 Open Access Article
Open Access ArticleIdentification of the best-suited leaving group for the diastereoselective synthesis of glycidic amides from stabilised ammonium ylides and aldehydes†
Richard
Herchl
a,
Martin
Stiftinger
b and
Mario
Waser
*a
aInstitute of Organic Chemistry, Johannes Kepler University Linz, Altenbergerstraße 69, 4040 Linz, Austria. E-mail: Mario.waser@jku.at; Fax: +43 732 2468 8747; Tel: +43 732 2468 8748
bInstitute of Analytical Chemistry, Johannes Kepler University Linz, Altenbergerstraße 69, 4040 Linz, Austria
First published on 11th August 2011
Abstract
In comparison to the use of sulfur ylides, the use of ammonium ylides for the synthesis of epoxides is significantly less developed. As a part of our systematic investigations concerning the use of amide-stabilised ammonium ylides for the synthesis of glycidic amides we have focused on the identification of the best-suited amino leaving group for this purpose. Whereas tertiary amines like quinuclidine or cinchona alkaloids were found to be not suited for epoxide formation, trimethylamine was found to be the leaving group of choice, yielding trans-glycidic amides in excellent yields of > 90%.
Introduction
The use of ylides for the (dia-)stereoselective synthesis of epoxides starting from carbonyl compounds has attracted considerable interest over the last decades.1–8 The straightforward use of easily available carbonyl electrophiles in combination with ylide nucleophiles makes this method a highly useful alternative to other epoxide forming reactions like oxidations of double bonds9 or the classical Darzens reaction.10The synthesis of an oxirane ring by addition of a sulfur ylide to an aldehyde or ketone was introduced in the 1960s1 and a variety of applications using chiral sulfonium ylides either as auxiliaries or in a catalytic fashion have been reported so far.2–4 Besides the use of sulfur ylides, ammonium ylides have also been successfully employed in different (dia-)stereoselective C–C bond forming reactions over the last years.5–8,11–15 Gaunt et al. showed that the addition of α-halo carbonyl compounds to α,β-unsaturated Michael acceptors in the presence of catalytic amounts of cinchona alkaloids gives access to almost enantiopure cyclopropanes via in situ formation of a chiral ammonium ylide.11 However, with respect to the syntheses of epoxides or aziridines only a few examples have been reported so far. Whilst the diastereoselective synthesis of aziridines can be accomplished using DABCO-derived stabilised ammonium ylides,14 the synthesis of epoxides has so far mainly been limited to semi-stabilised benzylic ammonium ylides7 and cyano-stabilised5 ammonium ylides. In addition, it was shown that more stabilised ester-derived ammonium ylides do not undergo epoxide formation.15
We have recently developed a highly trans-selective protocol for the synthesis of glycidic amides by reacting aromatic aldehydes with stabilised amide-derived ammonium ylides.8 Key to success in this approach was the use of biphasic conditions together with a two-fold excess of aldehyde to give the corresponding glycidic amides in moderate to good yields (Scheme 1).
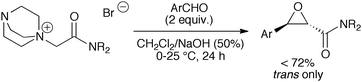 | ||
| Scheme 1 Amide-based ammonium ylides for epoxide formation.8 | ||
However, although it was shown for the first time that ammonium ylides bearing an α-carbonyl group can successfully be employed in oxirane syntheses, it must be clearly pointed out that this reaction is very close to the limit of what is possible with ammonium ylides. This can be illustrated by the fact that the yields were strongly dependent on the electronic properties of the aldehydes.8 Whereas electron neutral aldehydes reacted in moderate to good yields (up to 72%), more electron rich aldehydes reacted much more slowly. On the other hand, electron deficient aldehydes were prone to rapid Cannizzaro decomposition under the highly basic reaction conditions.
Due to the high potential of this type of reaction for the diastereoselective synthesis of glycidic amides we therefore decided to carry out additional investigations to get a clearer picture about the potential and the limitations of this methodology.
