Chemoenzymatic synthesis of a mixed phosphine–phosphine oxide catalyst and its application to asymmetric allylation of aldehydes and hydrogenation of alkenes†
Derek R.
Boyd
*a,
Mark
Bell
a,
Katherine S.
Dunne
b,
Brian
Kelly
b,
Paul J.
Stevenson
*a,
John F.
Malone
a and
Christopher C. R.
Allen
c
aSchool of Chemistry and Chemical Engineering, Queen's University, Belfast, BT9 5AG. E-mail: dr.boyd@qub.ac.uk; Fax: (+44)-28-9097-4687; Tel: (+44)-28-9097-4421
bCeltic Catalysis Ltd., Nova Centre, Belfield Innovation Park, Dublin 4, Ireland
cSchool of Biological Sciences, Queen's University, Belfast, BT9 5AG, UK
First published on 16th November 2011
Abstract
The chemoenzymatic synthesis of a Lewis basic phosphine–phosphine oxide organocatalyst from a cis-dihydrodiol metabolite of bromobenzene proceeds via a palladium-catalysed carbon–phosphorus bond coupling and a novel room temperature Arbuzov [2,3]-sigmatropic rearrangement of an allylic diphenylphosphinite. Allylation of aromatic aldehydes were catalysed by the Lewis basic organocatalyst giving homoallylic alcohols in up to 57% ee. This compound also functioned as a ligand for rhodium-catalysed asymmetric hydrogenation of acetamidoacrylate giving reduction products with ee values of up to 84%.
1 Introduction
The enzyme-catalysed cis-dihydroxylation of arenes using toluene dioxygenase-expressing mutant strains of the bacterium Pseudomonas putida, e.g., UV4 or 39/D, or E. coli recombinant strains, e.g., JM109(pDTG601), JM109(pKST11) or CL-4t (Scheme 1) has been widely investigated. The resulting enantiopure cis-dihydrodiol metabolites have proved to be useful additions to the chiral pool with several hundred having been reported to date.1 While these cis-diols continue to be widely used in the synthesis of natural and unnatural products,2 their application in the context of chiral organocatalysis or chiral ligand formation and asymmetric synthesis has received much less attention. In this context, we have recently shown how cis-dihydrodiol metabolites derived from substituted quinolines, by using bacterial whole cells containing different types of dioxygenases, can be used in the synthesis of chiral 2,2′-bipyridines,3 2,2′-bipyridine N-oxides4 and 4,4′-bipyridines.5 These chiral 2,2-bipyridines have then proved to be useful ligands in asymmetric allylic oxidations (→ 97% ee), cyclopropanations (→ 95% ee) and amminolysis of meso-epoxides (→ 84% ee). The corresponding 2,2′-bipyridine N-oxides were also found to act as organocatalysts in the asymmetric allylation of aldehydes (→ 86% ee).
The cis-dihydrodiol metabolites obtained from the biotransformation of monocyclic arenes with Pseudomonas putida UV4 in particular, are very suitable for use as precursors of chiral ligands, as they are generally obtained with very high enantiomeric excess values (>98% in the case of bromobenzene 1) and are perfectly functionalised for further chemistry. Due to the relative instability of the benzene cis-dihydrodiols, e.g., 2, which readily dehydrate to form phenols, an improved approach to partial hydrogenation of the unsubstituted double bond was recently found to give stable cis-tetrahydrodiols, e.g., 3, in good yield (Scheme 1).6 These partially hydrogenated derivatives of monohalogenated benzenes are of particular value where the halogen (e.g., Br or I) can be replaced with a boron, nitrogen or phosphorus atom.7 The current report is a natural extension of our recent programme of installing ligating centres7 and demonstrates that chiral organocatalysts/ligands bearing two phosphorus atoms can also be obtained from cis-tetrahydrodiol 3 and can be applied to asymmetric allylation of aldehydes and to asymmetric alkene hydrogenation reactions.
The use of Lewis basic organocatalysts to activate allyltrichlorosilanes, forming hypervalent silicon intermediates, for the allylation of aldehydes has been known since the pioneering work of Kobayashi in 1993.8 Since the first chiral Lewis bases for use in this reaction were reported by Denmark,9 there has been a large amount of work in developing different chiral Lewis basic systems for reaction of allyltrichlorosilanes including chiral phosphoramides,10 formamides,11N-oxides,12,13 phosphine oxides,14 sulfoxides15 and ureas.16 To date there have been no reports of the use of a chiral mixed-chelating phosphine–phosphine oxide Lewis base as an organocatalyst for this important asymmetric transformation.
2 Results and discussion
Herein we report the synthesis of a novel chiral phosphine–phosphine oxide Lewis basic organocatalyst 7 using the readily available diol 37 as a chiral starting material. This highly functionalised phosphorus compound 7 was then evaluated as an organocatalyst in the asymmetric allylation of aromatic aldehydes with allyltrichlorosilanes and also screened as a chiral ligand for rhodium-catalysed hydrogenation of alkenes.Typically an organocatalyst for facilitating addition of allyltrichlorosilanes to aldehydes uses mixed chelating sites and the installation of each ligating unit must be carried out independently. Compound 3 is an ideal substrate for the preparation of new chiral chelating ligands since there are three adjacent points at which to introduce additional functionality and since it is now readily available. Our group has recently demonstrated that the bromine atom in this substrate can be readily substituted with a range of nucleophiles, including phosphorus, under palladium catalysis.7 Knochel has established that a phosphine oxide functionality can be conveniently introduced into chiral cyclic allylic alcohols using an Arbuzov [2,3]-sigmatropic rearrangement17 of the corresponding allylic diphenylphosphinite giving enantiomerically pure products. The initial goal of this study was to combine these two technologies to prepare chiral mixed phosphine–phosphine oxide ligands as potential new catalysts for use in asymmetric transformations. Our initial synthetic approach is outlined in (Scheme 2).
 | ||
| Scheme 2 Reagents and conditions: (i) TBDPSCl, imidazole, DMF (67%); (ii) ClPPh2, DMAP, THF; (iii) toluene, reflux (93%, two steps); (iv) Pd(OAc)2, DPPF, Cs2CO3, HPPh2, toluene, 80 °C. | ||
It is well documented that in crystalline cis-tetrahydrobenzene diols of type 3, the allylic hydroxyl group prefers to occupy a pseudo axial position to minimise allylic 1,2-strain with the bromo substituent.18 Indeed with diol 3 it was confirmed that the homoallylic hydroxyl group was equatorial due to the presence of the large diaxial vicinal coupling constant of 9.3 Hz from H-1 to H-6. Because axial alcohols are in a more crowded position, they are much less reactive than equatorial alcohols in reactions which lead to an increase in steric bulk i.e. reactions in which the hydrogen on the hydroxyl group is replaced with a larger substituent. Consequently, in diol 3 the homoallylic equatorial hydroxyl is more reactive than the pseudo axial allylic hydroxyl. Reaction of diol 3 with the very bulky TBDPSCl silylating agent, and imidazole as base, in DMF resulted in chemoselective protection of the homoallylic hydroxyl group and gave alcohol 4 in 67% yield. Using Knochel's conditions, reaction of allylic alcohol 4 with chlorodiphenylphosphine proceeded smoothly in the presence of DMAP in THF to give diphenylphosphinite 5. Due to potential problems of stability (involving hydrolysis or oxidation), diphenylphosphinite 5 was not isolated or purified. However, 31P-NMR spectroscopy on the crude reaction mixture showed a new peak with chemical shift δP 119.4 ppm indicating that the desired compound had indeed been cleanly formed. Replacement of THF with toluene, as solvent, and subsequent heating at 80 °C for 18 h induced an Arbuzov thermal [2,3]-sigmatropic rearrangement, of the allylic diphenylphosphinite 5, to give the desired phosphine oxide 6 as the cis-diastereoisomer in 93% yield for the two steps. The reaction was conveniently monitored by 31P-NMR spectroscopy and the signal for the diphenylphosphinite 5 at δP 119.4 ppm, was replaced by a new peak for phosphine oxide 6 at δP 32.1 ppm on rearrangement. Assuming that the rearrangement involved a concerted suprafacial process, the stereochemistry at the new C4-chiral centre was tentatively assigned as R, as depicted in (Scheme 2).
