Degenerate [2]rotaxanes with electrostatic barriers†
Hao
Li‡
a,
Yan-Li
Zhao‡
ac,
Albert C.
Fahrenbach
a,
Soo-Young
Kim§
b,
Walter F.
Paxton
a and
J. Fraser
Stoddart
*a
aDepartment of Chemistry, Northwestern University, 2145 Sheridan Road, Evanston, Illinois 60208-3113, USA. E-mail: stoddart@northwestern.edu
bDepartment of Chemistry and Biochemistry, University of California, Los Angeles, 405 Hilgard Avenue, Los Angeles, California 90095, USA
cDivision of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, 21 Nanyang Link, 637371, Singapore
First published on 23rd February 2011
Abstract
A synthetic approach to the preparation of [2]rotaxanes (1–5·6PF6) incorporating bispyridinium derivatives and two 1,5-dioxynaphthalene (DNP) units situated in the rod portions of their dumbbell components that are encircled by a single cyclobis(paraquat-p-phenylene) tetracationic (CBPQT4+) ring has been developed. Since the π-electron-deficient bispyridinium units are introduced into the dumbbell components of the [2]rotaxanes 1–5·6PF6, there are Coulombic charge–charge repulsions between these dicationic units and the CBPQT4+ ring in the [2]rotaxanes. Thus, the CBPQT4+ rings in the degenerate [2]rotaxanes exhibit slow shuttling between two DNP recognition sites on the 1H NMR time-scale on account of the electrostatic barrier posed by the bispyridinium units, as demonstrated by variable-temperature 1H NMR spectroscopy. Electrochemical experiments carried out on the [2]rotaxanes 1·6PF6 and 2·6PF6 indicate that the one-electron reduced bipyridinium radical cation in the dumbbell components of the [2]rotaxanes serves as an additional recognition site for the two-electron reduced CBPQT2(˙+) diradical cationic ring. Under appropriate conditions, the ring components in the degenerate rotaxanes 1·6PF6 and 2·6PF6 can shuttle along the recognition sites – two DNP units and one-electron reduced bipyridinium radical cation – under redox control.
Introduction
Molecular switches and machines1–3 incorporating donor–acceptor [2]catenanes1a–e and [2]rotaxanes1a–e have been the focus of extensive experimental investigations in solution1a,1c–e as well as in solid-state devices.1b,3a,3e In particular, switchable [2]rotaxanes, containing π-electron-rich tetrathiafulvalene (TTF) and/or 1,5-dioxynaphthalene (DNP) recognition sites, located in the rod portions of their dumbbell components and encircled by a single cyclobis(paraquat-p-phenylene) (CBPQT4+) ring have been investigated3a,3b,3e as leading candidates for expressing the relative intramolecular translational motion1m between its dumbbell and ring components.![(a) Structural formulae for the five degenerate [2]rotaxanes 1–5·6PF6. (b) The two different kinds of dumbbell components present in these five degenerate [2]rotaxanes. In one set (1–3·6PF6) of rotaxanes, the two positive charges are part of a conjugate aromatic system, while, in the other set (4·6PF6 and 5·6PF6), the charges are associated with separate aromatic systems.](/image/article/2011/OB/c0ob00937g/c0ob00937g-f1.gif) | ||
| Fig. 1 (a) Structural formulae for the five degenerate [2]rotaxanes 1–5·6PF6. (b) The two different kinds of dumbbell components present in these five degenerate [2]rotaxanes. In one set (1–3·6PF6) of rotaxanes, the two positive charges are part of a conjugate aromatic system, while, in the other set (4·6PF6 and 5·6PF6), the charges are associated with separate aromatic systems. | ||
The constitution of the spacers between the recognition sites on the dumbbell components of the degenerate [2]rotaxanes is an important factor governing the movement of the CBPQT4+ ring. Previously, we investigated the switching behaviour of the CBPQT4+ ring in degenerate [2]rotaxanes with spacers such as polyethylene glycol chains,4terphenyl units,5 rigid arylmethyl and butadiynyl rods,6 and azobenzene units.7 In the case of degenerate [2]rotaxanes4c,6c,6d,7 containing two equivalent recognition sites – e.g., two DNP units – the CBPQT4+ ring shuttles back and forth between the two sites usually under the influence