First total synthesis of Papilistatin†
Received
20th December 2010
, Accepted 25th January 2011
First published on 26th January 2011
Abstract
Papilistatin has been isolated recently and found to have good anticancer and antibacterial activity. Papilistatin is a unique phenanthrene-1,10-dicarboxylic acid. The first total synthesis of papilistatin is described here with radical cyclisation as the key step.
Introduction
Papilistatin (Fig. 1) was isolated in 2010 by Pettit from a Taiwan butterfly, and was found to have anticancer and antibacterial activity.1 Against a panel of six human and the murine P388 leukemia cancer cell lines, papilistatin exhibited cancer cell growth inhibition with GI50’s of 0.093–3.5 μg mL−1. We have been continuously studying the synthesis and biological activities of phenanthroindolizidine alkaloids.2 Papilistatin is a unique phenanthrene-1,10-dicarboxylic acid which is similar to the intermediates used to construct phenanthroindolizidine alkaloids. Papilistatin's relatively simple structure and good biological activities evoked our interest. In order to explore if it can be used as an alternative to phenanthroindolizidine alkaloids, we developed the first total synthesis of papilistatin.
 |
| | Fig. 1 Structure of papilistatin. | |
Results and discussion
Our retrosynthetic analysis is shown in Scheme 1. Apparently, the construction of the phenanthrene ring would be the key step to synthesize papilistatin. To the best of our knowledge, the most convenient way to construct the phenanthrene ring system is intramolecular oxidative coupling of diphenylethene using oxidative coupling reagents such as Pb(OAc)4,3 VOF3,4 FeCl3,2a,5 MnO26 and m-CPBA.7 However, although we tried many oxidative coupling reagents (such as VOF3, FeCl3, MnO2, m-CPBA and PIFA), no oxidative coupling product 5 was formed from 4a. We believed that the electron-withdrawing methylcarbonyl group at the benzene ring of diphenylethene 4a made the oxidative coupling impossible. We then introduced bromine into the compound 2a to get 2b and then to afford 4b. An intramolecular coupling reaction of 4b mediated by a palladium catalytic reaction was tried. But when we used Pd(PPh3)2Cl2 and Pd(OAc)2 as the palladium catalyst, we also failed to construct the phenanthrene ring. Then we turned to radical cyclisation with Bu3SnH and AIBN as reagents which were reported to be used to construct the phenanthrene ring system efficiently.8 Using this method, we finally succeeded to get 5 from 4b.
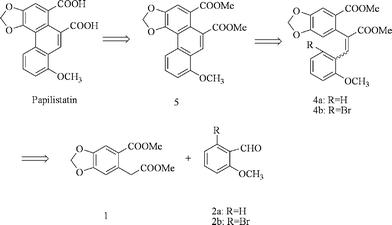 |
| | Scheme 1 Retrosynthetic analysis of papilistatin. | |
2-Methoxycarbonyl-4,5-methylenedioxyphenyl acetic acid methyl ester 1 was obtained employing piperonal 6 as the starting material. Condensation of commercially available piperonal 6 and malonic acid in the presence of piperidine and subsequent catalytic hydrogenation gave (3,4-methylenedioxy)phenylpropionic acid 8 in 74.8% yield over two steps.98 was treated with oxalyl choride in dichloromethane to give the corresponding acyl chloride, which formed 5,6-(methylenedioxy)-l-indanone 9via intramolecular Friedel-Crafts acylation in the presence of SnCl4 in 95% yield.10 Treating indanone 9 with amyl nitrite in methanol with HC1 afforded the 2-(hydroxyimino)-5,6-(methylenedioxy)-1-indanon 10 in 75% yield.10 Then treating the hydroxyimino ketone 10 with p-toluenesufonyl chloride in sodium hydroxide solution afforded 2-carboxy-4,5-methylenedioxyphenylacetic acid 11via esterification, Beckmann rearrangement and subsequent hydrolysis in 65% yield.11 Acid 11 reacted with diazomethane to give 1 in 98% yield (Scheme 2).
