Synthesis and properties of anion conductive ionomers containing fluorenyl groups for alkaline fuel cell applications
Manabu
Tanaka
a,
Masaki
Koike
b,
Kenji
Miyatake
*ab and
Masahiro
Watanabe
*a
aFuel Cell Nanomaterials, University of Yamanashi, 4 Takeda, Kofu, 400-8510, Japan. E-mail: m-watanabe@yamanashi.ac.jp; Fax: +81-55-220-8707; Tel: +81-55-220-8707
bClean Energy Research Center, University of Yamanashi, 4 Takeda, Kofu, 400-8510, Japan. E-mail: miyatake@yamanashi.ac.jp; Fax: +81-55-220-8707; Tel: +81-55-220-8707
First published on 23rd September 2010
Abstract
A series of anion conductive aromatic ionomers, poly(arylene ether)s containing different polymer backbones and quaternized ammonio-substituted fluorenyl groups, were synthesized via nucleophilic substitution polycondensation, chloromethylation, quaternization, and the subsequent ion exchange reactions. The ion exchange capacity (IEC) of the ionomers was controlled to be from 0.68 to 2.54 meq. g−1 by the chloromethylation reaction conditions. The designed chemical structures were well-characterized by the 1H NMR spectra. The ionomers provided ductile and transparent membranes. The ionomer membranes were thermally stable up to 180 °C under nitrogen and mechanically stable with 48 MPa of the maximum stress at 80 °C and 60% RH (relative humidity). High hydroxide ion conductivity up to 50 mS cm−1 was achieved at 30 °C in water for the ionomer membrane bearing sulfone/ketone structures and the highest IEC (2.54 meq. g−1). The membranes were durable in hot water (80 °C) for 1000 hours. These properties of the ionomer membranes seem promising as an anion exchange membrane for alkaline fuel cells.
Introduction
There have been great demands for clean energy or renewable energy sources in the past decade. Fuel cells have attracted considerable attention due to their high energy efficiency and low pollution levels.1 Among the various kinds of fuel cells, polymer electrolyte fuel cells (PEFCs) using proton exchange membranes (PEMs) have been developed especially for residential, portable, and electric vehicle applications.2 Perfluorinated ionomers such as Nafion (DuPont) are state-of-the-art polymer electrolyte membranes for PEM fuel cells. Although the perfluorinated ionomers exhibit excellent chemical, mechanical, and thermal stability along with high proton conductivity, there are several obstacles that impede the commercialization of PEM fuel cells such as high cost of the perfluorinated PEMs and platinum group metal (PGM) electrocatalysts, and insufficient durability of these materials under fuel cell operating conditions.2 One possible solution to overcome these problems would be the use of an anion exchange membrane (AEM) instead of the PEM. The alkaline fuel cells using AEM have several advantages over PEM fuel cells on both cost and performance.3 In the basic environment within AEM fuel cells, sluggish oxygen reduction reaction at the cathode can be significantly improved, leading to high fuel cell efficiency. Besides, such less corrosive environment allows us to use non-precious metals (such as nickel and silver4) as the cathode catalysts and thus, potentially reducing the cost of the fuel cells.Since the existing AEMs are not as conductive and stable as PEMs, a growing effort has been consumed in developing better AEM materials. A variety of AEMs based on poly(olefine)s,5 cross-linked poly(styrene)s,6poly(phenylene oxide),7 poly(phenylene)s,8 poly(ether imide)s,9 poly(arylene ether)s,10 or organic–inorganic hybrid composites11 have been reported. Poly(arylene ether sulfone)s and poly(arylene ether ketone)s fall into a class of aromatic polymers that show good solubility in organic solvents, and have been widely used as a backbone of hydrocarbon PEMs.12 Therefore, many research groups have attempted to apply poly(arylene ether)s as AEMs by introducing quaternized ammonio groups. These AEMs are typically prepared by chloromethylation on aromatic rings or bromination on methyl groups of the poly(arylene ether)s, followed by exposure to trimethylamine to form benzyltrimethylammonio groups.10
Various types of poly(arylene ether)s have been investigated for the AEMs, however, fluorenyl groups have gained little publicity as AEM polymer segments. Over the years, we have synthesized a series of sulfonated poly(arylene ether)s and sulfonated poly(arylene imide)s containing fluorenyl groups as PEMs, and have demonstrated the effectiveness of the fluorenyl groups not only on the chemical, thermal, and mechanical stability but also on the proton conductivity.13Sulfonic acid groups can be effectively introduced at the specific positions of fluorenyl groups, which causes high local concentration of ionic groups and high proton conductivity of the resulting polymer membranes. Since the sulfonic acid groups on fluorenyl groups are distanced from the polymer backbones, polymers are less susceptible to the hydrolytic and/or oxidative degradation triggered from the sulfonic acid groups. We have demonstrated that the poly(arylene ether sulfone)s containing sulfofluorenyl groups are highly proton conductive and durable for 5000 h in operating fuel cells.13a,b
Our idea is that the fluorenyl groups should function in the AEMs as well as PEMs. In our preliminary communication, we reported that poly(arylene ether sulfone ketone)s containing quaternized ammonio-substituted fluorenyl groups showed high hydroxide anion conductivity based on the polymer structures.14 In this article, we describe detailed syntheses and properties of our quaternized poly(arylene ether)s containing fluorenyl groups. Different main chain structures, biphenyl sulfone, biphenyl ketone, benzonitrile, and perfluorobiphenyl, have been investigated. Chloromethyl groups, which are precursors of quaternized ammonio groups, were successfully introduced at the specific positions of the fluorenyl groups of the poly(arylene ether)s via Friedel–Crafts reaction. The chloromethylation reaction was well-controlled by optimizing the reaction conditions. The obtained quaternized poly(arylene ether)s were evaluated as an AEM in terms of ion conductivities, thermal and mechanical properties, and long-term durability.
