 Open Access Article
Open Access ArticlePhotoinduced formation of reversible dye radicals and their impact on super-resolution imaging†
Sebastian
van de Linde
a,
Ivan
Krstić
b,
Thomas
Prisner
b,
Sören
Doose
a,
Mike
Heilemann
c and
Markus
Sauer
*a
aBiotechnology & Biophysics, Julius-Maximilians-University, Am Hubland, 97074, Wuerzburg, Germany. E-mail: m.sauer@uni-wuerzburg.de; Fax: +49-931-888-4509; Tel: +49-931-888-4507
bInstitute for Physical and Theoretical Chemistry, J. W. Goethe-University Frankfurt, Max-von-Laue-Str. 7, 60438, Frankfurt, Germany
cApplied Laser Physics and Laser Spectroscopy and Bielefeld Institute for Biophysics and Nanoscience, Bielefeld University, Universitätsstrasse 25, 33615, Bielefeld, Germany
First published on 10th December 2010
Abstract
Radical ions of organic dyes are highly reactive species and have been studied for decades by transient absorption spectroscopy and pulse radiolysis experiments in oxygen-depleted solution. Here we show by continuous wave EPR, absorption, and fluorescence experiments that the triplet state of rhodamine dyes can be photoreduced by thiols to form stable radical anions in aqueous solution with a lifetime of up to several hours. Our data demonstrate that reduction of the triplet state and photoinduced oxidation of reactive intermediates by oxygen represents a general mechanism for reversible photoswitching of dyes in aqueous thiol-containing solutions highlighting the key role of molecular oxygen for super-resolution fluorescence imaging. Since cells contain the thiol glutathione at millimolar concentrations and reactive oxygen species are formed as side products our findings are of consequence for live cell fluorescence microscopy.
Introduction
Carbon-based free radicals are trivalent compounds with one unpaired electron in an open shell configuration. Radicals play an important role in combustion, atmospheric chemistry, polymerization, plasma chemistry, biochemistry, and many other chemical processes, including human physiology.1 Although generally short-lived due to their reactivity, some organic radicals are relatively stable because of the delocalization of the unpaired electron in a conjugated π system. The first free radical, triphenylmethyl, was prepared by Gomberg more than 100 years ago by homolysis of triphenylmethyl chloride by a metal.2Triphenylmethyl is stable because the free electron is delocalized and the radical center is shielded by three phenyl groups making it difficult for the radical to react with other molecules. Since triphenylmethyl is rather unreactive, a solution of the radical usually survives for several days in the absence of oxygen. Such stable radicals can be easily detected by continuous wave electron paramagnetic resonance (EPR) spectroscopy.3–5Free radicals of organic dyes can be produced in a photoinduced process by excited state reactions with suitable electron donor or acceptor molecules. Rhodamine dyes, for example, are widely used as labels in fluorescence microscopy applications and produce the corresponding non-fluorescent semi-reduced form much more easily than the semi-oxidized form upon photoinduced electron transfer with suitable electron donors.6,7 Free radicals of organic dyes are usually short-lived (micro- to millisecond lifetimes) and are therefore studied by transient absorption spectroscopy and pulse radiolysis experiments in oxygen-depleted solutions.6,8,9 Since the photostability of fluorescent dyes sets detection limits, especially in single-molecule sensitive fluorescence microscopy applications, and ionized dye species are involved in major photobleaching pathways, their controlled formation and prevention attracted considerable interest in recent years.10–14
In most photostability improvement concepts the triplet state of the dye is quenched upon reduction or oxidation by suited electron donors or acceptors and a radical ion is formed. The singlet state of the dye is then restored by a second complementary redox reaction. Controlling the oxidation and reduction pathways through buffer conditions termed ROXS (reducing and oxidizing system) has been used successfully to reduce photobleaching of dyes.15,16 The photoinduced formation of charged intermediates plays also a vital role in photobleaching, photoconversion (e.g. oxidative ‘redding’), and reversible photoswitching of fluorescent proteins.17–21 Furthermore, charged intermediates of fluorophores are likewise the key for several single-molecule-based super-resolution imaging methods.22–26 However, the experimental verification of the involvement of dye radicals in photobleaching pathways and single-molecule photoswitching methods is still challenging due to the high reactivity of free radicals at room temperature in aqueous solution. Driven by the importance of radical formation for reversible photoswitching, photobleaching and photoconversion of fluorescent dyes we performed an EPR, absorption, and fluorescence spectroscopy study starting with the most commonly used rhodamine derivatives (most Alexa Fluor- and ATTO-dyes).
Materials and methods
The switching characteristics of thiazine, oxazine and rhodamine fluorophores were analyzed at the ensemble level using μM concentrated dye solutions containing 10–200 mM β-mercaptoethylamine (MEA-HCl; Sigma) in standard glass cuvettes (50–500 μL; Hellma, Germany). The pH-value was adjusted with KOH. Photoswitching experiments were carried out in a custom-made cuvette holder using laser irradiation at 405 (Cube 405–100C; Coherent, Santa Clara, CA) and 488, 514, 568, and 647 nm provided by an argon-krypton laser (Innova 70C, Coherent) and fluorescence detection by a fiber optic spectrometer (Ocean Optics, USB2000). The argon-krypton laser lines were selected by an acousto-optical transmission filter (AOTF; AAOptics, Paris, France) and optionally combined with 405 nm using a dichroic beam splitter. Absorption spectra were recorded with a standard absorption spectrometer (Lambda 25, PerkinElmer, USA). For recording the radical anionsvia absorption spectroscopy (Fig. 1 and S2, ESI†), 500 μL cuvettes were completely filled with μM dye solution containing MEA. Prior to measurement the cuvette was covered with a small glass slice, sealed with nail polish and completely irradiated for 2–10 min with 100–500 mW cm−2. For photoswitching experiments as shown in Fig. 2 and Fig. 5 50 μL cuvettes containing 50 μL of a μM dye solution were used to ensure that the whole volume was irradiated. Standard fluorescence spectra were recorded with a fluorescence spectrometer (FluoSpec: Cary Eclipse, Varian, USA).ATTO dyes (ATTO 488, ATTO 532, ATTO 565, ATTO 655, ATTO 680) were purchased from ATTO Tec GmbH (Siegen, Germany), Alexa Fluor dyes (Alexa 488, Alexa 532, Alexa 568) as well as Rhodamine 6G and Rhodamine 123 from Invitrogen GmbH (Darmstadt, Germany), and Dy505 and Dy530 from Dyomics GmbH (Jena, Germany). Methylene blue was purchased from Sigma Aldrich (St. Louis, USA).