of heat. In a few instances,7 the rate of shuttling has been controlled by means of light when, for example, azobenzene units are the spacers. In the case of bistable [2]rotaxanes,4,5 like those mentioned above, the CBPQT4+ ring can be induced to move between TTF and DNP units by means of chemical5 or electrochemical4,5 stimuli. The bistability of the switchable [2]rotaxanes relies (Fig. 2) on the ability of the CBPQT4+ ring to encircle much more strongly the TTF unit in the so-called ground state co-conformation (GSCC) than it does the considerably less π-electron rich DNP unit in the metastable state co-conformation (MSCC) in the dumbbell components. Switching is achieved by the reversible oxidation of the TTF unit, firstly to the radical cation4d,4e,8 (TTF+˙) and then to its dication (TTF2+), thus producing Coulombic charge–charge repulsion between the oxidised TTF+˙ or TTF2+ species and the CBPQT4+ ring, leading to the movement of the CBPQT4+ ring to the DNP unit. After reduction of the TTF+˙ radical cation or TTF2+ dication to their neutral form, the MSCC becomes populated, and the CBPQT4+ ring begins to migrate back onto the TTF recognition unit. The barrier to this relaxation process (ΔG‡) is 3 kcal mol−1 higher10b in the presence of the bipyridinium unit (BIPY2+).
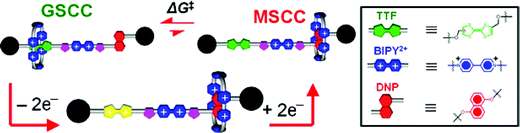 | ||
| Fig. 2 Graphical representation of a rotaxane undergoing redox-stimulated switching employing TTF and DNP recognition units, with a bipyridinium unit (BIPY2+) in the middle of the dumbbell component as a “speed bump”. The GSCC and the MSCC represent the ground state co-conformation and the metastable state co-conformation, respectively. | ||
In addition to the traditional TTF and/or DNP recognition sites for the CBPQT4+ ring in the [2]rotaxanes, of particular interest is the introduction of potential electrostatic barriers – that is, positively charged entities – which would potentially curtail the translational motion undergone by the CBPQT4+ ring. In order to investigate the influence of placing “speed bumps” in the shape of positively charged entities between two DNP units in degenerate molecular shuttles, the [2]rotaxanes 1–5·6PF6 (Fig. 1) have been synthesised (Schemes 1, 2 and 3). In 1·6PF6 and 2·6PF6, the speed bumps are BIPY2+ units. In 3·6PF6, the speed bump is a 1,4-bis(pyridinium)benzene9 unit. In the case of 4·6PF6 and 5·6PF6, the conjugation between the two pyridinium rings is broken by spacers containing saturated chains of atoms. All of the five molecular shuttles have been investigated by variable temperature (dynamic) 1H NMR spectroscopy: they all exhibit slow shuttling on the 1H NMR time-scale, at least up to +70 °C in CD3CN solutions.
![Synthesis of the [2]rotaxane 1·6PF6.](/image/article/2011/OB/c0ob00937g/c0ob00937g-s1.gif) | ||
| Scheme 1 Synthesis of the [2]rotaxane 1·6PF6. | ||
![Syntheses of the [2]rotaxanes 2·6PF6, 4·6PF6 and 5·6PF6.](/image/article/2011/OB/c0ob00937g/c0ob00937g-s2.gif) | ||
| Scheme 2 Syntheses of the [2]rotaxanes 2·6PF6, 4·6PF6 and 5·6PF6. | ||
![Synthesis of the [2]rotaxane 3·6PF6.](/image/article/2011/OB/c0ob00937g/c0ob00937g-s3.gif) | ||
| Scheme 3 Synthesis of the [2]rotaxane 3·6PF6. | ||
Recently, we introduced10BIPY2+ units into the dumbbell components of the CBPQT4+ ring-containing [2]rotaxanes and demonstrated10 that the one-electron reduced radical cation (BIPY˙+) serves as an additional recognition site for the two-electron reduced CBPQT2(˙+) ring. Electrochemical experiments carried out on 1·6PF6 and 2·6PF6 indicate that the BIPY˙+ radical cation does act as an additional recognition site for the CBPQT2(˙+) ring. Thus, ring shuttling can be induced by reduction in the case of 1·6PF6 and 2·6PF6. This paper describes how speed bumps can be introduced into molecular shuttles and how, in some instances, they can be modified by replacing the spacers. Establishing this kind of control in the switching of bistable [2]rotaxanes could have important consequences for the development of molecular flash memory using molecules of this kind.