Results and discussion
Recent studies by Aggarwal et al. clearly showed that a key factor in ylide-based epoxide formation reactions is the leaving group quality of the onium group, which decreases in the order O > S > N > P.13 Accordingly, ammonium ylides are less reactive in epoxide forming reactions than sulfur ylides. Furthermore, the presence of a carbonyl group α to the leaving group (stabilised ylides) significantly increases the barrier to ring closure.It is noteworthy that the high trans-selectivity observed in the reaction of amide-stabilised ammonium ylides with aldehydes (Scheme 1)8 is in full accordance with results published for epoxidations using amide-stabilised sulfur ylides.4 Thus, in analogy to studies by Aggarwal et al.,3d it can be assumed that addition of the ylide to the aldehyde gives the synclinal anti-betaine A first (Scheme 2). Bond rotation then leads to the antiperiplanar anti-betaine B, which, upon ring closure, yields the trans-oxirane. Systematic investigations by Aggarwal et al. revealed that for stabilised sulfonium ylides formation of syn- and anti-betaines is reversible, but bond rotation towards antiperiplanar betaines B is more hindered for syn-betaines (which would give cis-oxiranes) than for anti-betaines, thus favouring trans-epoxide formation.3d,13 Although we have no direct experimental or computational evidence yet, it seems reasonable that the high trans-selectivity using amide-derived ammonium ylides might be due to similar reasons.
 | ||
| Scheme 2 Formation of trans-oxiranes via the anti-betaines A and B. | ||
During our initial investigations, the biphasic combination of CH2Cl2 and aqueous NaOH (50%) (100 equiv.) was found to be superior over monophasic or liquid/solid conditions.8 During these investigations, aldehyde- and ylide-derived by- or decomposition products were observed. Whereas activated aldehydes (e.g. p-nitro) mainly gave Cannizzaro disproportionation byproducts, more electron neutral or electron rich aldehydes were found to be more stable under the chosen conditions.16 With respect to the ylide, a reasonable consumption was observed in all experiments, even if no product was formed. Although the formed by-products could not be clearly identified, it seems reasonable that not only the ylides themselves decompose, but also the intermediate betaines (A and B) undergo some decomposition. This is also supported by the fact that it was never possible to quantitatively re-isolate the aldehyde (respectively benzyl alcohol and benzoic acid), even if no epoxide product was formed. It can therefore be rationalized that some betaine is always formed but the reduced leaving group ability of the amino group makes ring closure be the limiting step in this reaction.
Therefore, we decided to investigate the influence of the leaving group on the reaction outcome and to determine the best-suited amino group for our approach. As summarised in Table 1, several tertiary amines were used to synthesise the corresponding quaternary ammonium ylides 3–7 and tested in the reaction with benzaldehyde (1) under biphasic conditions.
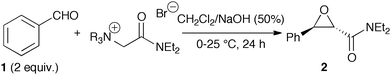
Entry 1 represents the recently developed standard reaction using DABCO-derived ammonium salt 3 as the nucleophile (67% yield).8 Replacing DABCO for quinuclidine (salt 4), the yield dropped significantly (32%). This significant decrease in yield is in contrast to recent reports by Aggarwal et al.7c and Kimachi et al.7a describing either similar or only slightly reduced yields in the syntheses of stilbene oxides using DABCO- or quinuclidine-derived benzylic ammonium ylides. Furthermore, using cinchona alkaloid based ylides (e.g. O-methyl protected quinine based salt 5, entry 3) absolutely no product formation was observed.
Employing triethylamine as the amine component (entry 4) the yield was still significantly lower compared to DABCO. Of note, in all these lower yielding experiments (entries 2–4), NMR spectra of the crude products showed significant amounts of unidentified decomposition products. However, when we changed to trimethylamine (Me3N) (salt 7, entry 5) almost quantitative conversion of 7 to the epoxide product 2 was observed. Using just 1.2 equiv. of 1 the yield decreased to 58% illustrating that decomposition of 1 is the limiting factor hereby.