X-Band (9.54 GHz) cw-EPR measurements were performed at room temperature using a Bruker E500 spectrometer equipped with a TE102 cavity. Experimental parameters were set at 100 kHz modulation frequency, 5 Gauss modulation amplitude (1 Gauss for the rhodamine 6G sample), 1 mW microwave power, 40.96 ms time constant, 40.96 ms conversion time, 1024 points, 60–80 G sweep width, typically 1–4 scans. In order to investigate the lifetime of created radicals, we performed 2D experiments, in which field sweep was kept on x-axis and time was varied on y-axis. Sweep time for individual field sweep was 41 s with usually 50–100 slices in the time domain. The EPR signal was recorded as the first derivative of the absorption signal. The radical lifetime was then determined by fitting the decrease of the signal intensity over time applying an exponential decay function. Individual samples of 20 μL of mixtures containing 30–100 μM dye solution and 5–200 mM MEA were placed into quartz EPR tubes with 1 mm inner diameter and spectra were recorded prior and after irradiation with laser light of appropriate wavelength. To facilitate sample purging with air, the solution was transferred to Eppendorf safe-lock tubes using a Hamilton syringe, flushed briefly with air and returned into the EPR tube. With the purpose of resolving the hyperfine interaction of the stable organic radical induced by illumination of the fluorophore, we reduced the modulation amplitude to 0.1 G, increased the number of accumulated scans to 10, and decreased the sweep width to 30 G.
Subdiffraction fluorescence imaging according to the dSTORM principle was performed as described earlier.25 Briefly, an inverted microscope (Olympus IX-71; Olympus, Japan) in total internal reflection fluorescence (TIRF) configuration equipped with an oil-immersion objective (60×, NA 1.45, Olympus) was used. Wavelengths at 641 nm (Cube 640–100C; Coherent), 488 nm (Sapphire 488 LP; Coherent, USA) and 405 nm (Cube 405–100C; Coherent) were sequentially combined by dichroic mirrors (LaserMUX; AHF Analysentechnik, Germany; HC 410/504/582/669; Semrock, Rochester, NY), and focused onto the back focal plane of the microscope lens. The fluorescence light in the detection path was filtered using a bandpass filter (HQ 535/50 or ET700/75; AHF Analysentechnik), before being projected on an electron-multiplying CCD camera (Andor Ixon DV897; Belfast, Northern Ireland, UK). Additional lenses were used to generate a final resolution of 91 nm per pixel. Reconstructed dSTORM images were generated from widefield microscopy images using the rapid2STORM software (Picoquant, Berlin, Germany). Spots appearing too wide or too elliptical, as well as spots with a fluorescence signal containing less than 500 or more than 3000 photons were discarded from further analysis.
COS-7 and U373 cells were grown at 37 °C and 5% CO2 in DMEM high glucose medium (Gibco) supplemented with 10% FCS (Gibco). Cells were plated in LabTek eight well chambered coverglass (Nunc). After 12–24 h, cells were fixed using 3.7% paraformaldehyde in PBS for 10 min. The fixed cells were washed with PBS, permeabilized (PBS containing 0.5% v/v Triton X-100) for 10 min, and treated with blocking buffer (PBS containing 5% w/v BSA (Bovine Serum Albumin; Sigma) for 30 min. For actin staining (Fig. 3) cells were incubated with Alexa Fluor 488 Phalloidin (A12379; Invitrogen) for 60 min at a concentration of 10−8 mol L−1 followed by three washing steps using PBS containing 0.1% v/v Tween 20 (Sigma). Prior to dSTORM measurements cells were embedded in 0.2 M carbonate buffer supplemented with β-mercaptoethylamine (MEA, Sigma) with a final concentration of 100 mM MEA at pH 8.