Results and discussion
The synthesis of the [2]rotaxane 1·6PF6 is summarised in Scheme 1. Reaction of the tosylated half dumbbell106 with 4,4′-bipyridine afforded the dumbbell 7·2PF6 after counterion exchange. The [2]rotaxane 1·6PF6 was obtained from 7·2PF6, the 8·2PF6 salt,11 and 1,4-bis(bromomethylbenzene) using a template-directed protocol12 in DMF under 10 kbar pressure at room temperature for 3 days. After the reaction was complete, the crude mixture was purified by preparative TLC on silica gel using Me2CO/NH4PF6 (100/1 v/w) as the mobile phase to afford 1·6PF6 in an overall yield of 37%. The syntheses of the [2]rotaxanes 2·6PF6,4·6PF6 and 5·6PF6 are summarised in Scheme 2.In a manner reminiscent of the synthesis of the [2]rotaxane 1·6PF6, the dumbbell compounds 13·2PF6–15·2PF6 were obtained from the tosylated half dumbbell 12 by reactions, in turn, with 4,4′-bipyridine, 1,5-bis(pyridinyloxy)pentane, and 1,2-bis(2-(pyridinyloxy)ethoxy)ethane. The [2]rotaxanes 2·6PF6, 4·6PF6 and 5·6PF6 were prepared from corresponding dumbbells by means of a templation protocol,12 similar to that employed in the synthesis of 1·6PF6, in yields ranging from 30% to 40%. The synthesis of the [2]rotaxane 3·6PF6 is summarised in Scheme 3. In contrast with the synthetic strategies associated with the clipping methodology,13 a strategy of threading-followed-by-stoppering14 was utilised in the synthesis of the [2]rotaxane 3·6PF6. Reaction (Scheme 3) of the azide derivative 17 with 1,4-bis(4-pyridyl)benzene afforded the dumbbell 18·2PF6 after counterion exchange. The [2]rotaxane 3·6PF6 was isolated in 27% yield, following the reaction of 17·2PF6 with the alkyne derivative 19 in Me2CO using the copper(I)-catalyzed azide-alkyne cycloaddition in the presence of CBPQT·4PF6 salt. The [2]rotaxanes 1–5·6PF6 were all fully characterized by 1H and 13C NMR spectroscopies and by electrospray ionization mass spectrometry.
In the 1H NMR spectra of these five [2]rotaxanes recorded in CD3CN at 298 K, the protons of the DNP units and stoppers resonate as two sets of peaks, demonstrating that the CBPQT4+ rings in the [2]rotaxanes encircle one of the two equivalent DNP units in their dumbbell components, i.e., the CBPQT4+ ring is not shuttling rapidly between the two DNP units on the 1H NMR time-scale. As a result of this slow shuttling on the 1H NMR time-scale, two sets of the resonances are observed for the DNP protons for all five [2]rotaxanes – one DNP unit that is “free” and one that is encircled by the CBPQT4+ ring. This fact is further substantiated by the observation that the set of resonances corresponding to the protons of the free DNP units have very similar δ values to those recorded for the DNP protons in their corresponding dumbbell compounds. The resonances for the protons on the other DNP units encircled by the CBPQT4+ ring experience large upfield shifts that can be observed at ca. δ = 2.3, 5.9, and 6.2 ppm as a result of [π⋯π] stacking interactions with the encircling CBPQT4+ ring.