Unfortunately, despite extensive experimentation, the desired palladium-catalysed coupling of the cyclohexenyl bromide 6, with diphenyl phosphine was unsuccessful and only starting material was recovered from this reaction. It appears that the combined steric effect of the bulky TBDPS–ether and diphenyl phosphine oxide groups are such that the desired coupling reaction was precluded.
Changing the installation sequence of the two phosphorus components led to a successful alternative synthetic route and this is outlined in Scheme 3. Palladium-catalysed coupling of cycloalkenyl bromide 8 with diphenyl phosphine proceeded efficiently as previously reported to give the phosphine 9.7 Evidence for formation of the unstable phosphine was provided by the 31P-NMR signal at δP −6.9 ppm. However, phosphine 9 was prone to oxidation, difficult to handle, and in the original study this compound was characterised as the corresponding phosphine oxide.7 In the current study, in order to facilitate handling, purification and characterization, compound 9 was reacted with diborane to give the more stable Lewis salt 10. On formation of intermediate 10 the 31P signal changed from being a sharp singlet at δP −6.9 ppm to a very broad multiplet at δP 22.7 ppm due to boron coupling and quadrupolar broadening. Furthermore, the olefinic proton changed chemical shift from δH 5.8 ppm to δH 6.80 ppm, on reaction with diborane, reflecting the fact that the phosphorus substituent was now strongly electron-withdrawing. Acid-catalysed removal of the acetonide gave the cis-tetrahydrodiol 11. 31P-NMR analysis indicated that the boron was still attached to the phosphine. This was shown by the presence of the broad multiplet at δP 19.5 ppm, though the olefinic proton had dropped back to δH 6.12 ppm. Chemoselective protection of the homoallylic hydroxyl group, as before, as a TBDPS–ether, proceeded smoothly and gave compound 12 with the desired free allylic hydroxyl group. Surprisingly, on treatment of allylic alcohol 12 with chlorodiphenylphosphine no 31P signal could be detected for the intermediate allylic diphenylphosphinite 13. Instead, new peaks were observed with chemical shifts at δP 30.5 (sharp) and 24.5 (broad) which were consistent with the phosphine oxide and phosphine–borane signals respectively of compound 14. In this case the Arbuzov [2,3]-sigmatropic rearrangement, to yield phosphine oxide 14, was extremely fast, even at room temperature. This astounding reactivity was in marked contrast to that of diphenylphosphinite 5 where heating at 80 °C for 18 h was required to effect the rearrangement. Similar or harsher conditions were employed by Knochel for comparable rearrangements of allylic diphenylphosphinites.17 It is known that amines, present in the preparation of the diphenylphosphinite, can catalyse Arbuzov [2,3]-sigmatropic rearrangements of strained alkenes.19 However, as diphenylphosphinite 5, (Scheme 2), required heating (80 °C for 18 h) to rearrange, in the presence of amine, it was likely that the catalytic effect of amine was negligible in this case. It seemed more likely that the phosphine borane functionality on the alkene in compound 13 was having a profound influence on the kinetics of the Arbuzov [2,3]-sigmatropic rearrangement. It was known that oxophilic Lewis acids catalyse normal Arbuzov reactions, allowing them to proceed at room temperature, in a process that involves binding to the oxygen atom facilitating cleavage of the carbon oxygen bond.20 More detailed mechanistic studies have confirmed that, in some cases, the mechanism of the catalysed reaction may be radically different to that of the uncatalysed Arbuzov reaction.21 In the current case it was possible that the borane in compound 12 was reversibly transferred to the phosphite oxygen and that this then facilitated the subsequent [2,3]-sigmatropic rearrangement. As there was the possibility of the Arbuzov [2,3]-sigmatropic rearrangement going through an ionic pathway it was important to firmly establish the stereochemistry of the rearrangement. On removal of the TBDPS-protecting group from compound 14, crystals of alcohol 15 were obtained and a single crystal X-ray structure analysis (Fig. 1) provided confirmation of both its gross phosphine–phosphine oxide structure as well as its relative (2,5-cis) and absolute (2R,5S) configuration, including the newly generated C2-chiral centre containing a phosphine oxide. The X-ray crystal structure contained one molecule of ethanol, the crystallisation solvent, which was hydrogen bonded to both the secondary alcohol and the phosphine oxide. It was reassuring that the room temperature Arbuzov [2,3]-sigmatropic rearrangement had the same stereochemical outcome as the high temperature counterpart, which makes it likely that both reactions are proceeding in a concerted fashion.
 | ||
| Scheme 3 Reagents and conditions: (i) 2,2-dimethoxypropane, acetone, PTSA (97%); (ii) Pd(OAc)2, Cs2CO3, toluene, 80 °C, DPPF, HPPh2; (iii) BH3-THF (84%, two steps); (iv) THF/H2O/TFA (66%); (v) TBDPSCl, imidazole, DMF (89%); (vi) ClPPh2, DMAP, THF (80%, two steps); (vii) CH3COCl, MeOH, 6 h 25 °C (89%); (viii) Et2NH (99%). | ||
 | ||
| Fig. 1 Single crystal X-ray structure of mixed phosphine Lewis salt–phosphine oxide 15 displaying hydrogen bonding with crystallisation solvent ethanol. | ||
The borane was removed from the phosphine group in compound 14 by reaction with diethylamine and gave the mixed phosphine–phosphine oxide compound 7. The 31P-NMR spectrum of the resulting sample indicated that it was relatively pure. Thus, the signal for the Lewis salt at δP 24.6 ppm was replaced with a new signal for the free phosphine at δP −4.1 ppm. However, on attempted chromatographic purification additional new signals appeared in the 31P-NMR spectrum so compound 7 was used crude for subsequent catalytic studies.