Results and discussion
Photoinduced formation of stable rhodamine radicals
In order to understand the photophysics of rhodamine dyes the redox reactivity has to be considered. With reduction potentials varying between E0 = − 0.90 V and E0 = − 0.70 V vs.SCE7 rhodamine derivatives exhibit a pronounced electron affinity and are efficiently quenched by potent electron donors such as tryptophan and guanosine with one-electron oxidation potentials Eox between 0.6 V and 1.3 V versusSCE.6–9,27 As compared to aromatic amines, thiol compounds such as β-mercaptoethylamine (MEA) or glutathione (GSH) are weaker electron donors at neutral pH.28,29 Therefore, thiol (RSH/RS−) concentrations ≥50 mM and pH values >8 are required to noticeably quench the first excited singlet state of rhodamine dyes with a lifetime of a few nanoseconds. The triplet state of rhodamine derivatives, however, exhibits lifetimes in the microsecond range in aqueous buffer6,8,9,27,30 and is therefore already quenched by millimolar concentrations of MEA and GSH at pH ∼ 8. Accordingly, photo-reduction of the rhodamine Alexa Fluor 488 by thiols in aqueous solutions can be described by the scheme shown in Fig. 1. The photoreaction is initiated by intersystem crossing from the excited singlet state to the triplet state. In the presence of molecular oxygen the triplet state is efficiently quenched via energy transfer from the dye triplet to triplet oxygen to produce singlet molecular oxygen.9 The thiol competes with molecular oxygen in triplet quenching, naturally present at 200–250 μM concentration in aqueous solvents at room temperature, forming the thiyl and dye radical (Fig. 1).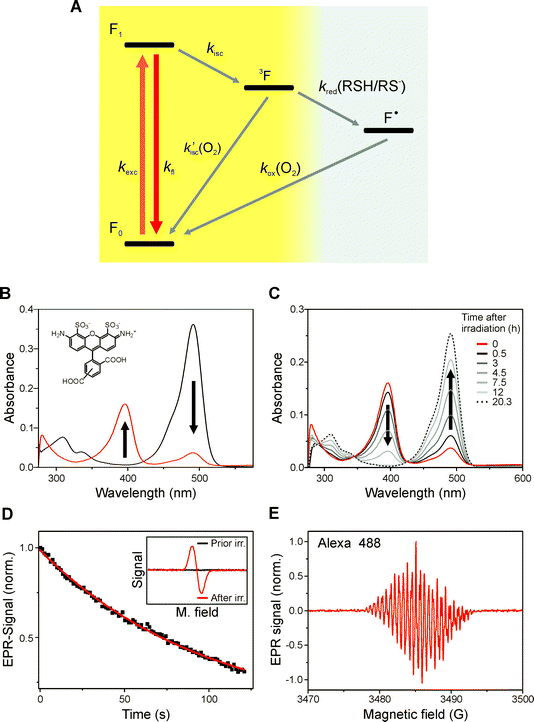 | ||
| Fig. 1 Reaction scheme, absorption, and EPR spectra of stable rhodamine radicals. (A) The dye is excited with rate kexc, fluoresces with kfl, or enters the triplet state (3F) dependent on its intersystem crossing rate kisc. The triplet state of the fluorophore can react with oxygen (k′isc(O2)) to repopulate the singlet ground state F0 and produce singlet oxygen 1O2, or react with the thiol with rate k1red(RSH/RS−) to form the semireduced dye radical (F˙) and the thiyl radical (RS˙). The semireduced dye radical can react with oxygen to repopulate the fluorescent singlet state with rate kox(O2). (B) Absorption spectra of an aqueous Alexa Fluor 488 solution containing 100 mM MEA, pH 9.3 before and after irradiation at 488 nm for 5 min. The radical anion exhibits an absorption maximum at 396 nm with an extinction coefficient of ε = 5.1 × 104 l mol−1 cm−1. (C) Absorption spectra of Alexa Fluor 488 measured in a sealed cuvette with time after photoreduction. The lifetime of the radical anion was determined to be about seven hours. (D, E) EPR spectra and time dependent behavior of the signal of a 10−4 M aqueous solution of Alexa Fluor 488 in the presence of 100 mM MEA, pH 9.3 after irradiation at 488 nm for several minutes. The exponential decay of the EPR signal reveals a radical lifetime of ∼100 min in non-sealed EPR capillary tubes. The pH of the solution was increased to 9.3 to facilitate photoreduction. The radical anion disappears immediately upon purging of the capillary with air. | ||
The produced thiyl radicals (RS˙) scavenge molecular oxygen as well. The main reactions of thiyl radicals in aqueous solution are conjugation with thiols or thiolates (RS˙ + RS− ↔ RSSR˙−), dimerization (RS˙ + RS˙ → RSSR) to the corresponding disulfides or reaction with molecular oxygen (RS˙ + O2 → RSOO˙). The free radical reactions can generate superoxide radicals (RSSR˙− + O2 → RSSR + O2˙−) and hydrogen peroxide (O2˙− + O2˙− + 2H+ → H2O2 + O2).31,32
In summary a consequence of the reaction mechanism is oxygen consumption by ‘quenching’ of the triplet, dye radical, and thiyl radical. Thus, with proceeding irradiation of an aqueous solution of a rhodamine dye in the presence of thiols, oxygen is consumed and the radical anion is formed with an efficiency limited by the amount of residual oxygen present in the sample. Upon re-dissolving oxygen, e.g. by shaking a cuvette with air-filled headspace, the radical anion is immediately oxidized (Fig. 1, S2†).
Irradiation of an aqueous solution of rhodamine dyes in the presence of 100 mM thiols (e.g. MEA or GSH) induces the formation of stable radical anions with lifetimes of minutes to several hours at room temperature in standard non-sealed EPR capillary tubes (Fig. 1D, 1E, and S1). Transient absorption spectroscopy and pulse radiolysis with nitrogen saturated aqueous solutions (pH 7) already showed that the radical anion of different rhodamine dyes exhibit absorption maxima at 380–430 nm with an extinction coefficient of 4.2 × 104 l mol−1 cm−1 at 420 nm (as measured for rhodamine B) and a lifetime of several tens of milliseconds.6,9,27 We now found that all rhodamine dyes investigated in this study (Alexa Fluor 488, Alexa Fluor 532, Alexa Fluor 568, Rh 123, Rh 6G, Dy 505, Dy 530, ATTO 488, ATTO 532, ATTO 565) form stable radical anions upon irradiation in the presence of thiols with lifetimes prolonged by three to six orders of magnitude and absorbing at 380–430 nm (Table 1, Fig. 1 and S1–S3). The absorption maxima, extinction coefficients and EPR spectra with partially resolved hyperfine interaction unequivocally demonstrate the formation of stable dye radicals in aqueous solution. This result is intriguing and surprising at the same time since rhodamine dyes have been investigated extensively for decades without any study having uncovered the formation of such stable dye radicals.