A series of variable-temperature (VT) 1H NMR spectroscopic experiments have been performed in CD3CN or d7-DMF in order to investigate6d,7 the shuttling behaviour of the CBPQT4+ rings in the degenerate [2]rotaxanes. We did not observe any substantial changes in the 1H NMR spectra of 1–5·6PF6 in the temperature range 233–343 K, indicating that the CBPQT4+ rings are not shuttling rapidly back and forth between the two DNP units on the 1H NMR time-scale in this temperature range. Two examples are presented in Fig. 3 which illustrates the partial VT 1H NMR spectra of the degenerate [2]rotaxane 1·6PF6 and 3·6PF6 recorded in CD3CN in the temperature range 233–343 K. The methyl protons (HMe) on the 2,6-diisopropylphenyl stoppers, which can be employed in order to probe the shuttling process, feature two doublets at 273 K. While the two doublets in each case undergo temperature-dependent shifts, they were not observed to coalesce. These observations indicate that the shuttling of the CBPQT4+ ring in the case of all five [2]rotaxanes is slow on the 1H NMR time-scale, even at 343 K.
![Partial 1H NMR spectra of the [2]rotaxane a) 1·6PF6 and b) 3·6PF6 recorded in CD3CN at different temperatures showing the signals for the probe protons (HMe) in the form of the methyl groups on the 2,6-diisopropylphenyl stoppers. No coalescence could be observed.](/image/article/2011/OB/c0ob00937g/c0ob00937g-f3.gif) | ||
| Fig. 3 Partial 1H NMR spectra of the [2]rotaxane a) 1·6PF6 and b) 3·6PF6 recorded in CD3CN at different temperatures showing the signals for the probe protons (HMe) in the form of the methyl groups on the 2,6-diisopropylphenyl stoppers. No coalescence could be observed. | ||
The location of the BIPY2+ unit in the [2]rotaxanes 1·6PF6 and 2·6PF6 allows for a reduction-based switching process to occur which also eradicates the electrostatic barrier to shuttling of the ring between the two DNP units in the degenerate [2]rotaxane 1·6PF6 and 2·6PF6. We have shown recently10b that, upon a two-electron reduction of the CBPQT4+ ring to its diradical dication CBPQT2(˙+) and a one-electron reduction of the BIPY2+ unit – located in either the dumbbell component of a rotaxane or in the thread component of a pseudorotaxane – to its radical cation BIPY˙+, encirclement of the ring around the dumbbell or thread occurs as a result of radical-pairing interactions (Fig. 4). These interactions have been characterized by cyclic voltammetry (CV) which provides evidence for the bistability of the [2]rotaxanes 1·6PF6 and 2·6PF6 under the influence of reductive potentials.
![Schematic of the switching of the CBPQT4+ ring in the [2]rotaxane 1·6PF6 or 2·6PF6 upon redox control.](/image/article/2011/OB/c0ob00937g/c0ob00937g-f4.gif) | ||
| Fig. 4 Schematic of the switching of the CBPQT4+ ring in the [2]rotaxane 1·6PF6 or 2·6PF6 upon redox control. | ||
The [2]rotaxane 1·6PF6 displays the characteristic CV behaviour observed previously10 for the reduction-induced switching of the CBPQT4+ ring in relation to the BIPY2+ unit in its dumbbell component. The first feature of significance in the CV (Fig. 5) is a three-electron reduction process at −0.29 V, the potential at which two electrons are gained by the CBPQT4+ ring component while the third electron is taken up by the BIPY2+ unit in the dumbbell component. The three-electron reduction results in the encirclement of the CBPQT2(˙+) diradical cationic ring component around the BIPY˙+ radical cationic unit in the dumbbell component. In the radical pairing interactions which are driving this intramolecular reaction to occur, only one of the two BIPY˙+ radical cations on the CBPQT2(˙+) ring is engaged intimately with BIPY˙+ present in the dumbbell component. This biased interaction results in the splitting of the second reduction cycle of the ring component into two one-electron processes at peak potentials of −0.73 and −1.03 V, the more negative of which is coupled to the second reduction of the BIPY2+ unit in the dumbbell component. The return scan shows (Fig. 5) that these two redox processes are completely reversible. Re-oxidation of the trisradical species shows one broad anodic process at −0.18 V (200 mV s−1 scan rate) corresponding to a potential that is considerably more positive than would be expected for a totally reversible process with respect to the initial three-electron reduction, an observation which reflects the highly stabilizing interactions that result as a consequence of a BIPY˙+ radical cation when the dumbbell component is encircled by a CBPQT2(˙+) ring. The [2]rotaxane 2·6PF6 also exhibited similar bistability and switching behaviour when investigated by CV. It follows that the two [2]rotaxanes 1·6PF6 and 2·6PF6, where bipyridinium radical cations can be generated in conjugated systems such that electron-delocalization can occur, may also be regarded as the precursors to tristable [2]rotaxanes when one of the two DNP units is replaced, for example, by a TTF unit.