Results and discussion
Synthesis of precursor poly(arylene ether)s (PEs)
A series of fluorenyl group-containing precursor poly(arylene ether)s, PE-a to PE-e, were synthesized by nucleophilic substitution polycondensation of 9,9-bis(4-hydroxyphenyl)fluorene and difluoro monomers in the presence of potassium carbonate in dry N,N′-dimethylacetamide (DMAc) solution (Scheme 1). 4,4′-Difluorophenyl sulfone, equimolar mixture of 4,4′-difluorophenyl sulfone and 4,4′-difluorobenzophenone, 4,4′-difluorobenzophenone, 3,5-difluorobenzonitrile, and decafluorobiphenyl were used as difluoro monomers to provide PE-a, PE-b, PE-c, PE-d, and PE-e, respectively. The PEs were obtained as a white fiber. PE-a, PE-b, PE-d, and PE-e were soluble in some organic solvents such as chloroform, dichloromethane, 1,1,2,2-tetrachloroethane, N,N-dimethylformamide (DMF), DMAc, and dimethyl sulfoxide (DMSO), while PE-c was only partly soluble in these solvents probably due to its higher crystallinity than the others. The molecular weights of the PEs were measured by gel permeation chromatography (GPC) and the results are summarized in Table 1. The PEs were of high molecular weights (weight average molecular weight: Mw > 111 kg mol−1 and number average molecular weight: Mn > 38 kg mol−1) to provide transparent and ductile membranes by solution casting except PE-c. Due to the insufficient solubility, PE-c did not provide self-standing membranes and was not suitable for the subsequent reactions.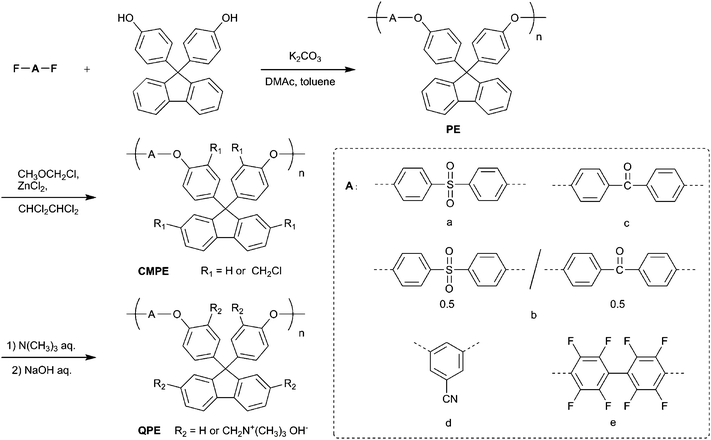 | ||
| Scheme 1 Synthesis of polyethers (PEs), chloromethylated polyethers (CMPEs), and quaternized ammonio-substituted polyethers (QPEs) containing (a) biphenyl sulfone, (b) biphenyl sulfone/biphenyl ketone, (c) biphenyl ketone, (d) benzonitrile, and (e) perfluorobiphenyl moieties. | ||
Synthesis and characterization of chloromethylated poly(arylene ether)s (CMPEs)
The Friedel–Crafts chloromethylation reactions of the PEs were carried out with chloromethyl methyl ether (CMME) and Lewis acid catalyst. Since this reaction easily causes gelation of the polymer mixtures due to undesirable cross-linking reactions of chloromethyl groups, the reaction conditions had to be carefully controlled. The reaction conditions were examined with PE-a from various angles of the reaction factors, such as concentration of CMME, kinds and amount of Lewis acid catalysts, reaction time and temperature (Table 2). The solvent and concentration of PE-a were fixed as 1,1,2,2-tetrachloroethane and 0.10 M, respectively, based on the previous report.10a Several Lewis acid catalysts, aluminium chloride, zinc chloride and tin chloride, were investigated. The catalytic activity to accelerate the chloromethylation and the following cross-linking reactions corresponded to their acidity (tin chloride > aluminium chloride > zinc chloride).15 In the present case, zinc chloride was selected as the catalyst because stronger Lewis acid caused difficulty in controlling the reaction. The amount of zinc chloride was changed from 0, 0.01, 0.1, and 1.0 equivalents to the polymer repeating unit. The other important parameters were temperature and equivalents of CMME to the polymer repeating unit. The reaction proceeded faster but was less controllable at higher temperature and higher equivalents of zinc chloride and CMME. We concluded that the optimum conditions were 35 °C, equimolar amount of zinc chloride, and 25 or 50 equivalents of CMME. Under the conditions, the degree of chloromethylation (introduced number of chloromethyl groups per polymer repeating unit) was controlled by changing the reaction time without gelation.| Run | PE | Chloromethylation reaction conditions | ||||
|---|---|---|---|---|---|---|