 |
| | Scheme 2 Synthesis of compound 1. | |
2-bromo-6-methoxybenzaldehyde 2b could be easily synthesized from commercially available 2-methoxybenzaldehyde 2a according to reported literature (Scheme 3).12
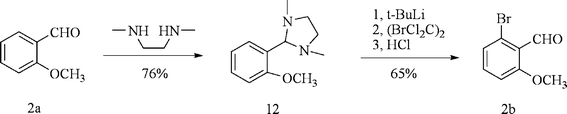 |
| | Scheme 3 Synthesis of compound 2. | |
With 1 and 2b in hand, we started to conduct reactions following our synthetic route (Scheme 4). The base-mediated condensation of aldehydes or ketones with dialkyl succinate to afford alkylidene succinic monoesters is known as Stobbe condensation. Stobbe condensation of 1 and 2b in the presence of sodium methoxide in methanol afforded 3-(2-bromo-6-methoxyphenyl)-2-(2-carboxyl-4,5-methylenedioxyphenyl)-acrylic acid methyl ester 3 in a yield of 92% as a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of Z and E isomers (as determined by 1H NMR). Then 3 was treated with diazomethane to afford the corresponding dimethyl ester 4b (Z/E = 1:1) in a yield of 98%. As we know, the E isomer of 4b is more suitable than the Z isomer for intramolecular radical cyclisation, therefore we tried to improve the percentage of the E isomer in 3 and 4b. But we found the percentage of the E isomer of 3 decreased when potassium tert-butoxide or sodium hydride was used as the base in Stobbe condensation. The effort to turn the Z isomers of 3 and 4b to their corresponding E isomers also failed. The Z and E isomers of 3 and 4b were very difficult to separate and only a little of the pure Z isomer of 4b was obtained. Therefore, the mixture 4b was used for the next step. As we got the pure Z isomer of 4b, Z and E of 4b can be easily assigned comparing their alkene-H chemical shift in NMR. According to the reference and chemdraw, the chemical shift of alkene-H in E-4b is more down-field than Z-4b, since in E-4b the ester group is at the cis-position of alkene-H, which can provide a greater deshielding effect. In the same way, we could assign the Z and E isomer of 3. The intramolecular radical cyclisation of 4b (Z/E = 1:1) with Bu3SnH and AIBN proved to be a little complicated, and 3,4-methylenedioxy-8-methoxyphenanthrene-1,10-dicarboxylic acid dimethyl ester 5 was separated in a yield of 30%. The main by-product separated was 4a. Treating the Z isomer of 4b in the same condition could afford 5 only in 10%, which also accounted for the low yield of the reaction above. Nonetheless, 5 was obtained easily and then converted to its corresponding dicarboxylic acid papilistatin through saponification in a yield of 85%.
:1 mixture of Z and E isomers (as determined by 1H NMR). Then 3 was treated with diazomethane to afford the corresponding dimethyl ester 4b (Z/E = 1:1) in a yield of 98%. As we know, the E isomer of 4b is more suitable than the Z isomer for intramolecular radical cyclisation, therefore we tried to improve the percentage of the E isomer in 3 and 4b. But we found the percentage of the E isomer of 3 decreased when potassium tert-butoxide or sodium hydride was used as the base in Stobbe condensation. The effort to turn the Z isomers of 3 and 4b to their corresponding E isomers also failed. The Z and E isomers of 3 and 4b were very difficult to separate and only a little of the pure Z isomer of 4b was obtained. Therefore, the mixture 4b was used for the next step. As we got the pure Z isomer of 4b, Z and E of 4b can be easily assigned comparing their alkene-H chemical shift in NMR. According to the reference and chemdraw, the chemical shift of alkene-H in E-4b is more down-field than Z-4b, since in E-4b the ester group is at the cis-position of alkene-H, which can provide a greater deshielding effect. In the same way, we could assign the Z and E isomer of 3. The intramolecular radical cyclisation of 4b (Z/E = 1:1) with Bu3SnH and AIBN proved to be a little complicated, and 3,4-methylenedioxy-8-methoxyphenanthrene-1,10-dicarboxylic acid dimethyl ester 5 was separated in a yield of 30%. The main by-product separated was 4a. Treating the Z isomer of 4b in the same condition could afford 5 only in 10%, which also accounted for the low yield of the reaction above. Nonetheless, 5 was obtained easily and then converted to its corresponding dicarboxylic acid papilistatin through saponification in a yield of 85%.
 |
| | Scheme 4 Synthesis of papilistatin. | |
Conclusion
Papilistatin was first synthesized from easily available materials 1 and 2b under mild conditions in only 4 steps with an overall yield of 23%. The chemistry described here provides a practical synthetic method of papilistatin, with which we could also prepare diversities of papilistatin for biochemical and pharmaceutical studies.