The striking differences in reactivity should mainly be due to the different leaving group abilities of the different amines and less due to different nucleophilicities as all ammonium ylides are supposed to be highly nucleophilic.7,13 As species with lower basicity are supposed to have a higher leaving group ability17 it is obvious why quinuclidine (pKa = 11.4)18a and triethylamine (pKa = 10.7)18b are not as good leaving groups as DABCO (pKa = 8.9)18a and trimethylamine (pKa = 9.8).18b However, this fact alone does not explain why Me3N is by far the best leaving group whereas cinchona alkaloids (pKa of quinine = 8.7)18c do not yield any product at all. The missing reactivity of the cinchona-derived salt 5 is not fully understood yet as these compounds should be good nucleophiles as demonstrated by Gaunt et al.11 However, one might reason that bond rotation towards the required antiperiplanar betaine B has a much higher activation barrier than in the case of small amine groups due to repulsion between the large quinoline group of quinine and the phenyl group of the benzaldehyde,19 which would explain why we just observed formation of decomposition products. On the other hand, the high yields using 7 may therefore be attributed to the smallest steric hindrance among the employed amino groups, thus facilitating bond rotation towards the required antiperiplanar betaine B.
It is noteworthy that even less stabilised cinchona-derived benzylic ammonium ylides, which should be more reactive, did not yield any epoxides under the biphasic conditions. This is in full accordance with a recent report by Kimachi et al. on the syntheses of stilbene oxides using cinchona-derived benzylic ammonium ylides in THF (t-BuOK as a base).7b Surprisingly, this group identified brucine as a suitable chiral amine for the syntheses of enantioenriched stilbene oxides.7b
Accordingly, these results clearly point out the high importance of the appropriate leaving group for this reaction.
Having identified trimethylamine as the amino group of choice, we next screened the reaction of 7 with several aldehydes (2 equiv.)16 under standard conditions (Table 2).
|
|
||||
|---|---|---|---|---|
| Entry | R | Aldehyde | Product | Yielda,b (%) |
| a Isolated yields. b Yields obtained using DABCO salt 3 are given in brackets (taken from Ref. 8) c Determined by 1H NMR of the crude product. d Large amounts of Cannizzaro decomposition products. e Decomposition of aldehyde. | ||||
| 1 | Ph- | 1 | 2 | 92 (67)b |
| 2 | 4-MeC6H4- | 8 | 9 | 95 (68)b |
| 3 | 2-MeC6H4- | 10 | 11 | 90 |
| 4 | 4-ClC6H4- | 12 | 13 | 90 (72)b |
| 5 | 4-BrC6H4- | 14 | 15 | 97 |
| 6 | 4-Biphenyl- | 16 | 17 | 93 |
| 7 | 4-MeOC6H4- | 18 | 19 | 83 (47)b |
| 8 | 2-MeOC6H4- | 20 | 21 | 98 |
| 9 | 4-Me2NC6H4- | 22 | 23 | <5c |
| 10 | 4-NO2C6H4- | 24 | 25 | 17d (<10)b |
| 11 | 3-NO2C6H4- | 26 | 27 | 12d |
| 12 | 3-Pyridyl- | 28 | 29 | <10c,d |
| 13 | n-Undecyl- | 30 | 31 | 0e |
| 14 | Cyclohexyl- | 32 | 33 | 54 |

Reactions with a variety of different aromatic aldehydes (entries 1–8) gave the corresponding trans-configured glycidic amides in good to excellent yields. In average the yields were about 20 to 30% higher compared to the DABCO-based epoxidations.8 In addition, it was found that the Me3N-derived salt 7 is also superior upon reaction with electron rich aldehydes like the anisaldehydes 18 and 20 (entries 7 and 8). Whereas the DABCO salt 3 gave the p-methoxy substituted glycidic amide 19 in 47% only under standard conditions,8 reaction of salt 7 with aldehyde 18 gave the oxirane 19 in 83% isolated yield (entry 7). Furthermore, the o-methoxy aldehyde 20 was found to be even more reactive furnishing epoxide 21 almost quantitatively. However, using the even more electron rich aldehyde 22 as an electrophile, only traces of product were observed in the NMR spectrum of the crude product. Also carrying out the reaction at higher temperature did not improve the conversion significantly (<10% in situ). Furthermore, the starting aldehyde was reisolated almost quantitatively. This is absolutely identical to our recent result using DABCO-derived salts and therefore suggests that the electron rich aldehyde 22 is not electrophilic enough for betaine formation. As the results for DABCO- and Me3N-derived ylides are the same it can be reasoned that both ylides are similarly nucleophilic. This also implies that the higher reactivity of 7 with more electron neutral aldehydes (entries 1–8, Table 2) is really due to an enhanced leaving group ability of Me3N.