| Reaction temperature/°C | Equivalents of ZnCl2 | Equivalents of CMME | Reaction time/h | Number of chloromethyl groups per repeating unit | ||
| 1 | PE-a | 60 | 0 | 25 | 3 | 0 |
| 2 | 60 | 0.01 | 25 | 3 | 0 | |
| 3 | 60 | 0.1 | 25 | 3 | 0.08 | |
| 4 | 60 | 1 | 25 | 3 | 1.32 | |
| 5 | 10 | 1 | 25 | 48 | 0 | |
| 6 | 25 | 1 | 25 | 48 | 0 | |
| 7 | 35 | 1 | 25 | 168 | 1.46 | |
| 8 | 40 | 1 | 25 | <72 | Gelation | |
| 9 | 60 | 1 | 25 | <6 | Gelation | |
| 10 | 35 | 1 | 50 | 168 | 1.82 | |
| 11 | 35 | 1 | 75 | <168 | Gelation | |
| 12 | PE-b | 35 | 1 | 25 | 168 | 1.34 |
| 13 | 35 | 1 | 40 | 168 | 1.80 | |
| 14 | 40 | 1 | 25 | 72 | 1.35 | |
| 15 | PE-d | 35 | 1 | 40 | <24 | Gelation |
| 16 | PE-e | 35 | 1 | 40 | 96 | 1.17 |
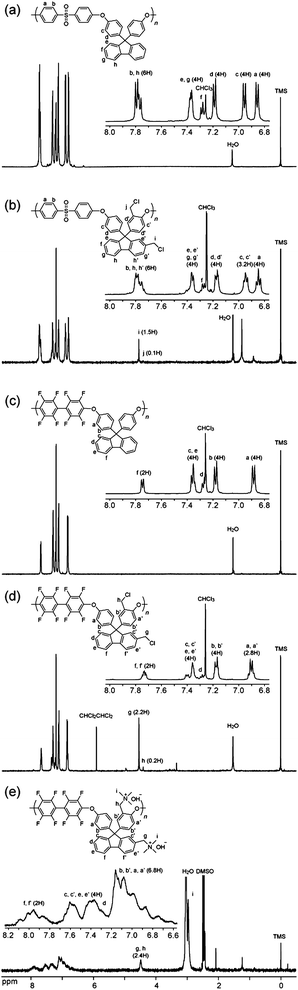
The chemical structure and the degree of chloromethylation were analyzed by comparing the proton nuclear magnetic resonance (1H NMR) spectra of the precursor PEs and the obtained chloromethylated PEs (CMPEs). Fig. 1a and b show 1H NMR spectra of PE-a and CMPE-a, respectively. In Fig. 1b, a new peak assignable to the methylene protons in the chloromethyl groups was observed at 4.59 ppm. The introduction of the chloromethyl groups altered several peaks between 6.9 and 7.4 ppm, which were derived from the fluorenyl biphenylene protons. The integral value of the peaks at 7.25 ppm, which correspond to the protons at 2- and 7-positions of the fluorenyl groups (denoted as f in Fig. 1a and b), was smaller than that of the corresponding original peaks in Fig. 1a. The integral value of the newly appeared peak at 4.59 ppm, which was assigned to the chloromethyl groups on the fluorenyl groups, was coincident with the integral value of the decreasing peaks at 7.25 ppm by replacing the 2- and 7-positional protons with the chloromethyl groups. Adjacent peak at 4.43 ppm was assigned to the methylene protons of the chloromethyl groups but on the polymer backbone (denoted as j in Fig. 1b). The 1H NMR spectra revealed that the chloromethyl groups were preferentially substituted at the specific positions: 2- and 7-positions of the fluorenyl groups (denoted as f in Fig. 1a) and ortho-positions to ether bond in fluorenylidene biphenylene unit (denoted as c in Fig. 1a). The degree of chloromethylation in CMPE was estimated from the integral ratio of the methylene protons of chloromethyl groups (4.43–4.59 ppm) to the protons at 7.75–7.86 ppm, which were assigned to 4-positional protons at the fluorenyl groups (denoted as h in Fig. 1b) and ortho-positional phenylene protons in biphenyl sulfone (denoted as b in Fig. 1b) and were intact during the chloromethylation reaction. As shown in Fig. 2, the degree of chloromethylation of CMPE-a increased with the reaction time. The highest value (1.82) was achieved by 168 h reaction.
 | ||
| Fig. 1 NMR spectra of (a) PE-a, (b) CMPE-a, (c) PE-e, (d) CMPE-e, and (e) QPE-e (the numbers in brackets indicate the integral values of the peaks). | ||
 | ||
| Fig. 2 Time dependence of the degree of chloromethylation. | ||
The chloromethylation reaction conditions optimized for PE-a were applied to PE-b, PE-d, and PE-e. The reaction temperature and equivalents of CMME were finely adjusted for PE-b to find that 35 °C and 40 eq. of CMME provided the maximum degree of chloromethylation (45%), which corresponded to 1.80 chloromethyl groups per repeating unit.14CMPE-b bearing different number of chloromethyl groups, 1.56, 1.17, 0.66, 0.23 and 0.06 chloromethyl groups per repeating unit, was also obtained by changing the reaction time (Fig. 2). The degree of chloromethylation of PE-d was not successful and only gave gel products within 24 h. PE-e gave a soluble CMPE-e under the similar reaction conditions to PE-b. The CMPE-e contained 1.17 chloromethyl groups per repeating unit after 96 h of reaction, as confirmed by the 1H NMR spectrum.