Experimental
The melting points were determined with an X-4 binocular microscope melting-point apparatus (Beijing Tech Instruments Co, Beijing, China) and were uncorrected. 1H NMR spectra were obtained by using a Bruker AV 400 spectrometer. Chemical shifts (δ) were given in parts per million (ppm) and were measured downfield from internal tetramethylsilane.13C NMR spectra were recorded by using a Bruker AV 400 (100 MHz) and CDCl3, C5D5N or DMSO-d6 as a solvent. Chemical shifts (δ) are reported in parts per million using the solvent peak. High-resolution mass spectra were obtained with an FT-ICR MS spectrometer (Ionspec, 7.0 T). All anhydrous solvents were dried and purified by standard techniques just before use.
2-Methoxycarbonyl-4,5-methylenedioxyphenylacetic acid methyl ester (1)
Compound 11 (1.81 g, 8.10 mmol) in dry dichloromethane (15 mL) was cooled to 0 °C and then carefully treated with diazomethane until the reaction had finished. The excess diazomethane was decomposed with a few drops of glacial acetic acid. The resulting mixture was evaporated to afford 2.00 g (98%) of 1 as a white solid. mp 87–88 °C; 1H NMR (400 MHz, CDCl3): δ 7.49 (s, 1 H, Ph-H), 6.71 (s, 1 H, Ph-H), 6.03 (s, 2 H, OCH2O), 3.93 (s, 2 H, Ph-CH2-), 3.83 (s, 3 H, COOCH3), 3.70 (s, 3 H, COOCH3); 13C NMR (100 MHz, CDCl3) δ 172.0, 166.6, 150.8, 146.8, 132,3, 122.7, 112.1, 110.7, 101.9, 51.9, 40.4. HRMS (ESI): calcd. for C12H12O6 [M + Na]+ 275.0526, found 275.0527.
(Z/E)-3-(2-Bromo-6-methoxyphenyl)-2-(2-carboxyl-4,5-methylenedioxyphenyl)-acrylic acid methyl ester (3)
Sodium (0.19 g, 8.10 mmol) was stirred in methanol (80 mL) until it disappeared. 1 (2.00 g, 7.94 mmol) and 2b (1.73 g, 8.10 mmol) were added to the solution. The mixture was heated at reflux for 4 h and then rotary evaporated to remove most of the methanol. The resulting mixture was cooled to 0 °C and acidified with 2 mol L−1 hydrochloric acid, then filtered, washed with water and dried to give 3.17 g (92%) of 3 (Z/E = 1:1) as a white solid. mp 176–179 °C; 1H NMR (400 MHz, CDCl3): δ 7.59 (s, 1 H, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 7.56 (s, 1 H, CCH), 7.55 (s, 1 H, Ar–H), 7.22 (d, J = 8.0 Hz, 1 H, Ar–H), 7.12–7.17 (m, 2 H, Ar–H), 7.06 (t, J = 8.0 Hz, 1 H, Ar–H), 7.02 (s, 1 H, Ar–H), 6.86 (d, J = 8.0 Hz, 1 H, Ar–H), 6.75 (s, 1 H, Ar–H), 6.62 (d, J = 8.4 Hz, 1 H, Ar–H), 6.42 (s, 1 H, Ar–H), 6.11 (s, 2 H, OCH2O), 5.97 (s, 2 H, OCH2O), 3.80 (s, 6 H, COOCH3), 3.51 (s, 3 H, OCH3), 3.46 (s, 3 H, OCH3); 13C NMR (100 MHz, CDCl3) δ 171.0, 170.5, 167.1, 166.3, 157.8, 156.9, 147.5, 147.1, 137.8, 137.5, 135.0, 134.2, 133.9, 130.0, 129.4, 126.9, 125.2, 124.4, 124.3, 123.6, 122.0, 112.3, 110.7, 110.6, 109.5, 102.2, 101.9, 56.0, 55.2, 52.2, 51.4. HRMS (ESI): calcd. for C19H15BrO7 [M − H]− 432.9928, found 432.9922.