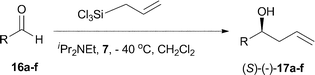
The phosphine–phosphine oxide Lewis basic organocatalyst 7 was evaluated in the asymmetric allylation of aromatic aldehydes 16a–f with allyltrichlorosilane (Scheme 4 and Table 1). Initial reactions using benzaldehyde 16a, allyltrichlorosilane (1.2 eq.), diisopropylethylamine (0.1 eq.) and 10 mol% of the catalyst 7 at −78 °C in dichloromethane yielded no product. Raising the temperature to −40 °C allowed the reaction to proceed, giving the desired homoallylic alcohol in 52% yield and 54% ee (Table 1, entry 1). Increasing the catalyst loading to 15 mol% improved the yield to 73% without greatly affecting the enantiomeric excess (Table 1, entry 2, 57%). Accepting 15 mol% of catalyst in dichloromethane at − 40 °C as standard, attention was turned to a number of other simple aldehydes (16b–f), to test the scope and limitations of the reaction. Although cinnamaldehyde 16b reacted to give the corresponding allylic alcohol 17b in 68% yield, disappointingly the product was racemic (Table 1, entry 3). The use of the more bulky 2-naphthaldehyde 16c gave the desired product 17c in 76% yield albeit with lower enantiomeric excess (40%). The electron deficient p-trifluoromethylbenzaldehyde 16d and p-nitrobenzaldehyde 16e both reacted well giving moderate levels of enantiocontrol (Table 1, entries 5 and 6) to give the corresponding alcohols 17d (45% ee) and 17e (30% ee). When the electron rich p-methoxybenzaldehyde 16f was used, the reaction was slow (34% yield) and the product 17f racemic (Table 1, entry 8).
| Entry | Aldehyde | Product | R | Yield (%)b | ee (%)c |
|---|---|---|---|---|---|
| a The reaction was carried out at 0.25 mmol scale in dichloromethane with 1.2 eq. of trichloroallylsilane, in the presence of the catalyst (15 mol%) for 18 h at −40 °C. b Isolated yield. c Determined by chiral stationary phase HPLC (CSP-HPLC). d (−)-(S)-configuration, established from the optical rotation and comparison with the literature data.12a e 10 mol% catalyst used. | |||||
| 1 | 16a | 17a | Ph | 52e | 54 |
| 2 | 16a | 17a | Ph | 73 | 57 |
| 3 | 16b | 17b | PhCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH CH |
68 | 0 |
| 4 | 16c | 17c | 2-Naphth | 76 | 40 |
| 5 | 16d | 17d | p-CF3–C6H4 | 78 | 45 |
| 6 | 16e | 17e | p-NO2–C6H4 | 81 | 30 |
| 7 | 16f | 17f | p-MeO–C6H4 | 34 | 0 |
 | ||
| Scheme 4 Mixed phosphine–phosphine oxide catalysed addition of allyl trichlorosilane to aldehydes 16a–f. | ||
The maximum ee value obtained when using this first member of a new chiral phosphine–phosphine oxide series 7 as an organocatalyst in the asymmetric allylation of benzaldehyde 16a was relatively modest (57% ee). However, it compares favourably with our earlier results obtained using the same reaction and a range of twelve chiral 2,2′-bipyridine N-oxides derived from quinoline cis-dihydrodiols where ee values were lower (14–35%).4 As these substituted chiral 2,2′-bipyridine organocatalysts proved to be much more stereoselective as chiral ligands (→ 97% ee) in other types of asymmetric reactions,5 so the chiral phosphine–phosphine oxide 7 was tested as a potential chiral ligand for an alternative reaction type i.e. asymmetric hydrogenation.
Among the first industrial scale catalytic asymmetric synthesis reactions was the hydrogenation of alkenes bearing coordinating groups, using a rhodium catalyst and a chiral phosphorus ligand. This discovery was driven by the need to produce L-dopa as a single enantiomer by reduction of acetamidoacrylates using a rhodium catalyst and DIPAMP, a bidentate P-chiral ligand.22,23 This technology remains a key reaction for the formation of enantioenriched functionalised chiral building blocks. However, P-chiral ligands are rather difficult to prepare and phosphorus ligands, both mono- and bi-dentate, with chirality in a carbon backbone are routinely used in asymmetric hydrogenation.24
A cationic rhodium complex encapsulating the chiral phosphine–phosphine oxide ligand 7 was evaluated as a catalyst for acetamidoacrylate hydrogenation as well as a series of more challenging alkenes (Scheme 5, 18a–e) and the results are summarised in Table 2.
 | ||
| Scheme 5 Mixed phosphine–phosphine oxide ligand catalysing the asymmetric reduction of alkenes (18a–f). | ||
| Entry | Alkene | Product | R | R1 | R2 | ee (%) |
|---|---|---|---|---|---|---|
| a ee values were determined by CSP-HPLC. b Products analysed as the corresponding methyl esters. | ||||||
| 1 | 18a | 19a | Ph | NHAc | CO2Hb | 84(S) |
| 2 | 18b | 19b | H | NHAc | CO2Me | 10(S) |
| 3 | 18c | 19c | 4-Cl–C6H4 | NHAc | CO2Hb | 20(S) |
| 4 | 18d | 19d | 4-Cl–C6H4 | NHAc | CO2Me | 22(S) |
| 5 | 18e | 19e | H | Ph | OAc | 40 |
| 6 | 18f | 19f | H | CH2CO2Me | CO2Me | 18 |
The activity of the catalyst was good and in each case the reaction went to completion with a low catalyst loading (1 mol%). With the more challenging alkenes (Table 2, entries 5 and 6), enantiomeric excesses were observed on reduction but with values which are too low to be considered useful (18–40%). However, with one of the acetamidoacrylate derivatives (Table 1, entry 1), the reaction proceeded with a workable value of 84% ee, a value comparable in magnitude to that achieved with monodentate P-chiral ligands which was found to increase and provide single enantiomers upon fractional recrystallization.23
3 Conclusion
In conclusion, Lewis basic organocatalyst 7 was synthesised from a chiral cis-dihydrodiol 2, available from a biotransformation of bromobenzene 1 followed by chemoselective hydrogenation. The synthesis was achieved by utilisation of a palladium-catalysed carbon–phosphorus bond coupling followed by a remarkable spontaneous Arbuzov [2,3]-sigmatropic rearrangement of a transient allylic diphenylphosphinite 13. The order in which the steps were carried out was crucial, with the diphenylphosphine borane moiety facilitating the subsequent sigmatropic rearrangement. The new organocatalyst 7 activates allyltrichlorosilane allowing the allylation of simple aromatic aldehydes 16a–f in often good yield (<81%) but modest enantioselectivities (<57% ee) and establishes the viability of the ligand choice. As a monodentate chiral ligand, the cationic rhodium complex derived from ligand 7 gives ee values on hydrogenation of acetamidoacrylate 18a comparable to some bidentate ligands (>80% ee). Studies of other asymmetric reactions catalysed by this mixed phosphine–phosphine oxide ligand and structural optimisation studies are underway in our laboratories.4 Experimental Section
Melting points were determined using a Kofler hot stage apparatus and are uncorrected. 1H-NMR spectra were recorded using 300 MHz (Bruker DPX 300), 400 MHz (Bruker Avance III 400) and 500 MHz (Bruker DRX 500) NMR spectrometers, in CDCl3 solvent unless stated otherwise. Chemical shifts are given in parts per million (δ) downfield from tetramethylsilane as the internal standard and coupling constants are given in Hertz (Hz). NMR splitting patterns are designated as singlet (s), doublet (d), triplet (t), quartet (q), multiplet (m), and broad signal (br). Mass spectra were recorded using a double focusing Triple Sector VG AutoSpec instrument, and accurate molecular masses were determined by the peak matching method using perfluorokerosene as a standard reference and were accurate to within ±0.006 amu. Analytical TLC was carried out on Merck Kielselgel 60254 plates and the spots visualised using a Hanovia Chromatolite UV lamp. Flash chromatography and preparative layer chromatography (PLC) was performed using Merck Kieselgel 60 (230–400 mesh) and PF254/366 respectively.Compounds 3, 8 and 9 were synthesised by our previously reported methods.7
(1S,6S)-2-Bromo-6-(tert-butyl-diphenyl-silyloxy)-cyclohex-2-enol 4
Imidazole (3.50 g, 51.3 mmol) and tert-butylchlorodiphenylsilane (5.33 mL, 20.5 mmol) were added to a solution of cis-tetrahydrodiol 3 (3.96 g, 20.5 mmol) in DMF (60 mL) and the resulting solution was stirred at room temperature for 18 h. Water (2.5 L) was added and the aqueous phase extracted twice with dichloromethane (400 mL). The combined organic extracts were concentrated under reduced pressure and flash chromatography (solvent: petroleum ether![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethyl acetate (20:1)) gave the titled compound as a clear oil, yield: (5.93 g, 67%). [α]D −30.0 (c 1.0, CH2Cl2); 1H-NMR (300 MHz, CDCl3): δH 7.67 (4 H, m), 7.56–7.33 (6 H, m), 6.13 (1 H, dd, J 4.9, 2.4), 4.20–4.07 (1 H, m), 3.91 (1 H, dt, J 10.6, 3.7), 2.90 (1 H, br s), 2.21–2.05 (1 H, m), 1.91–1.70 (2 H, m), 1.57–1.42 (1 H, m), 1.09 (9 H, s).13C-NMR (75.1 MHz, CDCl3): δC 136.2, 136.1, 133.9, 133.4, 132.9, 130.6, 130.5, 128.3, 128.2, 121.6, 72.9, 72.3, 27.4, 26.1, 24.8, 19.7.