Membrane formation and conversion to quaternized ammonio groups-substituted poly(arylene ether)s (QPEs)
The CMPEs were dissolved in 1,1,2,2-tetrachloroethane and cast on a flat glass plate to provide membranes. CMPE-a provided brittle membranes, which were not suitable for the further reaction. CMPE-b and CMPE-e provided colorless, transparent, tough, and ductile membranes, of which thickness was controlled to be 50 µm. The membranes of CMPE-b and CMPE-e were converted to quaternized ammonio forms, QPE-b and QPE-e, by treating with 30 wt% trimethylamine aqueous solution. The subsequent immersion of the membranes in a 1 M sodium hydroxide aqueous solution provided quaternized poly(arylene ether)s (QPEs) with hydroxide counter anions. The QPE-b and QPE-e membranes were slightly yellow and less soluble than the parent CMPE-b and CMPE-e.The chemical structure of QPE-e was characterized by 1H NMR spectrum as shown in Fig. 1e. The sharp peak of methylene protons at 4.58 and 4.44 ppm in CMPE-e (Fig. 1d) shifted to higher magnetic field at 4.51 ppm as a broad peak in QPE-e (Fig. 1e). A new peak at 2.97 ppm in QPE-e was assigned to methyl groups on the quaternized ammonio groups. The integral ratio of the proton peaks suggested quantitative quaternization reaction. The ion exchange capacity (IEC) of the QPE-e membrane was calculated to be 1.56 meq. g−1 from the integral ratio, which was in good agreement with the calculated value supposing that all of the chloromethyl groups were quaternized. The chemical structures of CMPE-b and QPE-b were also characterized by 1H NMR spectra as previously reported.14 The IECs of a series of QPE-b membranes were calculated to be 2.54, 1.88, 1.31, 1.23, and 0.68 meq. g−1 from the degree of chloromethylation in CMPE-b (1.80, 1.24, 0.81, 0.75, and 0.39 chloromethyl groups per repeating unit, respectively) obtained by the 1H NMR spectra (Table 3).
Water uptake and anion conductivity of the QPE membranes
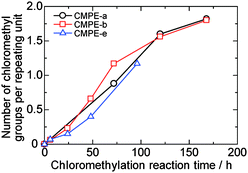
Water uptake and ion conductivity are particularly important properties for fuel cell applications. The in-plane hydroxide ion conductivity of the QPE membranes was measured at various temperatures by a four-probe impedance technique. Since hydroxide ions are easily transformed to carbonate and bicarbonate ions with the trace amount of carbon dioxide in the air,16 the measurement was carried out in degassed deionized water to obtain pure hydroxide ion conductivity. Water uptake was measured according to the previous reports.10,14 As expected, water uptake of the QPE-b membranes increased with increasing IEC. Water uptake of the QPE membranes summarized in Table 3 was reasonable compared with the other AEMs based on the aromatic polymers bearing quaternized ammonio groups (e.g., ca. 95% for IEC = 1.89 meq. g−110a and ca. 115% for IEC = 2.2 meq. g−110b).The hydroxide ion conductivity of the QPE membranes also increased with increasing IEC values. The QPE-b membrane with IEC = 2.54 meq. g−1 showed the highest conductivity of 51 mS cm−1 in water at 30 °C. Our QPE membranes showed very high conductivity compared with the other aromatic AEMs, of which hydroxide ion conductivity ranged from 2.3 to 35 mS cm−1 under similar conditions.10 The high ion conductivity is probably due to the high IECs of the QPE membranes based on the random sulfone-co-ketone structure with fluorenyl groups. The high molecular weights made them insoluble in water even with such high IEC (>2.5 meq. g−1). The QPE-e (IEC = 1.56 meq. g−1) membrane showed ca. twice the hydroxide ion conductivity (43 mS cm−1) than that (23 mS cm−1) of the QPE-b membrane (IEC = 1.88 meq. g−1) despite the former's lower IEC. It is assumed that the perfluorobiphenyl groups of the QPE-e facilitate ion conduction because more water in the membrane would selectively locate near the hydrophilic moieties and accordingly improve the ion conductivity.