| Dye | λ max/nm | ε/L mol−1 cm−1 | λ rad/nm | ε rad/L mol−1 cm−1 |
|---|---|---|---|---|
| Values were determined from data shown in Fig. 2 and S2 and do not consider reversible follow up reactions. | ||||
| Alexa Fluor 488 | 491 | 71000 | 396 | 51000 |
| Dy 505 | 500 | 80000 | 386 | 33000 |
| Rhodamine 123 | 500 | 101000 | 386 | 42000 |
| ATTO 488 | 501 | 90000 | 400 | 47000 |
| Rhodamine 6G | 526 | 105000 | 422 | 38000 |
| ATTO 532 | 532 | 115000 | 420 | 53000 |
| ATTO 565 | 564 | 120000 | 432 | 28000 |
The process of photo-reduction (by irradiation in the presence of thiols) and oxidation (by re-dissolving molecular oxygen) is reversible and can be repeated over many cycles for most rhodamine dyes with only minimal irreversible photodestruction (Fig. 2). In addition, photo-excitation of the radical anion at 405 nm induces recovery of the fluorescent form without any oxygen being required (Fig. 2). The production and quenching of dye radicals can thus occur in a photoinduced process and be related to a fluorescent off and on state. The first order rate constants for off- and on-switching (koff and kon, respectively) follow a linear dependence on the excitation intensities. While koff is controlled by the singlet-state excitation intensity (for Alexa 488 at 488 nm), the intersystem crossing yield, and the concentration of thiolate, kon is determined by the radical excitation intensity (for Alexa Fluor 488 at 405 nm) and the concentration of molecular oxygen.
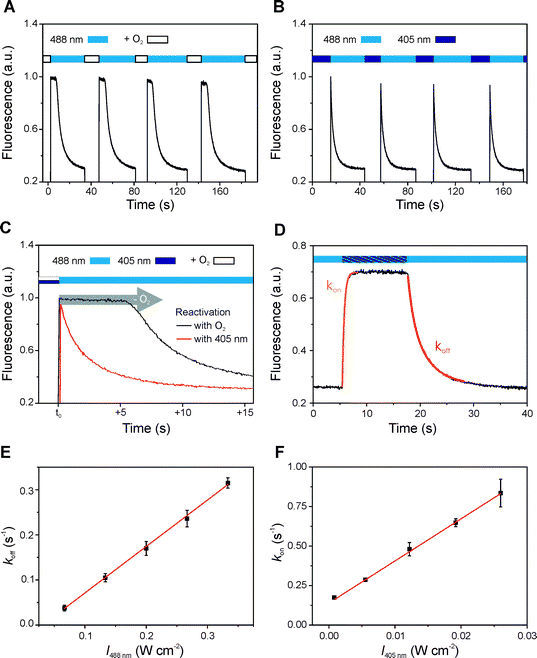 | ||
| Fig. 2 Reversible photoswitching of rhodamine dyes. Alexa Fluor 488 in the presence of 100 mM MEA at pH 7.4 (A–D) and pH 9.3 (E, F). (A) Irradiation of Alexa Fluor 488 at 488 nm with an excitation intensity of 650 mW cm−2. The fluorescence was recovered by gently agitating the solution to dissolve oxygen from the headspace of the cuvette. (B) Alternating irradiation at 488 nm (650 mW cm−2) and 405 nm (50 mW cm−2) without agitating, i.e. without oxygen balancing. (C) Direct comparison of the fluorescence decrease measured in the presence (black) and absence (red) of additional oxygen dissolved from the headspace. The delay in fluorescence decrease demonstrates that oxygen first has to be consumed before stable rhodamine radicals are formed. (D, E) To extract the photoswitching rate koff the cuvette was irradiated at 488 nm and the decrease in fluorescence intensity was monitored between 520–550 nm. The decay curve can be approximated by a monoexponential function. (D, F) The photoswitching rate kon was obtained by simultaneously irradiating the sample at 405 nm and at 488 nm (135 mW cm−2) fitting the increase in fluorescence intensity by an exponential growth model. koff and kon show a linear dependence on excitation intensity in the range of 60–340 and 1–28 mW cm−2, respectively. Error bars represent standard deviations of 5 measurements. | ||
Since the photoswitching rates and lifetimes of the underlying fluorescent and non-fluorescent radical state can be fine adjusted by the irradiation intensity of two different laser beams, stable dye radicals are of great significance for super-resolution fluorescence imaging based on single-molecule localization (Fig. 3).22–26 Fluorescence intermittency with stable and tunable off-state duration is a key requirement in super-resolution imaging and the ratio of the two photoswitching rates determines the achievable optical resolution.33,34 Excitation of the radical anion offers an elegant way to shorten the lifetime of the off-state, whereas reduction of the oxygen concentration of the sample, using, for example, an enzymatic oxygen scavenging system, prolongs the lifetime of the off-state. Tuning these parameters allows arbitrary control of the off-state duration from microseconds to minutes over several orders of magnitude.