:ethyl acetate (20:1)) gave the titled compound as a clear oil, yield: (5.93 g, 67%). [α]D −30.0 (c 1.0, CH2Cl2); 1H-NMR (300 MHz, CDCl3): δH 7.67 (4 H, m), 7.56–7.33 (6 H, m), 6.13 (1 H, dd, J 4.9, 2.4), 4.20–4.07 (1 H, m), 3.91 (1 H, dt, J 10.6, 3.7), 2.90 (1 H, br s), 2.21–2.05 (1 H, m), 1.91–1.70 (2 H, m), 1.57–1.42 (1 H, m), 1.09 (9 H, s).13C-NMR (75.1 MHz, CDCl3): δC 136.2, 136.1, 133.9, 133.4, 132.9, 130.6, 130.5, 128.3, 128.2, 121.6, 72.9, 72.3, 27.4, 26.1, 24.8, 19.7.
(1S,4R)-3-Bromo-4-(diphenylphosphoryl)-cyclohex-2-enyloxy]-tert-butyldiphenyl-silane 6
(1S,6S)-2-Bromo-6-(tert-butyl-diphenyl-silyloxy)-cyclohex-2-enol 4 (1.0 g, 2.3 mmol) and dimethylaminopyridine (295 mg, 2.4 mmol) were dissolved in THF (20 mL) in a flame-dried Schlenk tube equipped with a magnetic stirring bar. Chlorodiphenylphosphine (436 μL, 2.36 mmol) was added dropwise and the resulting suspension allowed to stir at room temperature for 1 h. The THF was removed under reduced pressure and toluene (20 mL) was added. The resulting solution was heated to 80 °C for 16 h. Toluene was removed under reduced pressure and flash chromatography (solvent: petroleum ether:ethyl acetate, 2:1) gave the titled compound as an amorphous solid, yield: (1.3 g, 93%), m.p. 56–58 °C. [α]D −69.2 (c 1.0, CH2Cl2); HR-MS: m/z = 637.1336, calcd. for C34H36BrO2SiPNa (M + Na)+: 637.1309; 1H-NMR (300 MHz, CDCl3): δH 7.96 (ddd, J 11.1, 7.8, 1.57, 2H), 7.83 (ddd, J 11.4, 8.0, 1.30, 2H), 7.70–7.24 (m, 16H), 6.14 (dd, J 3.3, 2.4, 1H), 4.14 (dd, J 8.7, 5.5, 1H), 3.44 (ddd, J 9.4, 5.1, 3.8, 1H), 2.58–2.26 (m, 1H), 1.95–1.46 (m, 3H), 0.99 (s, 9H). 13C-NMR (75.1 MHz, CDCl3): δC 136.6 (d, J 8.6), 134.7 (4C), 132.7 (2C), 132.6 (2C), 131.3 (d, J 8.7, 2C), 131.0 (d, J 2.7), 130.8 (d, J 2.7), 130.3 (d, J 9.2, 2C), 128.7 (2C), 127.5 (d, J 11.8, 2C), 127.2 (d, J 11.8, 2C), 126.6 (2C), 126.5 (2C), 118.3 (d, J 7.5), 67.7 (d, J 2.2), 44.8 (d, J 67.8), 27.5 (d, J 3.3) 25.8 (3C), 22.8, 18.1; 31P-NMR (121.5 MHz, CDCl3): δP 32.1 (s).
(3aS,7aS)-(2,2-Dimethyl-3a,6,7,7a-tetrahydro-benzo[d][1,3]diox-4-yl)diphenylphosphine borane 10
(1S, 2S)-1,2-Isopropylidenedioxy-3-bromocyclohex-3-ene 8 (1.5 g, 6.4 mmol), Pd2(dba)3 (87.9 mg, 0.19 mmol), Cs2CO3 (4.17 g, 12.8 mmol) and DPPF (80.2 mg, 0.38 mmol) were added to a Schlenk tube equipped with a magnetic stirring bar. Toluene (40 mL) and diphenylphosphine (1.03 mL, 6.7 mmol) were added and the mixture heated to 80 °C for 44 h. The reaction mixture was cooled to ambient temperature and 31P NMR-analysis of the crude mixture showed a peak at δP −6.87. BH3–THF 1 M (10.9 mL, 10.9 mmol) was added and this was stirred at room temperature for five minutes. Water (100 mL) was added and the aqueous phase extracted with CH2Cl2 (2 × 50 mL). The combined organic phases were concentrated under reduced pressure and flash chromatography on SiO2 eluting with petroleum ether:dichloromethane (1:1) gave the titled compound 10 as a clear oil yield: (1.9 g, 84%); [α]D + 19.4 (c 1.0, CH2Cl2); HR-MS m/z = 351.1688, calcd. for C21H25BO2P (M − H)+: 351.1689; 1H-NMR (300 MHz, CDCl3): δH 7.78 (2 H, ddd, J 10.9, 8.1, 1.4), 7.63 (2 H, ddd, J 11.0, 8.1, 1.5), 7.55–7.32 (6 H, m), 6.80 (1 H, dt, J 18.0, 4.2), 4.47 (1 H, dd, J 5.7, 4.6), 4.26 (1 H, apparent q, J 5.6), 2.41 (1 H, m), 2.22–2.06 (1 H, m), 1.82 (2 H, apparent q, J 5.9), 1.27 (3 H, s), 1.16 (3 H, s) 0.81–1.22 (3 H, br m); 13C-NMR (125.1 MHz, CDCl3): δC 147.5 (d, J 9.5), 132.6 (d, J 9.6, 2C), 132.1 (d, J 9.7, 2C), 129.9 (d, J 64.5, 2C), 128.5 (d, J 59.0), 127.3 (d, J 10.7, 2C), 127.2 (d, J 10.6, 2C), 126.6 (d, J 58.2), 126.3 (d, J 52.2), 107.3, 72.1 (d, J 6.4), 69.7 (d, J 5.2), 26.6, 24.6, 24.5, 22.2 (d, J 12.0); 31P-NMR (121.5 MHz, CDCl3): δP 21.9–23.5 (br m).