CH), 7.56 (s, 1 H, CCH), 7.55 (s, 1 H, Ar–H), 7.22 (d, J = 8.0 Hz, 1 H, Ar–H), 7.12–7.17 (m, 2 H, Ar–H), 7.06 (t, J = 8.0 Hz, 1 H, Ar–H), 7.02 (s, 1 H, Ar–H), 6.86 (d, J = 8.0 Hz, 1 H, Ar–H), 6.75 (s, 1 H, Ar–H), 6.62 (d, J = 8.4 Hz, 1 H, Ar–H), 6.42 (s, 1 H, Ar–H), 6.11 (s, 2 H, OCH2O), 5.97 (s, 2 H, OCH2O), 3.80 (s, 6 H, COOCH3), 3.51 (s, 3 H, OCH3), 3.46 (s, 3 H, OCH3); 13C NMR (100 MHz, CDCl3) δ 171.0, 170.5, 167.1, 166.3, 157.8, 156.9, 147.5, 147.1, 137.8, 137.5, 135.0, 134.2, 133.9, 130.0, 129.4, 126.9, 125.2, 124.4, 124.3, 123.6, 122.0, 112.3, 110.7, 110.6, 109.5, 102.2, 101.9, 56.0, 55.2, 52.2, 51.4. HRMS (ESI): calcd. for C19H15BrO7 [M − H]− 432.9928, found 432.9922.
(Z/E)-3-(2-Bromo-6-methoxyphenyl)-2-(2-methoxycarbonyl-4,5-methylenedioxyphenyl)-acrylic acid methyl ester (4b)
Compound 3 (3.17 g, 7.30 mmol) was dissolved in dry dichloromethane (40 mL) and cooled to 0 °C and then carefully treated with diazomethane until the reaction had finished. The excess diazomethane was decomposed with a few drops of glacial acetic acid. The resulting mixture was evaporated to afford 3.31 g (98%) of 4b as a white oil. A little of the Z isomer of 4b could be obtained through column chromatography (petroleum ether–EtOAc, 8:1, v/v). For 4b (Z/E = 1:1): 1H NMR (400 MHz, CDCl3): δ 7.52 (s, 1 H, CCH), 7.48 (s, 1 H, CCH), 7.46 (s, 1 H, Ar–H), 7.20 (d, J = 8.0 Hz, 1 H, Ar–H), 7.12 (t, J = 8.8 Hz, 1 H, Ar–H), 7.11 (d, J = 8.4 Hz, 1 H, Ar–H), 7.02 (t, J = 8.0 Hz, 1 H, Ar–H), 6.98 (s, 1 H, Ar–H), 6.84 (d, J = 8.0 Hz, 1 H, Ar–H), 6.69 (s, 1 H, Ar–H), 6.59 (d, J = 8.4 Hz, 1 H, Ar–H), 6.38 (s, 1 H, Ar–H), 6.06 (s, 2 H, OCH2O), 5.92 (s, 2 H, OCH2O), 3.84 (s, 3 H, COOCH3), 3.79 (s, 6 H, COOCH3), 3.78 (s, 3 H, COOCH3), 3.52 (s, 3 H, OCH3), 3.41 (s, 3 H, OCH3); For the Z isomer of 4b: 1H NMR (400 MHz, CDCl3): δ 7.48 (s, 1 H, CCH), 7.20 (d, J = 8.0 Hz, 1 H, Ar–H), 7.13 (t, J = 8.0 Hz, 1 H, Ar–H), 6.99 (s, 1 H, Ar–H), 6.84 (d, J = 8.0 Hz, 1 H, Ar–H), 6.69 (s, 1 H, Ar–H), 6.07 (s, 2 H, OCH2O), 3.79 (s, 3 H, COOCH3), 3.78 (s, 3 H, COOCH3), 3.52 (s, 3 H, OCH3); 13C NMR (100 MHz, CDCl3) δ 167.2, 166.0, 156.9, 150.2, 147.0, 137.8, 133.9, 133.7, 130.0, 125.1, 124.3, 124.2, 122.9, 110.4, 109.9, 109.5, 101.9, 55.1, 52.1, 51.8. HRMS (ESI): calcd. for C20H17BrO7 [M + Na]+ 471.0050, found 471.0052.