Hydroxide ion conductivity of QPE-b (IEC = 1.88 meq. g−1) and QPE-e (IEC = 1.56 meq. g−1) is plotted as a function of temperature in Fig. 3. The hydroxide ion conductivity of QPE membranes showed approximate Arrhenius-type temperature dependency. The apparent activation energy estimated from the slopes was 11.5 and 7.6 kJ mol−1 for QPE-b and QPE-e, respectively. The apparent activation energy of QPE was similar or somewhat lower than those of the other reported AEMs (9.92–23.03 kJ mol−1).10 The results imply that these AEMs share similar hydroxide conducting mechanism involving hydrated anions. The ion conductivity of QPE membranes was also measured in bicarbonate or chloride forms to investigate the effect of ionic species on the conductivity. The conductivities of QPE-b (IEC = 1.88 meq. g−1) were 5.6 (Cl− form) and 4.2 (HCO3− form) mS cm−1 at 40 °C, respectively, which were approximately one fifth of the hydroxide ion conductivity (23 mS cm−1) of the same QPE-b membrane (Fig. 3). The QPE-e membrane also showed lower Cl− (16) and HCO3− (13) conductivity at 40 °C than OH− conductivity (45 mS cm−1 at 40 °C). Taking into account the mobility of each ion species (νOH− = 2.0 × 10−7, νCl− = 7.9 × 10−8, and νHCO3− = 4.6 × 10−8 m2 s−1 V−1 in infinitely diluted aqueous solution at 298 K),17 the differences in the ion conductivities were reasonable. The apparent activation energy of QPE-b (Cl− form), QPE-b (HCO3− form), QPE-e (Cl− form), and QPE-e (HCO3− form) was calculated from the data in Fig. 3 (except at 30 °C) to be 18.5, 21.0, 12.7, and 10.9 kJ mol−1, respectively. The lower apparent activation energies of the QPE-e membranes than those of the QPE-b membranes might be related to the differences in their main chain structure.
 | ||
| Fig. 3 Temperature and counter anion dependence of ion conductivity of QPE-b (IEC = 1.88 meq. g−1) and QPE-e (IEC = 1.56 meq. g−1) membranes bearing OH−, Cl−, and HCO3− anions in water. | ||
Thermal and mechanical properties of the QPE membranes
Thermal properties of QPE-b (IEC = 1.23 meq. g−1), QPE-e (IEC = 1.56 meq.g−1), and the precursor polymers PE-b and CMPE-b were tested by thermogravimetric analyses (TGA) coupled with mass spectroscopy in nitrogen atmosphere. As shown in Fig. 4a, PE-b showed only minor weight loss up to 400 °C. CMPE-b showed initial weight loss around 100 °C with mass numbers of 35 (Cl) and 49 (CH2Cl) m/z, indicating decomposition of the chloromethyl groups (Fig. 4b). Similar degradation with mass numbers of 35 and 49 was also observed above 220 °C. The weight loss of CMPE-b between 60 °C and 340 °C measured by TG was 6.2%, which was in good agreement with the value of chloromethyl groups (6.4%) calculated from the number of chloromethyl groups (0.75 groups per repeating unit, determined by 1H NMR spectrum) and the molecular weights of PE-b (546.62 g per repeating unit mol) and CMPE-b (546.62 + 48.48 × 0.75 = 582.98 g per repeating unit mol). In the TG curve of QPE-b, there were several steps of weight loss. The first step was from r.t. to 100 °C with mass number of 18, which indicates desorption of water from the membrane. The second step of the weight loss was above 160 °C and assignable to the degradation of the ammonio groups as confirmed by the mass spectra and chromatograms. The observed mass numbers were 30 (NHCH3), 58 (CH2N(CH3)2), and 59 (N(CH3)3) m/z. In addition to the ammonio groups, mass number of 64 (SO2) m/z was also observed above 270 °C, suggesting the main chain degradation. The decomposition temperature of the polymer backbone was lower than that of the parent PE-b. It is assumed that the decomposition of the ammonio groups triggered degradation of the polymer backbone. QPE-e showed similar tendency in TG curves where the weight loss was more than that of QPE-b because of the former's higher IEC value (more ammonio groups). All of the QPEs and their precursor polymers did not show any obvious glass transition behavior in the DTA curves.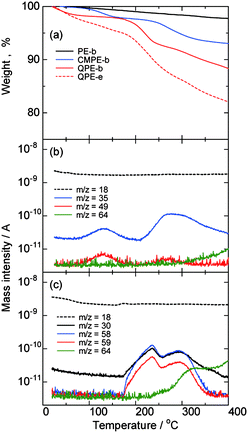 | ||
| Fig. 4 (a) TGA curves of PE-b, CMPE-b, and QPE-b (IEC = 1.23 meq. g−1), and QPE-e (IEC = 1.56 meq. g−1). (b) Mass chromatogram of CMPE-b. (c) Mass chromatograms of QPE-b (IEC = 1.23 meq. g−1). | ||
Tensile strength of QPE-b and QPE-e membranes was measured at 80 °C and 60% RH by a universal testing instrument (Fig. 5). Both membranes were tough with maximum stress higher than 30 MPa and strain lower than 70%, which were much better than those of the conventional PEM, Nafion NRE 212 (Table 4). The mechanical properties of the QPE membranes were more similar to those of our sulfonated poly(arylene ether)s containing fluorenyl groups.13d It is considered that the mechanical properties of the poly(arylene ether) ionomer membranes depend more on the IEC values than the nature of ionic groups.