(5S,6S)-(5,6-Dihydroxycyclohexenyl)diphenylphosphine borane 11
Acetonide 10 (1.87 g, 5.31 mmol) was dissolved in THF:H2O:trifluoroacetic acid (22 mL, 8:2:1) and the resulting solution was stirred for 18 h at room temperature. The solution was cooled to 0 °C and neutralised with triethylamine. Saturated sodium bicarbonate solution (100 mL) was added and the aqueous phase extracted twice with dichloromethane (50 mL). The combined organic phases were concentrated under reduced pressure and flash chromatography (solvent: petroleum ether:ethyl acetate, 3:1) gave the titled compound 10 as a white solid, m.p. 53–56 °C, yield: (1.09 g, 66%) together with recovered starting material (591 mg, 32%). [α]D −84.9 (c 1.0, CH2Cl2); HR-MS m/z = 335.1365, calcd. for C18H22BO2PNa (M + Na)+: 335.1352; 1H-NMR (500 MHz, CDCl3): δH 7.76–7.37 (10 H, m), 6.12 (1 H, dt, J 16.4, 3.6), 4.30 (1 H, dd, J 5.7, 3.8), 3.79 (1 H, dt, J 10.7, 3.6), 3.01 (2 H, br s), 2.45–2.38 (1 H, m), 2.29–2.22 (1 H, m), 1.97–1.89 (1 H, m), 1.79 (1 H, m) 0.61–1.62 (3 H, br m); 13C-NMR (125.1 MHz, CDCl3): δC 146.5 (d, J 3.4), 133.6 (2C), 133.5(2C), 131.9 (d, J 2.4), 131.8 (d, 2.4), 129.7 (d, J 52.8), 129.3 (d, J 10.2, 2C), 129.2 (d, J 10.3, 2C), 128.0 (d, J 59.0, 2C), 69.3 (d, J 8.1), 66.5 (d, J 10.7), 26.7 (d, J 11.4), 24.9; 31P-NMR (121.5 MHz, CDCl3): δP 18.9–20.2 (br m).
(5S,6S)-5-tert-Butyldiphenylsilyloxy-6-diphenylphosphoryl-cyclohexenyl diphenylphosphine borane 12
Imidazole (558 mg, 8.20 mmol) and tert-butylchlorodiphenylsilane (838 μL, 3.22 mmol) were added to a solution of cis-tetrahydrodiol 11 (1.00 g, 3.22 mmol) in DMF (10 mL) and the solution was stirred at room temperature for 18 h. Water (1 L) was added and the aqueous phase extracted twice with dichloromethane (100 mL). The combined organic phases were concentrated under reduced pressure and flash chromatography (solvent petroleum ether:ethyl acetate, 40:1) gave the titled compound as a clear oil, yield: (1.58 g, 89%). [α]D −30.0 (c 1.0, CH2Cl2); HR-MS m/z = 573.2530, calcd. for C34H40BO2SiPNa (M + Na)+: 573.2532; 1H-NMR (500 MHz, CDCl3): δH 7.80–7.68 (2 H, m), 7.68–7.61 (2 H, m), 7.61–7.53 (4 H, m), 7.54–7.25 (12 H, m), 6.45 (1 H, dt, J 17.8, 3.5), 4.15 (1 H, apparent t, J 4.35), 3.88 (1 H, dt, J 10.2, 3.3), 2.58 (1 H, br s), 2.32 (1 H, dd, J 19.2, 3.8), 1.98 (1 H, m), 1.87(1 H, m) 1.48 (1 H, m), 1.04 (9 H, s); 13C-NMR (125.1 MHz, CDCl3): δC 147.6 (d, J 8.6), 136.1 (2C), 136.0 (2C), 135.2 (2C), 134.1 (2C), 133.8 (d, J 9.6, 2C), 133.4 (d, J 9.5, 2C), 131.6, 131.3, 130.4 (d, J 4.8, 2C), 129.0 (d, J 5.0, 2C), 128.9 (d, J 5.1, 2C), 128.3 (2C), 128.2 (2C), 128.1, 71.8 (d, J 7.4), 67.1 (d, J 6.1), 27.4 (3C), 27.0, 26.6 (d, J 12.9), 14.7; 31P-NMR (121.5 MHz, CDCl3): δP 21.6–22.9 (br s).
(2R,5S)-5-tert-Butyldiphenylsilyloxy-2-diphenylphosphoryl-cyclohexenyl diphenylphosphine borane 14
Compound 12 (1.00 g, 1.81 mmol) and dimethylaminopyridine (232 mg, 1.90 mmol) were dissolved in dry toluene (15 mL) in a flame dried Schlenk tube equipped with a magnetic stirring bar. Chlorodiphenylphosphine (344 μL, 1.86 mmol) was added dropwise and the resulting suspension allowed to stir at room temperature for 1 h. THF was removed under reduced pressure and flash chromatography (solvent: petroleum ether:ethyl acetate, 4:1) gave the titled compound 14 as white prisms, yield: (555 mg, 80%); m.p 112–114 °C; [α]D −120.7 (c 1.0, CH2Cl2); HR-MS m/z = 757.2983, calcd. for C46H49BO2SiP2Na (M + Na)+: 757.2976; 1H-NMR (400 MHz, CDCl3): δH 7.91–7.72 (2 H, m), 7.71–7.58 (2 H, m), 7.55 (2 H, dd, J 7.8, 1.5), 7.49 (2 H, dd, J 11.1, 1.3), 7.44–7.28 (13 H, m), 7.24 (2 H, dd, J 14.2, 1.2), 7.19–6.96 (7 H, m), 5.90 (1 H, dt, 17.7, 3.6), 4.04 (1 H, m), 3.95 (1 H, m), 2.51–2.42 (1 H, m), 2.0 (3 H, br m) 1.83–1.74 (2 H, 2 × m), 1.60–1.54 (1 H, m), 0.99 (9 H, s); 13C-NMR (125.1 MHz, CDCl3): δC 152.0 (d, J 8.1), 136.1–127.9 (inseparable signals, 36C) 125.0 (d, J 57.1), 70.0 (d, J 10.5), 36.5 (dd, J 64.6, 13.8), 28.2, 27.8 (d, J 7.0), 27.2 (3C), 27.02; 31P-NMR (121.5 MHz, CDCl3):δP 30.5 (d, J 1.73), 23.9–25.1 (br s).