 | ||
| Fig. 3 The effect of direct excitation of rhodamine radical anions. Super-resolution imaging of actin filaments in U373 cells with Alexa Fluor 488 phalloidin according to the dSTORM concept25,37. Photoswitching of Alexa Fluor 488 was performed in 100 mM MEA (pH 8.0) at room temperature exciting at 488 nm with an excitation intensity of 5 kW cm−2 and a frame rate of 60 Hz. (A–C) Demonstration of the influence of additional excitation at 405 nm with 0.1 kW cm−2 on the number of rhodamine dyes residing in their fluorescent on-state. (D) Reconstructed dSTORM image. The lower left part shows the conventional widefield image to emphasize the improvement in resolution. | ||
Photoreduction of other dyes
Radical formation was further observed for oxazine and thiazine dyes. Whereas rhodamine dyes are trapped in their radical state in the presence of reducing thiols, thiazine and oxazine dyes such as methylene blue (MB) and ATTO 655 can accept a second electron to form the colorless leuco-dye due to their higher lying one-electron reduction potentials of ERed = −0.30 V, and −0.42 V, respectively, measured versusSCE.7,8 This is corroborated by electrochemical studies which indicate that the semireduced radical of MB and ATTO 655 is unlikely to be oxidized by molecular oxygen (ERed < −0.44 V vs.SCE).35 Therefore, the semireduced radicals of MB and ATTO 655 tend to accept a second electron forming the leuco-dye. The leuco-dye remains stable in oxygen-depleted environments (Fig. 4, S4, and S5).![Reaction scheme describing photoreduction of oxazine and thiazine dyes by thiols in aqueous solution. (A) The fluorophore is excited with rate kexc, fluoresces with kfl, or enters the triplet state (3F) dependent on its intersystem crossing rate kisc. The triplet state of the fluorophore can react with oxygen (k′isc(O2)) to repopulate the singlet ground state F0 and produce singlet oxygen 1O2, or react with the thiol with rate k1red(RSH/RS−) (likely via a charge-transfer intermediate (F–RSH/RS−)) to form the semireduced fluorophore radical (F˙) and the thiyl radical (RS˙). In aqueous solvents radical ions are efficiently solvated and escape rapid recombination. The semireduced dye radical can either accept a second electron to form the corresponding leuco-dye with rate k2red(RSH/RS−) or react with oxygen to repopulate the fluorescent singlet state with rate k1ox(O2) producing superoxide radicals (O2˙−) and hydrogen peroxide (O2˙− + O2˙− + 2H+ → H2O2 + O2). Finally, the leuco-dye can react with oxygen with rate k2ox(O2) to restore the colored form. (B) EPR experiments of the colored (F) and non-colored leuco-dye (FH) in the presence of high thiol concentrations show no signature of unpaired electrons. (C) On the other hand, applying a low RSH/RS− and relatively high dye concentration (5 mM MEA and 5 × 10−4 M ATTO 655) at pH 7.4 corresponding to a reducer to dye ([RS−]/[F]) ratio of 0.1–1, the steady-state concentration of the intermediate radical is just high enough to be detectable by EPR. (D) The radical anion disappears immediately upon purging of the capillary with air.](/image/article/2011/PP/c0pp00317d/c0pp00317d-f4.gif) | ||
| Fig. 4 Reaction scheme describing photoreduction of oxazine and thiazine dyes by thiols in aqueous solution. (A) The fluorophore is excited with rate kexc, fluoresces with kfl, or enters the triplet state (3F) dependent on its intersystem crossing rate kisc. The triplet state of the fluorophore can react with oxygen (k′isc(O2)) to repopulate the singlet ground state F0 and produce singlet oxygen 1O2, or react with the thiol with rate k1red(RSH/RS−) (likely via a charge-transfer intermediate (F–RSH/RS−)) to form the semireduced fluorophore radical (F˙) and the thiyl radical (RS˙). In aqueous solvents radical ions are efficiently solvated and escape rapid recombination. The semireduced dye radical can either accept a second electron to form the corresponding leuco-dye with rate k2red(RSH/RS−) or react with oxygen to repopulate the fluorescent singlet state with rate k1ox(O2) producing superoxide radicals (O2˙−) and hydrogen peroxide (O2˙− + O2˙− + 2H+ → H2O2 + O2). Finally, the leuco-dye can react with oxygen with rate k2ox(O2) to restore the colored form. (B) EPR experiments of the colored (F) and non-colored leuco-dye (FH) in the presence of high thiol concentrations show no signature of unpaired electrons. (C) On the other hand, applying a low RSH/RS− and relatively high dye concentration (5 mM MEA and 5 × 10−4 M ATTO 655) at pH 7.4 corresponding to a reducer to dye ([RS−]/[F]) ratio of 0.1–1, the steady-state concentration of the intermediate radical is just high enough to be detectable by EPR. (D) The radical anion disappears immediately upon purging of the capillary with air. | ||
The classical analogue of this photoinduced reversible formation of a colorless leuco-dye is the so-called ‘blue bottle' experiment.36 It comprises the thiazine dye MB dissolved in an aqueous alkaline glucose solution in a sealed bottle with reasonable headspace, so that when shaken the solution is thoroughly aerated. Due to reduction of MB to its reduced form (leuco-MB) by deprotonated glucose (or thiols), the solution turns colorless and turns blue again when shaken due to oxidation of the leuco-dye by re-dissolved oxygen from the headspace (Fig. S6, ESI†).