| Membrane | IEC/meq. g−1 | Maximum stress/MPa | Maximum strain (%) | Young's module/GPa |
|---|---|---|---|---|
| QPE-b | 1.23 | 48.1 | 54 | 0.72 |
| QPE-e | 1.56 | 33.5 | 69 | 0.55 |
| NRE212 | 0.91 | 19.0 | 430 | 0.05 |
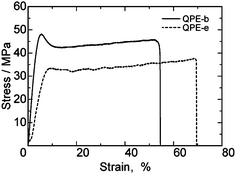 | ||
| Fig. 5 Stress vs. strain curves of QPE-b (IEC = 1.23 meq. g−1) and QPE-e (IEC = 1.56 meq. g−1) membranes at 80 °C and 60% RH. | ||
Durability of the QPE membranes
Durability of the QPE-b membranes of two different IECs (1.88 and 1.23 meq. g−1) was tested in water at 80 °C for 1000 h by immersing the membranes in degassed deionized water in sealed vessels, which were expected to get rid of the effect of contaminants such as carbon dioxide. The hydroxide ion conductivity of the QPE-b (1.88 and 1.23 meq. g−1) membranes are plotted as a function of time in Fig. 6. The conductivities of the membranes increased rapidly during the first several tens of hours, then increased moderately but did not level off. The ion conductivity of QPE-b (IEC = 1.88 meq. g−1) reached 125 mS cm−1 after 1000 h. The increase in the conductivities is considered to be derived from gradual morphological changes of the membranes to form better ionic channels. After 1000 h, the membranes were washed with deionized water and then analyzed by 1H NMR spectra. In the 1H NMR spectra, integral values of the peaks at 4.59 and 2.97 ppm changed while those of aromatic proton remained unchanged. The results suggest that ammonio groups were partly lost from the QPEs and that the polymer main chains were intact. The residual ratio of the ammonio groups in QPE-b (1.88 and 1.23 meq. g−1) after 1000 h durability test was ca. 50% and 42%, respectively. It is still unclear why the ion conductivity increased while the IEC decreased during the 1000 h durability test. More detailed morphological analyses will be needed. In any case, since ammonio group-substituted anion exchange membranes have been known for their low chemical stability under high temperature and hydrolytic conditions,3 improving the durability of the QPE membranes is our prior future objective.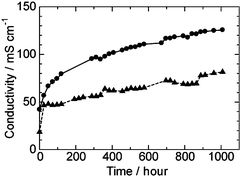 | ||
| Fig. 6 Time dependence of ion conductivity of QPE-b (IEC = 1.88 (●) and 1.23 (▲) meq. g−1) membranes in pure water at 80 °C. | ||
Experimental
Materials
4,4′-Difluorophenyl sulfone (FPS) and 4,4′-difluorobenzophenone were purchased from TCI Inc. and crystallized from toluene prior to use. 9,9-Bis(4-hydroxyphenyl)fluorene (BHF), 3,5-difluorobenzonitrile, and decafluorobiphenyl (TCI Inc.) were used as received. Chloromethyl methyl ether (CMME), zinc chloride, tin chloride, trimethylamine aqueous solution (30 wt%), potassium carbonate, sodium hydroxide, sodium bicarbonate, 1,1,2,2-tetrachloroethane, tetrahydrofurane (THF), toluene, methanol, chloroform, acetone, and N,N-dimethylformamide (DMF) were purchased from Kanto Chemical Co. and used as received. N,N-Dimethyl acetamide (DMAc, organic synthesis grade, 99%) was purchased from TCI and dried over 3 Å molecular sieves prior to use.Polymerization
A typical polymerization procedure (synthesis of PE-a) is as follows. A 100 mL round-bottomed flask was charged with FPS (2.7019 g, 10.625 mmol), BHF (3.7230 g, 10.625 mmol), and potassium carbonate (3.6710 g, 26.563 mmol). The reaction was carried out in 80 mL DMAc and 10 mL toluene with Dean–Stark trap. The reaction was maintained at 145 °C for 3 h before the Dean–Stark trap was removed. Then, the temperature was elevated to 160 °C. The reaction was continued for 1 h and light yellow viscous mixture was obtained. About 80 mL of additional DMAc was added to the mixture, which was poured into large excess of hot water. The obtained polymer was washed with deionized water and methanol several times. After collecting with glass filter (G2), the obtained white polymer was dried at 80 °C under vacuum. The polymer was dissolved in chloroform (200 mL), and then 300 mL of acetone were slowly added to the solution to precipitate the polymer with narrow molecular weight distribution. Finally, the polymer was dissolved in DMAc again, and reprecipitated from hot water and washed with deionized water and methanol several times. The product, PE-a, was collected with glass filter (G2) and dried at 80 °C in vacuum oven to remove the solvent.Chloromethylation of poly(arylene ether)s
A typical procedure of chloromethylation reaction is as follows. A 50 mL pressure glass bottle was charged with PE-a (1.0000 g, 1.771 mmol) and 1,1,2,2-tetrachloroethane (20 mL) and set in combinatorial reaction equipment (L-COS Pres., MORITEX Inc.). After stirring the solution at 35 °C for 30 min to dissolve the polymer, 1.0 mL THF solution of zinc chloride (0.2414 g, 1.771 mmol) and CMME (3.5646 g, 44.275 mmol) were added into the mixture. The reaction was carried out at 35 °C for 168 h to obtain yellow viscous mixture. About 20 mL of additional 1,1,2,2-tetrachloroethane were added to the mixture which was poured into large excess of methanol. The obtained polymer was washed with deionized water and methanol several times. After collecting with glass filter (G2), the obtained white polymer, CMPE-a, was dried at 80 °C under vacuum.Membrane preparation and quaternization