On the other hand, using more electron deficient aldehydes like the nitro aldehydes 24 and 26 or the pyridyl carbaldehyde 28, mainly Cannizzaro decomposition products were observed (entries 10–12). Although the nitroaldehydes 24 and 26 could be converted into the corresponding epoxides 25 and 27 in rather low yields only (entries 10, 11), a beneficial effect of the leaving group was observed as reaction of 24 with 3 was found to give epoxide 25 in trace amounts only.8 Nevertheless, these highly activated aldehydes (the same counts for 28) are too unstable under the highly basic reaction conditions and neither carrying out these reactions at lower temperatures nor using less base did improve the result.
Using the lauric aldehyde 30, full consumption of the aldehyde was observed but no epoxide 31 could be obtained. As an NMR spectrum of the crude product showed signals in the olefinic region, a self-aldol condensation under the highly basic conditions seems to be the dominant reaction of 30. In contrast, the reaction of 7 with cyclohexanecarboxaldehyde (32) gave the targeted trans-epoxide 33 in 54%. Although the yield was lower than for most aromatic aldehydes, it was clearly shown that α-branched aliphatic aldehydes, which cannot undergo self-condensation reactions, are suitable electrophiles for ylide-based epoxidations. Accordingly, despite the highly basic conditions, the scope of this reaction is not just limited to aldehydes without α-H only.
Next, a screening of other trimethylamine-derived amide-based ylides in the reaction with 1 (2 equiv.) was undertaken (Table 3).






Other tertiary amides (entries 2, 4, and 5) gave yields comparable to the test reaction between 1 and 7. Using the secondary amide 36, the corresponding glycidic amide 37 could be isolated in 49% (entry 3). This lower yield for secondary amides is in accordance with our recent results using DABCO-derived ammonium salts where a low yield of 24% only was obtained in the synthesis of 37.8 As an increased amount of Cannizzaro products was formed, the lower yield seems to be due to a significantly reduced reactivity of the secondary amide-derived salt 36 (similar results for sulfur ylides were reported by Aggarwal et al.4b). Unfortunately, the use of Weinreb amide 42 did not yield any epoxide 43. Hereby, the aldehyde 1 (besides Cannizzaro products) was almost fully recovered whereas the ammonium salt 42 totally decomposed.
Finally, we were also interested whether the scope of this methodology can be expanded to even more stabilised ester-derived ammonium ylides. However, the reaction of trimethylamine-derived ester-based ammonium ylides with benzaldehyde totally failed and not even traces of the epoxides could be obtained. Accordingly, although we have identified the best-suited leaving group and reaction conditions with respect to the synthesis of glycidic amides, the corresponding glycidic esters are still not accessible.