Using high thiolate concentrations, two-step photo-reduction proceeds very efficiently and the semireduced dye intermediate cannot be detected by continuous wave (cw) EPR spectroscopy. However, applying a low thiol to dye ratio ([RSH/RS−]/[F]) in the range of 1–10 corresponding to a reducer to dye ([RS−]/[F]) ratio of 0.1–1 at pH 7.4,31 conditions can be established that ensure the formation of a substantial steady-state concentration of semireduced dye radicals (Fig. 4, S4, and S5). Furthermore, in the case of the oxazine dye ATTO 655 the number of dyes residing in the fluorescent state can be substantially increased by additional irradiation at 405 nm corroborating the involvement of radical anions absorbing at ∼400 nm (Fig. 5).
 | ||
| Fig. 5 Light-induced reversible photoswitching of ATTO 655. (A, B) Upon irradiation of an aqueous solution of ATTO 655 at 647 nm (200 mW cm−2) in the presence of 100 mM MEA, pH 9.3 the non-fluorescent form is formed almost quantitatively within seconds. The proceeding reaction is accompanied by oxygen consumption. Upon agitation of the cuvette (marked by a gray bar), fresh oxygen is dissolved from the headspace and the singlet state is restored efficiently because oxygen oxidizes both, leuco-dye as well as radical anion (see right peak at ∼320 s in (A)). About 30% of the colored fluorescent form can be recovered upon irradiation of the sample at 405 nm (330 mW cm−2) without dissolving additional oxygen (marked by a blue bar). This implies that irradiation at 405 nm induces photooxidation of radical anions and disaranges the equilibrium between the three states colored dye, radical anion, and leuco-dye. Measurements were performed in microcuvettes with a sample volume of ∼50 μl irradiating the entire sample volume to minimize errors due to diffusion processes. (C) The effect of photoinduced restoration for ATTO 655 can also be observed at the single-molecule level (10 mM MEA, pH 7.4). In these experiments microtubulin filaments of a fixed COS-7 cell were labeled with ATTO 655 labeled antibodies. The widefield fluorescence images measured in the absence of additional irradiation at 405 nm show that the majority of dyes has been transferred to the off state. Only a few dyes reside in their fluorescent form. Upon additional irradiation at 405 nm the number of dyes (molecules) residing in the fluorescent state can be substantially increased in dSTORM experiments (D); a single frame corresponds to 37.5 ms. | ||
The finding of the involvement of radicals in reversible photoswitching of rhodamine, oxazine and thiazine dyes prompts us to reconsider reversible photoswitching of cyanine dyes in the presence of thiols and oxygen scavengers.19,23,37 In contrast to rhodamine dyes, cyanine dyes are less prone to photo-reduction7 and it is rational to presume that radical anions, intermediately formed upon irradiation of a thiol-containing aqueous dye solution, will be immediately quenched by molecular oxygen. If the oxygen concentration is reduced, e.g. by an enzymatic scavenging system, photo-reduction is facilitated since both, triplet state and the resulting semireduced radical anion exhibit longer lifetimes. It has previously been observed that the fluorescent state of cyanine dyes can be restored in a photoinduced process upon irradiation of what could be the intermediate radical at ∼500 nm.19,23,37 The fact that radical anions of structurally related cyanine dyes with similar length of the polymethine chain show strong absorption at shorter wavelengths with an absorption maximum at ∼500 nm38 strongly suggests the involvement of radical species in fluorescence experiments with cyanine dyes. Similar to the photoinduced leuco-dye formation of thiazine and oxazine dyes, a follow up reaction of photo-reduction of the triplet state could involve thiol-adduct formation to the polymethine bridge of the dye, as was recently observed by mass spectroscopy.39
Consequences for photoswitching and live cell fluorescence microscopy
The reaction mechanism unravels that the role of molecular oxygen in dye photophysics is twofold14 because it quenches not only the triplet state but likewise other reactive species such as radical anions and leuco-dyes. This finding demonstrates that molecular oxygen is essential for super-resolution imaging based on single-molecule localization. This is rationalized by two simple facts: first, in the absence of oxygen dyes can be trapped in reduced states and second, the triplet state can be quenched in the absence of oxygen by thiols or other reducing components. At the ensemble level the fluorescence intensity will therefore decrease exponentially upon irradiation in the absence of oxygen, whereas the fluorescence decrease is delayed in the presence of oxygen because oxygen first has to be consumed (Fig. 2C). That is, in the absence of oxygen the triplet state is substantially prolonged and quenched by the thiol. In other words, in the absence of oxygen and presence of millimolar thiol concentrations the number of fluorescence photons detected from a single dye molecule before it enters the stable radical anion state is solely determined by its intersystem crossing yield. With intersystem crossing yields of 0.2–1%6,8,9,27,30 single rhodamine dyes would emit at best 500 fluorescence photons before being reduced by the thiol. Hence, only ∼25–50 photons would be detected from a single molecule assuming an overall detection efficiency of ∼5–10% rendering precise single-molecule localization difficult. In other words, a minimum amount of oxygen is required to ensure the detection of enough fluorescence photons from the on state of single emitters before they enter a stable off state. These findings demonstrate that the triplet state of organic dyes has to be considered as part of the ‘on state’ in super-resolution imaging based on single-molecule localization whereas the off state is the charged radical or leuco-dye state.Furthermore, our data demonstrate that photoinduced dye radical formation by thiols is accompanied by thiyl radical formation and subsequent production of reactive oxygen species such as superoxide radicals and hydrogen peroxide. Since the tripeptide glutathione is the most abundant low-molecular-weight thiol protectant and antioxidant in mammalian biology and kept in a reduced state at millimolar concentration in animal cells25,40 and oxygen is naturally present, live-cell super-resolution imaging is feasible with rhodamine and oxazine dyes in combination with suitable chemical tags.41 However, whereas thiols and oxygen enable reversible photoswitching of dyes in living cells, our data also clearly indicate that cellular damage has to be considered in all live cell fluorescence microscopy experiments. The extent of cell damage might be negligible for low irradiation intensities and dye concentrations corresponding to low radical concentrations but certainly induces cell death with increasing concentration of fluorescent labels especially in combination with high irradiation intensities. The role of photoinduced radical and reactive oxygen species formation in live cell fluorescence microscopy with fluorescent proteins cannot be answered accurately yet. But the increasing number of reports demonstrating the involvement of charged species in photoswitching and photoconversion of fluorescent proteins21 supposes that related reaction mechanisms and consequences also have to be considered.