(2R,5S)-2-Diphenylphosphoryl-5-hydroxycyclohexenyl-diphenylphosphine borane 15
Compound 14 (404 mg, 0.55 mmol) was dissolved in methanol (2 mL) in a flame-dried Schlenk tube equipped with a magnetic stirring bar. Acetyl chloride (5.5 μL, 0.083 mmol) dissolved in methanol (1 mL) was added dropwise at room temperature. The solution was stirred for 6 h and saturated sodium bicarbonate solution (20 mL) was added and the aqueous phase extracted twice with dichloromethane (10 mL). The combined organic phases were concentrated under reduced pressure and flash chromatography (solvent: ethyl acetate) gave the titled compound as a white solid, yield: (242 mg, 89%); m.p. 111–113 °C; [α]D − 3.0 (c 1.0, CH2Cl2); HR-MS m/z = 519.1801, calcd. for C30H31BO2P2Na (M + Na)+: 519.1796; 1H-NMR (300 MHz, CDCl3): δH 7.84 (2 H, dd, J 8.0, 1.7), 7.61–7.48 (4 H, m), 7.47–7.39 (3 H, m), 7.39–7.26 (4 H, m), 7.26–7.17 (4 H, m), 7.14–7.07 (1 H, m, 1H), 7.00 (2 H, dd, J 7.5, 3.0), 6.35 (1 H, td, J 15.3, 5.3), 5.16 (1 H, br s), 4.51–4.14 (2 H, m), 2.14–1.99 (3 H, m), 1.95–1.86 (1 H, m), 1.14 (3 H, br m); 13C-NMR (125.1 MHz, CDCl3): δC 151.4 (d, J 7.5), 133.6 (d, J 9.7, 2C), 131.7–128.1 (inseparable signals, 20C), 128.1 (d, J 11.9, 2C), 124.4 (d, J 57.0), 61.5 (d, J 8.9), 35.1 (dd, J 63.5, 14.2), 27.9 (d, J 4.5), 27.4 (d, J 3.4); 31P-NMR (121.5 MHz, CDCl3):δP 30.5, 24.5-24.7 (br s).(1S,4R)-3-Diphenylphosphino-4-diphenylphosphoryl-cyclohex-2-enyloxy(tert-butyl)diphenylsilane 7
Lewis salt 14 (50 mg, 0.07 mmol) was dissolved in degassed diethylamine (3 mL) and stirred at 40 °C for two hours under a nitrogen atmosphere. The volatiles were then removed on a Schlenk line at 0 °C. Proton NMR analysis showed phosphine 7 contaminated with diborane diethylamine adducts. Attempted column chromatography led to decomposition so the material could not be purified or fully characterised. This compound was used crude for subsequent studies: 31P-NMR (121.5 MHz, CDCl3): δP 33.9, −4.10.General procedure for the asymmetric allylation of aromatic aldehydes 16a–f catalysed by organocatalyst 7
A solution of compound 7 (27.0 mg, 1.67 μmol), aldehyde (0.25 mmol) and diisopropylethylamine (4.7 μL, 27 μmol) in dichlormethane (2 mL) was stirred in a flame-dried Schlenk tube cooled to the required temperature. Allyltrichlorosilane (42.8 μL, 0.30 mmol) was added and the mixture was stirred at the same temperature for 20 h and then quenched with aqueous saturated sodium bicarbonate solution (10 mL). The aqueous layer was extracted with dichloromethane (3 × 10 mL), the combined organic layers were washed with brine, dried over sodium sulphate, and the solvent was removed in vacuo. Purification was effected by flash chromatography (solvent: petroleum ether:ethyl acetate, 4:1). 1H-NMR data of all products corresponded to those previously published.13
(S)-(−)-1-Phenyl-but-3-en-1-ol 17a
[α]D −35.8 (c 1.0, CH2Cl2); 1H-NMR (300 MHz, CDCl3): δH 2.06 (1 H, br s), 2.48–2.56 (2 H, m), 4.77 (1 H, dd, J 7.7, 5.2 Hz), 5.15–5.22 (2 H, m), 5.79–5.89 (2 H, m), 7.28–7.39 (5 H, m); CSP-HPLC (Chiralcel OD-H, hexane/2-propanol 9:1, 1.0 mL min−1) 57% ee (tR = 9.0 min, tS = 10.3 min).
1-Phenyl-hexa-1,5-dien-3-ol 17b
[α]D 0.0 (c 1.0, CH2Cl2)1H-NMR (300 MHz, CDCl3): δH 1.74 (1 H, br s), 2.28–2.40 (2 H, m), 4.27–4.32 (1 H, m), 5.09–5.15 (2 H, m), 5.74–5.84 (1 H, m), 6.18 (1 H, dd, J 15.9, 6.3), 6.55 (1 H, d, J 15.8), 7.15–7.33 (5 H, m).(S)-(−)-1-(Naphthalen-2-yl)-but-3-en-1-ol 17c
[α]D −31.8 (c 1.0, CH2Cl2); 1H-NMR (300 MHz, CDCl3): δH 2.09 (1 H, br s), 2.47–2.58 (2 H, m), 4.84 (1 H, dd, J 7.6, 5.3 Hz), 5.07–5.14 (2 H, m), 5.71–5.82 (1 H, m), 7.37–7.43 (3 H, m), 7.74–7.78 (4 H, m); CSPHPLC (Chiralcel OD-H, hexane/2-propanol, 9:1, 0.75 mL min−1) showed 40% ee (tS = 13.6 min, tR = 15.2 min).
(S)-(−)-1-(4-Trifluoromethyl-phenyl)-but-3-en-1-ol 17d
[α]D −17.6 (c 1.0, CH2Cl2); 1H-NMR (300 MHz, CDCl3): δH 2.07 (1 H, br s), 2.33–2.51 (2 H, m), 4.72 (1 H, dd, J 7.8, 3.1), 5.09–5.14 (2 H, m), 5.67–5.78 (1 H, m), 7.41(2 H, d, J 8.4), 7.54 (2 H, d, J 8.4); CSP-GC (Supelco β-DEX 225 column, oven: 110 °C for 40 min, then 5 °C min−1 to 200 °C showed 45% ee (tS = 24.8 min, tR = 26.2 min).(S)-(−)-1-(4-Nitro-phenyl)-but-3-en-1-ol 17e
[α]D −51.8 (c 1.0, CH2Cl2); 1H-NMR (300 MHz, CDCl3): δH 2.18 (1 H, br s), 2.76–2.26 (m, 2H), 4.80 (1 H, dd, J 7.6, 4.6), 5.11 (1 H, d, J 8.0), 6.03–5.50 (2 H, m), 7.47 (2 H, d, J 8.5), 8.14 (2 H, d, J 8.7); CSP-HPLC (Chiralcel AD-H, hexane/2-propanol, 97:3, 0.8 mL min−1) showed 30% ee (tR = 44.2 min, tS = 46.6 min).