The chloromethylated polymer, CMPE (1 g), was dissolved in 1,1,2,2-tetrachloroethane (25 mL) and filtrated with a 0.45 µm membrane filter. The filtrate was cast onto a flat glass plate. Drying the solution at 60 °C overnight gave a 50 ± 5 µm thick, transparent, and tough membrane. The membrane was immersed in 30 wt% trimethylamine aqueous solution at room temperature for 2 days and washed with water several times. The obtained quaternized polymer was immersed in 1 M sodium hydroxide aqueous solution at room temperature for 2 days to convert the counter anion from chloride to hydroxide anion. The obtained quaternized poly(arylene ether)s (QPE) membrane was washed with deionized water several times and stored in a closed vessel filled with deionized water.Measurements
1H NMR spectroscopy, gel permeation chromatography (GPC), water uptake measurement, and hydroxide ion conductivity measurement were carried out as previously reported.14Thermogravimetric analyses (TGA) were carried out using a RIGAKU Thermo Plus TG8120 coupled with mass spectroscopy Thermo Mass under nitrogen atmosphere. Polymer samples, which were dried under air at room temperature in advance, were heated from room temperature to 400 °C at a heating rate of 10 °C min−1. Tensile strength was measured by a universal test machine (AGS-J 500N, Shimadzu) attached with temperature and humidity controllable chamber (Bethel-3A, Toshin Kogyo). Stress versus strain curves were obtained for samples cut into a dumbbell shape (DIN-53504-S3, 35 mm × 6 mm (total) and 12 mm × 2 mm (test area)). Measurement was conducted at 60% RH and 80 °C at a stretching speed of 10 mm min−1.
Long-term durability tests were performed using a closed pressure vessel equipped with a four-probe cell holder at the bottom of the vessel. Membrane sample was immersed in a diluted sodium hydroxide aqueous solution for 12 h prior to use. The membrane was expeditiously washed with deionized water several times, set on the four-probe cell holder, and placed in the vessel filled with deionized water. The vessel was heated at 80 °C and degassed by bubbling nitrogen for at least 1 h in advance, to cover the membrane and the holder. The vessel was carefully capped with a pressure seal so as not to contain air into the vessel. The vessel was set in an oven at 80 °C and the four electrodes were connected to an impedance spectroscopy (Solartron 1255B and 1287, Solartron Inc.). The durability of the membranes with the passage of time was evaluated by measuring the conductivity change every day. After 1000 h durability test, the membrane samples were taken out and evaluated by 1H NMR spectra.
Conclusions
A series of novel high-molecular-weight anion exchange membranes containing fluorenyl groups were synthesized via chloromethylation of poly(arylene ether)s followed by quaternization. In the case of QPE-b bearing a sulfone/ketone structure, up to 1.80 chloromethyl groups were successfully introduced per repeating unit (or per fluorenylidene biphenylene unit) without significant side reactions by careful optimization of the chloromethylation reaction conditions. Quaternization and the subsequent ion exchange reactions were quantitative so that the obtained ionomer membranes had high IEC up to 2.5 meq. g−1. High hydroxide ion conductivity up to 50 mS cm−1 at 30 °C and 78 mS cm−1 at 60 °C was achieved with the membrane of the highest IEC. A QPE-e membrane bearing decafluorobiphenyl moieties showed higher hydroxide ion conductivity than that of the QPE-b membrane with a similar IEC. The effects of counter ions for the ion conductivity were also investigated as a function of temperature: the QPE-e bearing counter ions with lower ion mobility (νOH− > νCl− > νHCO3−) showed lower ion conductivity (45 (OH−) > 16 (Cl−) > 13 (HCO3−) mS cm−1 at 40 °C). Thermal and mechanical properties of the QPE membranes were superior as anion exchange membranes for alkaline fuel cells. Long-term durability of the QPE-b membranes was confirmed by monitoring the conductivity change for 1000 h in hot water. Though some of the ammonium groups were lost during the 1000 h durability test as confirmed by 1H NMR spectra, the conductivities of the QPE-b membranes increased. These results of the new poly(arylene ether) based AEMs make them promising as a membrane for alkaline fuel cells.Acknowledgements
This work was partly supported by the Ministry of Education, Culture, Sports, Science and Technology, Japan, through a Grant-in-Aid for Scientific Research (22760536).References
- (a) W. Vielstich, Handbook of Fuel Cells, Wiley, Chichester, England, 2009 Search PubMed; (b) L. Carrette, K. A. Friedrich and U. Stimming, Fuel Cells, 2001, 1, 5 CrossRef CAS.
