–2130 CrossRef CAS.
- J. Geng, J. Lindqvist, G. Mantovani, G. Chen, C. Sayers, G. Clarkson and D. Haddleton, QSAR Comb. Sci., 2007, 26, 1220–1228 CrossRef CAS.
- R. M. Dawson, M. R. Alderton, D. Wells and P. G. Hartley, J. Appl. Toxicol., 2006, 26, 247–252 Search PubMed.
- L. Zhang, T. Nguyen, J. Bernard, T. Davis, C. Barner-Kowollik and M. Stenzel, Biomacromolecules, 2007, 8, 2890–2901 CrossRef CAS.
- T. R. Chan, R. Hilgraf, K. B. Sharpless and V. V. Fokin, Org. Lett., 2004, 6, 2853–2855 CrossRef CAS.
- K. Lau, H. Chow, M. Chan and K. Wong, Angew. Chem., Int. Ed., 2008, 47, 6912–6916 CrossRef CAS.
- D. Balasubramanian, B. Raman and C. S. Sundari, J. Am. Chem. Soc., 1993, 115, 74–77 CrossRef CAS.
- L.-C. You, F.-Z. Lu, Z.-C. Li, W. Zhang and F.-M. Li, Macromolecules, 2003, 36, 1–4 CrossRef CAS.
- Y. P. Venkatesh and J. M. Lambert, Glycobiology, 1997, 7, 329–335 Search PubMed.
- E. Rutenber and J. D. Robertus, Proteins: Struct., Funct., Genet., 1991, 10, 260–269 Search PubMed.
- R. M. Dawson, B. M. Paddle and M. R. Alderton, J. Appl. Toxicol., 1999, 19, 307–312 Search PubMed.
- S. M. Dimick, S. C. Powell, S. A. McMahon, D. N. Moothoo, J. H. Naismith and E. J. Toone, J. Am. Chem. Soc., 1999, 121, 10286–10296 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1py00048a |
| This journal is © The Royal Society of Chemistry 2011 |