1-(4-Methoxy-phenyl)-but-3-en-1-ol 17f
[α]D 0.0 (c 1.0, CH2Cl2) 1H-NMR (300 MHz, CDCl3): δH 2.01 (1 H, br s), 2.49 (2 H, t, J 8.0), 3.80 (s, 3 H), 4.68 (1 H, t, J 6.5), 5.10–5.17 (2 H, m), 5.74–5.84 (1 H, m), 6.88 (2 H, d, J 8.7), 7.27 (d, J 8.7, 2 H).General procedure for the asymmetric hydrogenation of alkenes 18a–f catalysed by ligand 7
Lewis salt 14 (50 mg, 0.07 mmol) was dissolved in degassed diethylamine (3 mL) and stirred at 40 °C for two hours under a nitrogen atmosphere. The volatiles were then removed on a Schlenk line at 0 °C. The residue was dissolved in degassed methanol/ethanol 9:1 (2 mL). [Rh(cod)Cl]2 (9.0 mg, 0.018 mmol) was added in one portion to the mixture and this was stirred at room temperature for one hour. A degassed aqueous solution of sodium tetrafluoroborate (0.04 mL, 1.2 M, 0.05 mol) was added dropwise and the solution was stirred at room temperature for 1.5 h. The mixture was evaporated to dryness on the Schlenk line and additional methanol/ethanol 9:1 (1.5 mL) was added and evaporated again. The residue was taken up in dichloromethane (2 mL) filtered under nitrogen and the solvent removed. The catalyst (7, 1 mol%) was used to hydrogenate the alkenes 18a–f, Table 2, on a 0.2 mmol scale in methanol as solvent (3 mL) using a hydrogen pressure of 100 psi at 50 °C with stirring at 400 rpm for 10 h. In all cases the reactions went to completion, as determined by HPLC, and the products were identified by comparison of HPLC retention times with those of authentic racemic samples.
(S)-2-Acetamido-3-phenylpropanoic acid 19a
1H-NMR (300 MHz, CDCl3): δH 2.02 (3 H, s), 3.14 (1 H, dd, J 13.6, 6.0), 3.25 (1 H, dd, J 13.6, 6.0), 3.80 (1 H, br s), 4.87 (1 H, apparent q, J 6.0), 6.18 (1 H, br s), 7.08–7.69 (5 H, m); Trimethylsilyldiazomethane was added to the sample prior to HPLC analysis. CSP-HPLC, CHIRALPAK IB 5 μm 250 × 4.6 mm ID column, heptane/ethanol 9:1, 1 mL min−1 showed 84% ee (tR = 10.8 min, tS = 11.9 min).
(S)-Methyl-2-acetamidopropanoate 19b
1H-NMR (300 MHz, CDCl3): δH 1.41 (3 H, d, J 7.2), 2.02 (3 H, s), 3.75 (3 H, s), 4.59 (1 H, apparent quintet, J 7.2), 6.32 (1 H, br s); CSP-HPLC, CHIRALPAK IB 5 μm 250 × 4.6 mm ID column, heptane/ethanol 7:3, 1 mL min−1 showed 10% ee (tR = 4.2 min, tS = 4.9 min).
(S)-2-Acetamido-3-(3-chlorophenyl)-propanoic acid 19c
1H-NMR (300 MHz, CDCl3): δH 2.03 (3 H, s), 3.07 (1 H, dd, J 14.1, 6.0), 3.15 (1 H, dd, J 14.1, 6.0), 3.98 (1 H, br s), 4.86 (1 H, apparent q, J 6.0), 6.18 (1 H, br s), 7.03 (2 H, d, J 8.1), 7.26 (2 H, d, J 8.1); Trimethylsilyldiazomethane was added to the sample prior to HPLC analysis. CSP-HPLC, CHIRALPAK IB 5 μm 250 × 4.6 mm ID column, heptane/ethanol 4:1, 1 mL min−1 showed 20% ee (tR = 6.6 min, tS = 7.9 min).
(S)-Methyl-2-acetamido-3-(3-chlorophenyl)-propanoate 19d
1H-NMR (300 MHz, CDCl3): δH 1.98 (3 H, s), 3.04 (1 H, dd, J 13.9, 6.0), 3.13 (1 H, dd, J 13.9, 6.0), 3.72 (3 H, s), 4.85 (1 H, apparent q, J 6.0), 6.20 (1 H, br s), 7.04 (2 H, d, J 8.0), 7.26 (2 H, d, J 8.0); CSP-HPLC, CHIRALPAK IB 5 μm 250 × 4.6 mm ID column, heptane/ethanol 4:1, 1 mL min−1 showed 20% ee (tR = 6.6 min, tS = 7.9 min).
1-Phenylethyl acetate 19e
1H-NMR (300 MHz, CDCl3): δH 1.54 (3 H, d, J 6.6), 2.07 (3 H, s), 5.88 (1 H, q, J 6.6), 7.26–7.49 (5 H, m); CSP-HPLC, CHIRALPAK IB 5 μm 250 × 4.6 mm ID column, heptane/ethanol 99:1, 0.5 mL min−1 showed 40% ee (tR = 9.0 min, tS = 10.0 min).
Dimethyl 2-methylsuccinate 19f
1H-NMR (300 MHz, CDCl3): δH 1.22 (3 H, d, J 7.1), 2.41 (1 H, dd, J 16.5, 7.1), 2.75 (1 H, dd, J 16.5, 7.1), 2.93 (1 H, apparent sextet, J 7.1), 3.68 (3 H, s), 3.70 (3 H, s); CSP-HPLC, CHIRALPAK IB 5 μm 250 × 4.6 mm ID column, heptane/ethanol 7:3, 1 mL min−1 showed 18% ee (tR = 5.7 min, tS = 6.8 min).
Acknowledgements
Financial assistance was provided by an EU Marie Curie Host Fellowship for Transfer of Knowledge, NOVACAT, project no. 29846 (K. S. D. and M. B.).Notes and references
- D. A. Widdowson and D. W. Ribbons, Janssen Chim. Acta, 1990, 8, 3 Search PubMed; H. A. J. Carless, Tetrahedron: Asymmetry, 1992, 3, 795 CrossRef CAS; G. N. Sheldrake, Chirality in Industry, Wiley, Chichester, 1992 Search PubMed; S. M. Brown and T. Hudlicky, Organic Synthesis: Theory and Applications, 1993, 2, 113 Search PubMed; S. M. Resnick, K. Lee and D. T. Gibson, J. Ind. Microbiol., 1996, 17, 438 CrossRef; D. R. Boyd and G. N. Sheldrake, Nat. Prod. Rep., 1998, 15, 309 RSC; T. Hudlicky, D. Gonzalez and D. T. Gibson, Aldrichimica Acta, 1999, 32, 35 Search PubMed; D. T. Gibson and R. E. Parales, Curr. Opin. Biotechnol., 2000, 11, 236 CrossRef; D. R. Boyd, N. D. Sharma and C. C. R. Allen, Curr. Opin. Biotechnol., 2001, 12, 564 CrossRef; R. A. Johnson, Org. React., 2004, 63, 117 Search PubMed; D. R. Boyd and T. D. H. Bugg, Org. Biomol. Chem., 2006, 4, 181 Search PubMed; T. Hudlicky and J. W. Reed, Chem. Soc. Rev., 2009, 38, 3117 RSC; T. Hudlicky and J. W. Reed, Synlett, 2009, 685 CrossRef.