Conclusions
It was shown that the use of ammonium ylides bearing an α-carbonyl group for the syntheses of epoxides is highly dependent on the choice of the employed leaving group. Whereas trimethylamine and DABCO were found to be suitable amino groups giving access to trans-configured glycidic amides in good to excellent yields, other tertiary amines were found to be less suited. Especially cinchona alkaloid-derived ammonium ylides were found to be absolutely unreactive in the syntheses of glycidic amides as well as in the syntheses of stilbene oxides. The scope of the reaction was found to be rather broad for several aromatic aldehydes and different amides and excellent yields of >90% could be obtained. Only highly electron rich as well as highly electron deficient aldehydes were found to be less suited. Whereas electron rich aldehydes are not electrophilic enough for this reaction, more activated aldehydes were found to mainly undergo Cannizzaro decomposition mainly. On the other hand, it was also shown that enolisable α-branched aldehydes do undergo epoxide formation under the highly basic reaction conditions whereas unbranched ones do not yield any epoxides due to self-aldol condensation. On the other hand, ester-derived ammonium ylides were not reactive under the developed conditions.Experimental section
General
Melting points were measured on a Kofler melting point microscope (Reichert, Vienna). 1H- and 13C-NMR spectra were recorded on a Bruker Avance DRX 500 MHz spectrometer using a TXI cryoprobe with z-gradient coil and on a Bruker Avance DPX 200 MHz spectrometer. All NMR spectra were referenced on the solvent peak. High resolution mass spectra were obtained using an Agilent 6520 Q-TOF mass spectrometer with an ESI source and an Agilent G1607A coaxial sprayer. IR spectra were recorded on a Shimadzu IR Affinity-1 Fourier transform infrared spectrometer. All chemicals were purchased from commercial suppliers and used without further purification unless otherwise stated. All reactions were performed under an Ar-atmosphere.General procedure for the syntheses of Me3N-ammonium salts
One equivalent of Me3N (33% solution in EtOH) was added to a solution of one equivalent of the α-bromo amide in THF (10 mL g−1 amide) and stirred for 24 h at room temperature. The resulting solid was filtered off, washed with EtOAc (3×), and dried in vacuo to give the product in sufficient purity for the epoxide formation reaction.Ammonium salt 7
Prepared from 2-bromo-N,N-diethylacetamide20 (8.92 g, 45.9 mmol) in 88% (10.27 g, 40.5 mmol). White solid. M.p.: 186–188 °C; 1H NMR (500 MHz, δ, CDCl3, 298 K): 1.14 (t, J = 7.3 Hz, 3H), 1.27 (t, J = 7.3 Hz, 3H), 3.36 (q, J = 7.3 Hz, 2H), 3.47 (q, J = 7.3 Hz, 2H), 3.63 (s, 9H), 5.00 (s, 2H) ppm; 13C NMR (125 MHz, δ, CDCl3, 298 K): 12.9 (CH3-), 14.5 (CH3-), 41.0 (-CH2-), 42.0 (-CH2-), 54.7 (-N+(CH3)3), 63.2 (-CH2CO-), 162.4 (-CO-) ppm; IR (film):![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) = 3003, 2978, 2945, 1639, 1483, 1467, 1446, 1433, 1384, 1357, 1278, 1238, 1215, 1139, 1097, 1078, 1022, 975, 954, 927, 894 cm−1; HRMS (ESI): m/z calcd for C9H21N2O+: 173.1654 [M+]; found: 173.1651.
= 3003, 2978, 2945, 1639, 1483, 1467, 1446, 1433, 1384, 1357, 1278, 1238, 1215, 1139, 1097, 1078, 1022, 975, 954, 927, 894 cm−1; HRMS (ESI): m/z calcd for C9H21N2O+: 173.1654 [M+]; found: 173.1651.
General procedure for the preparation of epoxides
A vigorously stirred solution of ammonium salt (1 mmol) in CH2Cl2 (10 mL) was cooled to 0 °C, followed by addition of 50% NaOH (5 mL). After 5 min the aldehyde (2 mmol) was added in one portion. The biphasic mixture was warmed to 25 °C over 1 h and vigorously stirred for 23 h. After extraction with EtOAc, the organic layer was washed with brine, dried over Na2SO4 and evaporated to dryness. Column chromatography (silica gel, heptanes/EtOAc = 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) gave the glycidic amides in the reported yields.
:3) gave the glycidic amides in the reported yields.
trans-N,N-Diethyl-3-phenyloxirane-2-carboxamide (2)
Obtained in 92% as a white to yellow solid. Analytical data are in full accordance with those reported in literature.4a1H NMR (500 MHz, δ, CDCl3, 298 K): 1.16 (t, J = 7.3 Hz, 3H), 1.20 (t, J = 7.3 Hz, 3H), 3.40–3.51 (m, 4H), 3.58 (d, J = 1.4 Hz, 1H), 4.09 (d, J = 1.4 Hz, 1H), 7.32–7.39 (m, 5H) ppm; 13C NMR (125 MHz, δ, CDCl3, 298 K): 13.1, 15.0, 41.0, 41.6, 57.3, 57.7, 125.8, 128.6, 128.7, 135.9, 165.8 ppm; IR (film): = 2972, 2933, 2873, 1643, 1487, 1450, 1409, 1365, 1271, 1217, 1145, 1095, 1076, 893, 754, 698, 617 cm−1; HRMS (ESI): m/z calcd for C13H17NO2: 220.1332 [M + H]+; found: 220.1329.