3,4-Methylenedioxy-8-methoxyphenanthrene-1,10-dicarboxylic acid dimethyl ester (5)
To a solution of 4b (3.31 g, 7.16 mmol) in toluene (200 mL), AIBN (0.23 mg, 1.43 mmol) and Bu3SnH (5.00 g, 17.18 mmol) were added, and the mixture was heated under reflux for 6 h. The reaction mixture was concentrated by rotary evaporation, and purified by column chromatography (petroleum ether–EtOAc, 10:1, v/v) to give 0.82 g (30%) of 5 as a yellow solid. mp 212–213 °C; 1H NMR (400 MHz, CDCl3): δ 8.68 (d, J = 8.0 Hz, 1 H, H-5), 8.67 (s, 1 H, H-9), 7.71 (s, 1 H, H-2), 7.63 (t, J = 8.0 Hz, 1 H, H-6), 7.06 (d, J = 8.0 Hz, 1 H, H-7), 6.32 (s, 2 H, OCH2O), 4.04 (s, 3 H, OCH3), 3.92 (s, 3 H, COOCH3), 3.90 (s, 3 H, COOCH3); 13C NMR (100 MHz, CDCl3) δ 169.4, 168.8, 156.1, 146.6, 144.9, 130.4, 129.2, 127.2, 125.4, 124.2, 123.8, 121.2, 119.0, 117.8, 112.1, 107.3, 101.9, 55.7, 52.1, 52.0. HRMS (ESI): calcd. for C20H16O7 [M + Na]+ 391.0788, found 391.0796.
Papilistatin
Compound 5 (0.82 g, 2.15 mmol) was dissolved in methanol (35 mL) and dioxane (30 mL) and then 20% potassium hydroxide (55 mL) was added to the solution. The mixture was warm to 40 °C and stirred for 6 h. The mixture was rotary evaporated to remove dioxane and methanol, and the resulting mixture was cooled to 0 °C and acidified with 2 mol L−1 hydrochloric acid to afford 0.62 g (85%) of papilistatin as a yellow solid. mp > 300 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.89 (br, 1 H, COOH), 12.85 (br, 1 H, COOH), 8.65 (d, J = 8.4 Hz, 1 H, H-5), 8.45 (s, 1 H, H-9), 7.74 (t, J = 8.4 Hz, 1 H, H-6), 7.70 (s, 1 H, H-2), 7.30 (d, J = 8.0 Hz, 1 H, H-7), 6.44 (s, 2 H, OCH2O), 4.04 (s, 3 H, OCH3); 1H NMR (400 MHz, C5D5N) δ 9.29 (s, 1 H, H-9), 8.84 (d, J = 8.0 Hz, 1 H, H-5), 8.21 (s, 1H, H-2), 7.67 (t, J = 8.0 Hz, 1 H, H-6), 7.08 (d, J = 8.0 Hz, 1 H, H-7), 6.23 (s, 2 H, OCH2O), 3.83 (s, 3 H, OCH3); 13C NMR (100 MHz, C5D5N) δ 172.2, 171.7, 156.8, 146.7, 145.8, 131.9, 131.2, 129.6, 129.0, 125.3, 124.7, 122.3, 120.1, 118.8, 112.8, 108.3, 102.8, 56.2. HRMS (ESI): calcd. for C18H12O7 [M − H]− 339.0510, found 339.0501.
Acknowledgements
We are grateful to the National Key Project for Basic Research (2010CB126100) and the National Natural Science Foundation of China (20872072) for generous financial support for our programs.