- R. Borup, J. Meyers, B. Pivovar, Y.-S. Kim, R. Mukundan, N. Garland, D. Myers, M. Wilson, F. Garzon, D. Wood, P. Zelenay, K. More, K. Stroh, T. Zawodzinski, J. Boncella, J. E. McGrath, M. Inaba, K. Miyatake, M. Hori, K. Ota, Z. Ogumi, S. Miyata, A. Nishikata, Z. Siroma, Y. Uchimoto, K. Yasuda, K. Kimijima and N. Iwashita, Chem. Rev., 2007, 107, 3904 CrossRef CAS.
- (a) J. R. Vorcoe and R. C. T. Slade, Fuel Cells, 2005, 5, 187 CrossRef CAS; (b) T. Xu, J. Membr. Sci., 2005, 263, 1 CrossRef CAS.
- (a) K. Asazawa, T. Sakamoto, S. Yamaguchi, K. Yamada, H. Fujikawa, H. Tanaka and K. Oguro, J. Electrochem. Soc., 2009, 156, B509 CrossRef CAS; (b) S. Lu, J. Pan, A. Huang, L. Zhuang and J. Lu, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 20611 CrossRef CAS.
- (a) Q. H. Zeng, Q. L. Liu, I. Broadwell, A. M. Zhu, Y. Xiong and X. P. Tu, J. Membr. Sci., 2010, 349, 237 CrossRef CAS; (b) N. J. Robertson, H. A. Kostalik, IV, T. J. Clark, P. F. Mutolo, H. D. Abruña and G. W. Coates, J. Am. Chem. Soc., 2010, 132, 3400 CrossRef CAS.
- J. R. Varcoe, R. C. T. Slade and E. L. H. Yee, Chem. Commun., 2006, 1428 RSC.
- Y. Li and T. Xu, J. Appl. Polym. Sci., 2009, 114, 3016 CrossRef CAS.
- M. R. Hibbs, C. H. Fujimoto and C. J. Cornelius, Macromolecules, 2009, 42, 8316 CrossRef CAS.
- G. Wang, Y. Weng, D. Chu, D. Xie and R. Chen, J. Membr. Sci., 2009, 326, 4 CrossRef CAS.
- (a) M. R. Hibbs, M. A. Hickner, T. M. Alam, S. K. McIntyre, C. Y. Fujimoto and C. J. Cornelius, Chem. Mater., 2008, 20, 2566 CrossRef CAS; (b) J. Zhou, M. Unlu, J. A. Vega and P. A. Kohl, J. Power Sources, 2009, 190, 285 CrossRef CAS; (c) G. Wang, Y. Weng, D. Chu, R. Chen and D. Xie, J. Membr. Sci., 2009, 332, 63 CrossRef CAS; (d) J. Wang, Z. Zhao, F. Gong, S. Li and S. Zhang, Macromolecules, 2009, 42, 8711 CrossRef CAS; (e) J. Pan, S. Lu, Y. Li, A. Huan, L. Zhuang and J. Lu, Adv. Funct. Mater., 2010, 20, 312 CrossRef CAS; (f) J. Tan and M. A. Hickner, Macromolecules, 2010, 43, 2349 CrossRef CAS.
- Y. Wu, C. Wu, J. R. Varcoe, S. D. Poynton, T. Wu and Y. Fu, J. Power Sources, 2010, 195, 3069 CrossRef CAS.
- (a) M. A. Hickner and B. S. Pivovar, Fuel Cells, 2005, 5, 213 CrossRef CAS; (b) A. Roy, M. A. Hickner, B. R. Einsla, W. L. Harrison and J. E. McGrath, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 384 CrossRef CAS; (c) K. Matsumoto, T. Higashihara and M. Ueda, Macromolecules, 2009, 42, 1161 CrossRef CAS; (d) Y. Qi, Y. Gao, S. Tian, A. R. Hlil, J. Gaudet, D. Guay and A. S. Hay, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1920 CrossRef CAS.
- (a) M. Aoki, Y. Chikashige, K. Miyatake, H. Uchida and M. Watanabe, Electrochem. Commun., 2006, 8, 1412 CrossRef CAS; (b) K. Miyatake, Y. Chikashige, E. Higuchi and M. Watanabe, J. Am. Chem. Soc., 2007, 129, 3879 CrossRef CAS; (c) K. Miyatake, T. Shimura, T. Mikami and M. Watanabe, Chem. Commun., 2009, 6403 RSC; (d) B. Bae, T. Yoda, K. Miyatake, H. Uchida and M. Watanabe, Angew. Chem., Int. Ed., 2010, 49, 317 CAS.
- M. Tanaka, M. Koike, K. Miyatake and M. Watanabe, Macromolecules, 2010, 43, 2657 CrossRef CAS.
- C. F. Raley Jr, US Pat., 3311602, 1967.
- H. Yanagi and K. Fukuta, ECS Trans., 2008, 16, 257 Search PubMed.
- K. B. Oldham and J. C. Myland, Fundamentals of Electrochemical Science, Academic Press, Tronto, 1993, p. 12 Search PubMed.
| This journal is © The Royal Society of Chemistry 2011 |