Acknowledgements
This work was supported by the Austrian Science Funds (FWF): Project No. P22508-N17. We are grateful to Prof. Dr Norbert Müller for inspiring discussions and to Andrea Zollner and Martin Strnad for assistance with the lab work.Notes and references
- (a) E. J. Corey and M. Chaykovsky, J. Am. Chem. Soc., 1962, 84, 867–868 CrossRef CAS; (b) E. J. Corey and M. Chaykovsky, J. Am. Chem. Soc., 1965, 87, 1353–1364 CrossRef CAS; (c) A. W. Johnson and R. B. Lacount, Chem. Ind., 1958, 1440–1441 Search PubMed.
- (a) V. K. Aggarwal, in Comprehensive Asymmetric Catalysis, ed. E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Springer, New York, 1999, vol. 2, 679 Search PubMed; (b) E. M. McGarrigle, E. L. Myers, O. Illa, M. A. Shaw, S. L. Riches and V. K. Aggarwal, Chem. Rev., 2007, 107, 5841–5883 CrossRef CAS.
- (a) O. Illa, M. Arshad, A. Ros, E. M. McGarrigle and V. K. Aggarwal, J. Am. Chem. Soc., 2010, 132, 1828–1830 CrossRef CAS; (b) J. Zanardi, C. Leriverend, D. Aubert, K. Julienne and P. Metzner, J. Org. Chem., 2001, 66, 5620–5623 CrossRef CAS; (c) M. Davoust, J.-F. Briere, P.-A. Jaffres and P. Metzner, J. Org. Chem., 2005, 70, 4166–4169 CrossRef CAS; (d) V. K. Aggarwal and J. Richardson, Chem. Commun., 2003, 2644–2651 RSC.
- For glycidic amides from sulfur ylides see: (a) Y.-G. Zhou, X.-L. Hou, L.-X. Dai, L.-J. Xia and M.-H. Tang, J. Chem. Soc., Perkin Trans. 1, 1999, 77–80 RSC; (b) V. K. Aggarwal, J. P. H. Charmant, D. Fuentes, J. N. Harvey, G. Hynd, D. Ohara, W. Picoul, R. Robiette, C. Smith, J.-L. Vasse and C. L. Winn, J. Am. Chem. Soc., 2006, 128, 2105–2114 CrossRef CAS; (c) V. K. Aggarwal, G. Hynd, W. Picoul and J.-L. Vasse, J. Am. Chem. Soc., 2002, 124, 9964–9965 CrossRef CAS.
- For the use of cyano-stabilised ammonium ylides see: (a) A. Kowalkowska, D. Sucholbiak and A. Jonczyk, Eur. J. Org. Chem., 2005, 925–933 CrossRef CAS; (b) A. Jonczyk and A. Konarska, Synlett, 1999, 1085–1087 CrossRef CAS.
- A. Alex, B. Larmanjat, J. Marrot, F. Couty and O. David, Chem. Commun., 2007, 2500–2502 RSC.
- For benzylic ammonium ylides see: (a) T. Kimachi, H. Kinoshita, K. Kusaka, Y. Takeuchi, M. Aoe and M. Ju-ichi, Synlett, 2005, 842–844 CrossRef CAS; (b) H. Kinoshita, A. Ihoriya, M. Ju-ichi and T. Kimachi, Synlett, 2010, 2330–2334 CAS; (c) R. Robiette, M. Conza and V. K. Aggarwal, Org. Biomol. Chem., 2006, 4, 621–623 RSC.
- M. Waser, R. Herchl and N. Müller, Chem. Commun., 2011, 47, 2170–2172 RSC.