Conclusions
The accomplished spectroscopic study has revealed several unforeseen important results. First, the triplet state of rhodamine dyes can be reduced by thiols to form stable radical anions in aqueous solution with lifetimes of up to several hours. In case of oxazine and thiazine dyes with higher electron affinity the semireduced radical anion represents an intermediate that can be further reduced to the leuco-dye by two electron or hydrogen atom transfer steps. The lifetimes of the radical anions can be controlled by both, the oxygen concentration and direct excitation. Second, our data demonstrate that the reducing environment of cells induces cell damage in fluorescence microscopy especially at high dye concentrations under high irradiation intensities. Third, the reaction mechanism for radical anion formation and destruction involves several very efficient oxygen consuming steps. That is, an aqueous sample containing millimolar concentrations of thiols and oxazine, thiazine, or rhodamine dyes removes oxygen efficiently upon irradiation. Consequently, the sample could also be used as oxygen sensor announcing the presence of oxygen in an immediate color change. Furthermore, the interdependence between formation of stable dye radicals and oxygen depletion suggests a broad range of applications including production of reactive oxygen species as substantial ingredients in photodynamic therapy, or distance measurements by EPR based on radical spin interactions. Stable radicals might also serve as charge carriers in optoelectronic applications.Acknowledgements
We thank K. H. Drexhage for helpful discussion. This work was supported by the Biophotonics and the Systems Biology Initiative (FORSYS) of the German Ministry of Research and Education (BMBF, grants 13N9234 and 0315262).References
- P. Pacher, J. S. Beckman and L. Liaudet, Nitric oxide and peroxynitrite in health and disease, Physiol. Rev., 2007, 87, 315–424 CrossRef CAS.
- M. Gomberg, An instance of trivalent carbon triphenylmethyl, J. Am. Chem. Soc., 1900, 22, 757–771 CrossRef.
- G. M. Rosen, B. E. Britigan, H. J. Halpern and S. Pou, Free radicals: biology and detection by spin trapping, Oxford University Press, Oxford 1999 Search PubMed.
- M. D. E. Forbes and Time-Resolved, (CW) Electron paramagnetic resonance spectroscopy: an overview of the technique and its use in organic photochemistry, Photochem. Photobiol., 1997, 65, 73–81 CrossRef CAS.
- G. Grampp, et al., ESR and ENDOR investigations on various Wurster's radical cations in solution. Experimental results, theoretical ab initio, and DFT calculations, Monatsh. Chem., 2005, 136, 519–536 CrossRef.
- P. C. Beaumont, D. G. Johnson and B. J. J. Parsons, Excited state and free radical properties of rhodamine dyes in aqueous solution: A laser flash photolysis and pulse radiolysis study, J. Photochem. Photobiol., A, 1997, 107, 175–183 CrossRef CAS.
- S. Doose, H. Neuweiler and M. Sauer, Fluorescence quenching by photoinduced electron transfer: a reporter for conformational dynamics of macromolecules, ChemPhysChem, 2009, 10, 1389–1398 CrossRef CAS.
- R. H. Kayser, R. Young and The, photoreduction of methylene blue by amines - i. a flash photolysis study of the reaction between triplet methylene blue and amines, Photochem. Photobiol., 1976, 24, 395–401 CAS.
- H. Görner, Oxygen uptake induced by electron transfer from donors to the triplet state of methylene blue and xanthene dyes in air-saturated aqueous solution, Photochem, Photochem. Photobiol. Sci., 2008, 7, 371–376 RSC.
- H. Giloh and J. W. Sedat, Fluorescence microscopy: reduced photobleaching of rhodamine and fluorescein protein conjugates by n-propyl gallate, Science, 1982, 217, 1252–1255 CrossRef CAS.
- R. Zondervan, F. Kulzer, S. B. Orlinski and M. Orrit, Photoblinking of rhodamine 6G in poly(vinyl alcohol): radical dark state formed through the triplet, J. Phys. Chem., 2003, 107, 6770–6776 Search PubMed.
- I. Rasnik, S. A. McKinney and T. Ha, Nonblinking and long-lasting single-molecule fluorescence imaging, Nat. Methods, 2006, 3, 891–893 CrossRef CAS.
- J. Widengren, A. Chmyrov, C. Eggeling, P. A. Löfdahl and C. A. M. Seidel, Strategies to improve photostabilities in ultrasensitive fluorescence spectroscopy, J. Phys. Chem. A, 2007, 111, 429–440 CrossRef CAS.
- M. Orrit, Chemical and physical aspects of charge transfer in the fluorescence intermittency of single molecules and quantum dots, Photochem. Photobiol. Sci., 2010, 9, 637–642 RSC.
- J. Vogelsang, R. Kasper, B. Person, M. Heilemann, M. Sauer and P. Tinnefeld, A reducing and oxidizing system minimizes photobleaching and blinking of fluorescent dyes, Angew. Chem., Int. Ed., 2008, 47, 5465–5469 CrossRef CAS.
- R. Kasper, B. Harke, C. Forthmann, P. Tinnefeld, S. W. Hell and M. Sauer, Single-molecule STED microscopy with photostable organic fluorophores, Small, 2010, 6, 1379–1384 CrossRef.