![The first (black) and second (red) scans arising from the CV of the [2]rotaxane 1·6PF6 in the reduction region.](/image/article/2011/OB/c0ob00937g/c0ob00937g-f5.gif) | ||
| Fig. 5 The first (black) and second (red) scans arising from the CV of the [2]rotaxane 1·6PF6 in the reduction region. | ||
We have also investigated the reduction properties of the degenerate [2]rotaxanes 4·6PF6 and 5·6PF6 under the same conditions as for 1·6PF6 and 2·6PF6 and did not observe any interactions between the CBPQT2(˙+) diradical cationic ring and the flexible spacers containing two pyridinium units in their dumbbell components. Consequently, the CBPQT4+ ring cannot be induced to shuttle electrochemically between the two degenerate DNP units in the dumbbell component of 4·6PF6 and 5·6PF6. The reason for the inability to undergo well-defined switching upon reduction no doubt resides in the fact that the two pyridinium units are not conjugated and so cannot generate radical cations for binding with the CBPQT2(˙+) diradical cationic ring. In the case of the [2]rotaxane 3·6PF6, we are currently investigating its electrochemical behaviour, which is revealing interesting preliminary results – likely as a consequence of the aromatic conjugation between the two pyridinium units.
Conclusions
In degenerate donor–acceptor rotaxanes, the insertion of positively charged entities which can be reduced readily leads to electrostatic barriers to the shuttling of a cyclobis(paraquat-p-phenylene) ring. When, however, the positive charge is associated with a conjugated bipyridinium ring, or its higher homologues, then reduction to form radical cations leads to a dramatic stabilisation when interacting with the ring also in a partially reduced form. This interaction, not only removes the electrostatic barrier to shuttling, but also renders the [2]rotaxanes bistable. Investigation of the potential for the 1,4-bis(pyridinium)benzene unit in the degenerate [2]rotaxane 3·6PF6 to serve as a recognition site for a CBPQT4+ ring is still under investigation at this time. These redox-active bistable molecules involving direct and reversible control on oxidation, could come into their own as candidates for developing the next generation of molecular electronic devices.Experimental
General
All reagents were purchased from Aldrich and were used without further purification. The starting materials 6,108·2PF6,119,1510,1616,151,4-bis(4-pyridyl)benzene17 and CBQPT·4PF618 were prepared according to literature procedures. Deuterated solvents (Cambridge Isotope Laboratories) for nuclear magnetic resonance (NMR) spectroscopic analyses were used as received. NMR spectra were recorded on a Bruker Avance 500 MHz or 600 MHz NMR spectrometer. Chemical shifts were reported in parts per million (ppm) downfield from the Me4Si resonance which was used as the internal standard when recording 1H NMR spectra. High resolution mass spectra were measured on an Applied