Notes and references
- G. R. Pettit, Q. H. Ye, D. L. Herald, F. Hogan and R. K. Pettit, J. Nat. Prod., 2010, 73, 164–166 CrossRef CAS
 .
.
-
(a) K. L. Wang, M. Y. Lü, Q. M. Wang and R. Q. Huang, Tetrahedron, 2008, 64, 7504–7510 CrossRef CAS ;
(b) Z. W. Wang, Z. Li, K. L. Wang and Q. M. Wang, Eur. J. Org. Chem., 2010, 292–299 CrossRef ;
(c) M. B. Cui, H. J. Song, A. Z. Feng, Z. W. Wang and Q. M. Wang, J. Org. Chem., 2010, 75, 7018–7021 CrossRef CAS ;
(d) K. L. Wang, B. Su, Z. W. Wang, M. Wu, Z. Li, Y. N. Hu, Z. J. Fan, N. Mi and Q. M. Wang, J. Agric. Food Chem., 2010, 58, 2703–2709 CrossRef CAS ;
(e) K. L. Wang, Y. N. Hu, Y. X. Liu, N. Mi, Z. J. Fan, Y. Liu and Q. M. Wang, J. Agric. Food Chem., 2010, 58, 12337–12342 CrossRef CAS .
- K. S. Feldman and S. M. Ensel, J. Am. Chem. Soc., 1994, 116, 3357–3366 CrossRef CAS .
-
(a) B. Halton, A. I. Maidment, D. L. Officer and J. M. Warner, Aust. J. Chem., 1984, 37, 2119–2128 CAS ;
(b) D. A. Evans, C. J. Dinsmore, D. A. Evrard and K. M. DcVries, J. Am. Chem. Soc., 1993, 115, 6426–6427 CrossRef CAS ;
(c) A. Furstner and J. W. J. Kennedy, Chem.–Eur. J., 2006, 12, 7398–7410 CrossRef .
- K. L. Wang, M. Y. Lü, A. Yu, X. Q. Zhu and Q. M. Wang, J. Org. Chem., 2009, 74, 935–938 CrossRef CAS .
- K. L. Wang, Y. N. Hu, Z. Li, M. Wu, Z. H. Liu, B. Su, A. Yu, Y. Liu and Q. M. Wang, Synthesis, 2009, 7, 1083–1090 .
- K. L. Wang, Y. N. Hu, M. Wu, Z. Li, Z. H. Liu, B. Su, A. Yu, Y. X. Liu and Q. M. Wang, Tetrahedron, 2010, 66, 9135–9140 CAS .
-
(a) D. C. Harrowven, M. I. T. Nunn and D. R. Fenwick, Tetrahedron Lett., 2002, 43, 3185–3187 CrossRef CAS ;
(b) D. C. Harrowven, I. L. Guy and L. Nanson, Angew. Chem., Int. Ed., 2006, 45, 2242–2245 CrossRef CAS ;
(c) H. Suzuki, S. Aoyagi and C. Kibayashi, J. Org. Chem., 1996, 60, 6114–6122 ;
(d) A. Couture, E. Deniau, P. Grandclaudon and C. Hoarau, J. Org. Chem., 1998, 63, 3128–3132 CrossRef CAS .
- H. Yasushi, O. Momotoshi and S. Yoshihiro, Biosci., Biotechnol., Biochem., 2003, 67, 2183–2193 CrossRef CAS .
- E. N. David, K. B. William, P. J. Michael, O. Robert and M. R. Robert, J. Med. Chem., 1990, 33, 703–710 CrossRef .
- N. J. Srivastava and D. N. Chaudhury, J. Org. Chem., 1962, 4337–4341 CrossRef .
- M. Rawat, V. Prutyanov and W. D. Wulff, J. Am. Chem. Soc., 2006, 128, 11044–11053 CrossRef CAS .
Footnote |
| † Electronic Supplementary Information (ESI) available: 1H, 13C-NMR and HRMS spectra for the compouds 1, 3, 4b, 5, papilistatin. See DOI: 10.1039/c0ob01214a. |
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.