- J. Lippincott and G. H. Patterson, Development and use of fluorescent protein markers in living cells, Science, 2003, 300, 87–91 CrossRef.
- N. C. Shaner, G. H. Patterson and M. W. Davidson, Advances in fluorescent protein technology, J. Cell Sci., 2007, 120, 4247–4260 CrossRef CAS.
- M. Heilemann, P. Dedecker, J. Hofkens and M. Sauer, Photoswitches: key molecules for subdiffraction-resolution fluorescence imaging and molecular quantification, Laser Photonics Rev., 2009, 3, 180–202 Search PubMed.
- M. B. Elowitz, M. G. Surette, P. E. Wolf, J. Stock and S. Leibler, Curr. Biol., 1997, 7, 809–812 CrossRef CAS.
- A. M. Bogdanov, A. S. Mishin, I. V. Yampolsky, V. V. Belousov, D. M. Chudakov, F. V. Subach, V. V. Verkhusa, S. Lukyanov and K. A. Lukyanov, Green fluorescent proteins are light-induced electron donors, Nat. Chem. Biol., 2009, 5, 459–461 CrossRef CAS.
- S. W. Hell, Far-field optical nanoscopy, Science, 2007, 316, 1153–1158 CrossRef CAS.
- M. J. Rust, M. Bates and X. Zhuang, Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM), Nat. Methods, 2006, 3, 793–796 CrossRef CAS.
- E. Betzig, G. H. Patterson, R. Sougrat, O. W. Lidwasser, S. Olenych, J. S. Bonifacino, M. W. Davidson, J. Lippincott-Schwartz and H. F. Hess, Imaging intracellular fluorescent proteins at nanometer resolution, Science, 2006, 313, 1642–1645 CAS.
- M. Heilemann, S. van de Linde, A. Mukherjee and M. Sauer, Super-resolution imaging with small organic fluorophores, Angew. Chem., Int. Ed., 2009, 48, 6903–6908 CrossRef CAS.
- J. Vogelsang, T. Cordes, C. Forthmann, C. Steinhauer and P. Tinnefeld, Controlling the fluorescence of ordinary oxazine dyes for single-molecule switching and superresolution microscopy, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 8107–8112 CrossRef CAS.
- E. Klimtchuk, M. A. J. Rodgers and D. C. Neckers, Laser flash photolysis studies of novel xanthene dye derivatives, J. Phys. Chem., 1992, 96, 9817–9820 CrossRef CAS.
- E. Madej and P. Wardman, The oxidizing power of the glutathione thiyl radical as measured by its electrode potential at physiological pH, Arch. Biochem. Biophys., 2007, 462, 94–102 CrossRef CAS.
- P. Wardmann, Reduction potentials of one-electron couples involving free radicals in aqueous solution, J. Phys. Chem. Ref. Data, 1989, 18, 1637–1755 CrossRef CAS.
- R. Menzel and E. Thiel, Intersystem crossing rate constants of rhodamine dyes: influence of the amino-group substitution, Chem. Phys. Lett., 1998, 291, 237–243 CrossRef CAS.
- U. M. Burner, W. Jantschko and C. Obinger, Kinetics of oxidation of aliphatic and aromatic thiols by myeloperoxidase compounds I and II, FEBS Lett., 1999, 443, 290–296 CrossRef CAS.
- U. Burner and C. Obinger, Transient-state and steady-state kinetics of the oxidation of aliphatic and aromatic thiols by horseradish peroxidise, FEBS Lett., 1997, 411, 269–274 CrossRef CAS.
- H. Shroff, C. G. Galbraith, J. A. Galbraith and E. Betzig, Live-cell photoactivated localization microscopy of nanoscale adhesion dynamics, Nat. Methods, 2008, 5, 417–423 CrossRef CAS.
- S. van de Linde, S. Wolter, M. Heilemann and M. Sauer, The effect of photoswitching kinetics and labeling densities on super-resolution fluorescence imaging, J. Biotechnol., 2010, 149, 260–266 CrossRef CAS.
- D. T. Sawyer and J. S. Valentine, How super is superoxide?, Acc. Chem. Res., 1981, 14, 393–400 CrossRef CAS.
- J. A. Campbell, Kinetics - Early and often, J. Chem. Educ., 1963, 40, 578–583 CrossRef CAS.
- M. Heilemann, S. van de Linde, M. Schüttpelz, R. Kasper, B. Seefeldt, A. Mukherjee, P. Tinnefeld and M. Sauer, Subdiffraction-resolution fluorescence imaging with conventional fluorescent probes, Angew. Chem., Int. Ed., 2008, 47, 6172–6176 CrossRef CAS.
- J. R. Lenhard and A. D. Cameron, Electrochemistry and electronic spectra of cyanine dye radicals in acetonitrile, J. Phys. Chem., 1993, 97, 4916–4925 CrossRef CAS.
- G. T. Dempsey, M. Bates, W. E. Kowtoniuk, D. R. Liu, R. Y. Tsien and X. Zhuang, Photoswitching mechanism of cyanine dyes, J. Am. Chem. Soc., 2009, 131, 18192–18193 CrossRef CAS.
- H. Sies, Glutathione and its role in cellular functions, Free Radical Biol. Med., 1999, 27, 916–921 CrossRef CAS.
- R. Wombacher, M. Heidbreder, S. van de Linde, M. P. Sheetz, M. Heilemann, V. W. Cornish and M. Sauer, Live-cell super-resolution imaging with trimethoprim conjugates, Nat. Methods, 2010, 7, 717–719 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S6. See DOI: 10.1039/c0pp00317d |
| This journal is © The Royal Society of Chemistry and Owner Societies 2011 |