- For selected examples see: (a) M. J. Porter and J. Skidmore, Chem. Commun., 2000, 1215–1225 RSC; (b) T. Nemoto, H. Kakei, V. Gnanadesikan, S.-Y. Tosaki, T. Ohshima and M. Shibasaki, J. Am. Chem. Soc., 2002, 124, 14544–14545 CrossRef CAS; (c) S. Matsunaga, T. Kinoshita, S. Okada, S. Harada and M. Shibasaki, J. Am. Chem. Soc., 2004, 126, 7559–7570 CrossRef CAS.
- For stereoselective Darzens approaches see: (a) S. Arai, K. Tokumaru and T. Aoyama, Tetrahedron Lett., 2004, 45, 1845–1848 CrossRef CAS; (b) E. J. Corey and S. Choi, Tetrahedron Lett., 1991, 32, 2857–2860 CrossRef CAS; (c) P. Bakó, Z. Rapi, G. Keglevich, T. Szabó, P. Sóti, T. Vigh, A. Grün and T. Holczbauer, Tetrahedron Lett., 2011, 52, 1473–1476 Search PubMed.
- For the syntheses of cyclopropanes see: (a) M. J. Gaunt and C. C. C. Johansson, Chem. Rev., 2007, 107, 5596–5605 CrossRef CAS; (b) C. C. C. Johansson, N. Bremeyer, S. V. Ley, D. R. Owen, S. C. Smith and M. J. Gaunt, Angew. Chem., Int. Ed., 2006, 45, 6024–6028 CrossRef CAS; (c) C. D. Papageorgiou, M. A. Cubillo de Dios, S. V. Ley and M. J. Gaunt, Angew. Chem., Int. Ed., 2004, 43, 4641–4644 CrossRef CAS; (d) N. Bremeyer, S. C. Smith, S. V. Ley and M. J. Gaunt, Angew. Chem., Int. Ed., 2004, 43, 2681–2684 CrossRef CAS; (e) C. D. Papageorgiou, S. V. Ley and M. J. Gaunt, Angew. Chem., Int. Ed., 2003, 42, 828–831 CrossRef CAS.
- C.-Y. Zhu, X.-M. Deng, X.-L. Sun, J.-C. Zheng and Y. Tang, Chem. Commun., 2008, 738–740 RSC.
- V. K. Aggarwal, J. N. Harvey and R. Robiette, Angew. Chem., Int. Ed., 2005, 44, 5468–5471 CrossRef CAS.
- For aziridination see: L. D. S. Yadav, R. Kapoor and Garima, Synlett, 2009, 3123–3126 Search PubMed.
- Y. Wang, Z. Chen, A. Mi and W. Hu, Chem. Commun., 2004, 2486–2487 RSC.
- Although all reactions were carried out under inert conditions, varying amounts of benzoic acids were observed in several cases presumably due to autoxidation of the aldehydes. Thus 2 equiv. of aldehyde were used to obtain a reasonable compromise between yield and required amount of starting material.
- This means correlating a kinetic property (leaving group ability) with a thermodynamic property (basicity): K. P. C. Vollhardt and N. E. Schore, Organic Chemistry, W. H. Freeman, New York, 1998 Search PubMed.
- pKa values in H2O: (a) E. A. Castro, M. Aliaga, P. R. Campodonico, J. R. Leis, L. Garcia-Rio and J. G. Santos, J. Phys. Org. Chem., 2008, 21, 102–107 CrossRef CAS; (b) D. R. Lide, Handbook of Chemistry and Physics, 76th Ed. CRC Press, Boca Raton, 1995–1996 Search PubMed; (c) M. Srinivas, M. G. Hopperstad and D. C. Spray, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 10942–10947 Search PubMed.
- It has been shown that bond rotation towards betaines B is the crucial step in epoxidations using stabilised ylides (see Ref [3d]).
- T. Hama, X. Liu, D. A. Culkin and J. F. Hartwig, J. Am. Chem. Soc., 2003, 125, 11176–11177 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details including analytical and spectroscopic data of new compounds. See DOI: 10.1039/c1ob05721a |
| This journal is © The Royal Society of Chemistry 2011 |