- L. V. White, C. E. Dietinger, D. M. Pinkerton, A. C. Willis and M. G. Banwell, Eur. J. Org. Chem., 2010, 4365 Search PubMed; B. D. Schwartz, M. T. Jones, M. G. Banwell and I. A. Cade, Org. Lett., 2010, 12, 5210 CrossRef CAS; A. Lin, A. C. Willis and M. G. Banwell, Tetrahedron Lett., 2010, 51, 1044 CrossRef; T. Hudlicky, Pure Appl. Chem., 2010, 82, 1785 CrossRef; J. Collins, U. Rinner, M. Moser, T. Hudlicky, I. Ghiviriga, A. E. Romero, A. Kornienko, D. Ma, C. Griffin and S. Pandey, J. Org. Chem., 2010, 75, 3069 CrossRef; D. R. Boyd, N. D. Sharma, C. A. Acaru, J. F. Malone, C. R. O'Dowd, C. C. R. Allen and P. J. Stevenson, Org. Lett., 2010, 12, 2206 CrossRef; M. G. Banwell, X. H. Ma, O. P. Karunaratne and A. C. Willis, Aust. J. Chem., 2010, 63, 1437 CrossRef; D. Bon, M. G. Banwell and A. C. Willis, Tetrahedron, 2010, 66, 7807 CrossRef; A. D. Findlay and M. G. Banwell, Org. Lett., 2009, 11, 3160 CrossRef; J. Gilmet, B. Sullivan and T. Hudlicky, Tetrahedron, 2009, 65, 212 CrossRef; M. T. Jones, B. D. Schwartz, A. C. Willis and M. G. Banwell, Org. Lett., 2009, 11, 3506 CrossRef; H. Leisch, A. T. Omori, K. J. Finn, J. Gilmet, T. Bissett, D. Ilceski and T. Hudlicky, Tetrahedron, 2009, 65, 9862 CrossRef; M. Matveenko, A. C. Willis and M. G. Banwell, Tetrahedron Lett., 2009, 50, 2982 CrossRef; D. M. Pinkerton, M. G. Banwell and A. C. Willis, Aust. J. Chem., 2009, 62, 1639 CrossRef; D. M. Pinkerton, M. G. Banwell and A. C. Willis, Org. Lett., 2009, 11, 4290 CrossRef; B. Sullivan and T. Hudlicky, Tetrahedron Lett., 2008, 49, 5211 CrossRef; T. K. Macklin and G. C. Micalizio, J. Am. Chem. Soc., 2009, 131, 1392 CrossRef.
- D. R. Boyd, N. D. Sharma, L. Sbircea, D. Murphy, T. Belhocine, J. F. Malone, S. L. James, C. C. R. Allen and J. T. G. Hamilton, Chem. Commun., 2008, 5535 RSC.
- D. R. Boyd, N. D. Sharma, L. Sbircea, D. Murphy, J. F. Malone, S. L. James, C. C. R. Allen and J. T. G. Hamilton, Org. Biomol. Chem., 2010, 8, 1081 CAS.
- L. Sbircea, N. D. Sharma, W. Clegg, R. W. Harrington, P. N. Horton, M. B. Hursthouse, D. C. Apperley, D. R. Boyd and S. L. James, Chem. Commun., 2008, 5538 RSC.
- D. R. Boyd, N. D. Sharma, N. M. Llamas, C. R. O'Dowd and C. C. R. Allen, Org. Biomol. Chem., 2007, 5, 2267 CAS.
- D. R. Boyd, N. D. Sharma, M. Kaik, M. Bell, V. Berberian, P. Mc Intyre, B. Kelly, C. Hardacre and P. J. Stevenson, Adv. Synth. Catal., 2011, 353, 2455 CrossRef CAS.
- S. Kobayashi and K. Nishio, Tetrahedron Lett., 1993, 34, 3453 CrossRef CAS.
- S. E. Denmark, D. M. Coe, N. E. Pratt and B. D. Griedel, J. Org. Chem., 1994, 59, 6161 CrossRef CAS.
- S. E. Denmark and J. P. Fu, Chem. Rev., 2003, 103, 2763 CrossRef CAS; S. E. Denmark and J. P. Fu, Chem. Commun., 2003, 167 RSC; S. E. Denmark and E. N. Jacobsen, Acc. Chem. Res., 2000, 33, 324 CrossRef.
- K. Iseki, S. Mizuno, Y. Kuroki and Y. Kobayashi, Tetrahedron Lett., 1998, 39, 2767 CrossRef CAS; K. Iseki, S. Mizuno, Y. Kuroki and Y. Kobayashi, Tetrahedron, 1999, 55, 977 CrossRef.
- A. V. Malkov, M. Bell, F. Castelluzzo and P. Kocovsky, Org. Lett., 2005, 7, 3219 CrossRef CAS; A. V. Malkov, M. Bell, M. Vassieu, V. Bugatti and P. Kocovsky, J. Mol. Catal. A: Chem., 2003, 196, 179 CrossRef; M. Nakajima, M. Saito and S. Hashimoto, Chem. Pharm. Bull., 2000, 48, 306 CrossRef; M. Nakajima, M. Saito, M. Shiro and S. Hashimoto, J. Am. Chem. Soc., 1998, 120, 6419 CrossRef; T. Shimada, A. Kina, S. Ikeda and T. Hayashi, Org. Lett., 2002, 4, 2799 CrossRef.
- A. V. Malkov, M. Bell, M. Orsini, D. Pernazza, A. Massa, P. Herrmann, P. Meghani and P. Kocovsky, J. Org. Chem., 2003, 68, 9659 CrossRef CAS.
- C. Ogawa, M. Sugiura and S. Kobayashi, Angew. Chem., Int. Ed., 2004, 43, 6491 CrossRef CAS.
- A. Massa, A. V. Malkov, P. Kocovsky and A. Scettri, Tetrahedron Lett., 2003, 44, 7179 CrossRef CAS.
- I. Chataigner, U. Piarulli and C. Gennari, Tetrahedron Lett., 1999, 40, 3633 CrossRef CAS.
- F. Liron and P. Knochel, Chem. Commun., 2004, 304 RSC; S. Demay, K. Harms and P. Knochel, Tetrahedron Lett., 1999, 40, 4981 CrossRef CAS; A. Gavryushin, K. Polborn and P. Knochel, Tetrahedron: Asymmetry, 2004, 15, 2279 CrossRef.
- J. K. Gawronski, M. Kwit, D. R. Boyd, N. D. Sharma, J. F. Malone and A. F. Drake, J. Am. Chem. Soc., 2005, 127, 4308 CrossRef CAS.
- M. Rubina, E. W. Woodward and M. Rubin, Org. Lett., 2007, 9, 5501 CrossRef CAS.
- P. Y. Renard, P. Vayron, E. Leclerc, A. Valleix and C. Mioskowski, Angew. Chem., Int. Ed., 2003, 42, 2389 CrossRef CAS.
- W. Dabkowski, A. Ozarek, S. Olejniczak, M. Cypryk, J. Chojnowski and J. Michalski, Chem.–Eur. J., 2009, 15, 1747 CrossRef CAS.
- W. S. Knowles, Adv. Synth. Catal., 2003, 345, 3 CrossRef CAS; W. S. Knowles, Acc. Chem. Res., 1983, 16, 106 CrossRef.
- W. S. Knowles, Angew. Chem., Int. Ed., 2002, 41, 1998 CrossRef CAS.
- H. Fernandez-Perez, P. Etayo, A. Panossian and A. Vidal-Ferran, Chem. Rev., 2011, 111, 2119 CrossRef CAS; W. Li, G. H. Hou, X. F. Sun, G. Shang, W. C. Zhang and X. M. Zhang, Pure Appl. Chem., 2010, 82, 1429 CrossRef; J. F. Teichert and B. L. Feringa, Angew. Chem., Int. Ed., 2010, 49, 2486 CrossRef; W. J. Tang and X. M. Zhang, Chem. Rev., 2003, 103, 3029 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: CCDC reference number 842558. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1ob06599h |
| This journal is © The Royal Society of Chemistry 2012 |