Biosystems Voyager DE-PRO MALDI TOF mass spectrometer or a Micromass Q-TOF Ultima electrospray ionization mass spectrometer. The reported molecular mass (m/z) values were the most abundant monoisotopic mass. UV-Vis spectra were recorded at room temperature on a Shimadzu UV-3600 UV-Vis-NIR spectrophotometer. Electrochemical experiments were carried out at 298 K in Ar-purged MeCN, with a Gamry Multipurpose instrument (Reference 600) interfaced to a PC. Cyclic voltammetry (CV) experiments were performed using a glassy carbon working electrode (0.071 cm2, Cypress system). Its surface was polished routinely with 0.05 μm alumina-water slurry on a felt surface immediately before use. The counter electrode was a Pt coil and the reference electrode was an Ag/AgCl electrode. The concentration of the sample and supporting electrolyte tetrabutylammonium hexafluorophosphate (TBA·PF6) were 1.0 × 10−3 mol L−1 and 0.1 mol L−1, respectively.Syntheses
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/w) and the purple band was collected. Most of the solvent was removed in vacuo, followed by addition of H2O (15 mL). The resulting precipitate was collected by filtration, affording the dumbbell 7·2PF6 (24.2 mg, 42%). 1H NMR (500 MHz, (CD3)2CO, TMS): δ = 1.14–1.15 (d, 24H, J = 7.0 Hz), 3.35–3.37 (m, 4H), 3.66–3.79 (m, 20H), 4.47–4.51 (m, 12H), 6.89–6.91 (d, 4H, J = 7.6 Hz), 7.07–7.10 (d, 6H), 7.43–7.45 (d, 4H, J = 7.6 Hz), 7.89–7.90 (d, 4H, J = 7.6 Hz), 8.12–8.14 (d, 4H, J = 7.8 Hz), 9.09–9.11 ppm (d, 4H, J = 7.8 Hz). 13C NMR (125 MHz, (CD3)2CO, TMS): δ = 23.5, 28.4, 68.4, 69.9, 70.5, 108.1, 115.8, 121.7, 123.6, 124.3, 126.2, 128.6, 142.0, 144.8, 148.7, 150.8, 154.3 ppm. HRMS: calcd for C70H86F6N2O10P [M − PF6]+m/z = 1259.5924, found m/z = 1259.5914; calcd for C70H86N2O10 [M − 2PF6]2+m/z = 557.3141, found m/z = 557.3179.
:1 v/w) as the mobile phase. The product was recovered from the silica gel by washing with an excess of eluent. The solution was concentrated to a minimum volume and the product was precipitated from the solution by addition of H2O (12 mL). The resulting precipitate was collected by filtration, affording the pure [2]rotaxane 1·6PF6 (37.1 mg, 37%). 1H NMR (500 MHz, CD3CN, TMS): δ = 1.07–1.08 (d, 12H, J = 6.8 Hz), 1.16–1.17 (d, 12H, J = 6.8 Hz), 2.31–2.33 (m, 2H), 3.21–3.23 (m, 2H), 3.32–3.34 (m, 2H), 3.59–3.89 (m, 20H), 4.32–4.47 (m, 12H), 5.68–5.74 (m, 8H), 5.86–5.88 (m, 2H), 6.21–6.23 (m, 2H), 6.86–6.88 (d, 2H, J = 8.0 Hz), 7.03–7.16 (m, 14H), 7.44–7.46 (d, 2H, J = 8.0 Hz), 7.77–7.79 (d, 2H, J = 8.0 Hz), 7.88–7.92 (m, 8H), 8.08–8.14 (m, 4H), 8.89–8.91 (d, 2H, J = 7.0 Hz), 8.94–8.96 (d, 2H, J = 7.0 Hz), 9.06–9.11 ppm (m, 8H). 13C NMR (125 MHz, CD3CN, TMS): δ = 23.4, 28.6, 62.4, 68.7, 69.5, 70.7, 108.2, 115.7, 121.2, 121.7, 123.4, 124.6, 126.2, 127.5, 128.8, 134.7, 142.1, 144.9, 148.6, 150.8, 154.4 ppm. HRMS: calcd for C106H118F30N6O10P5 [M − PF6]+m/z = 2359.7118, found m/z = 2359.7467; calcd for C106H118F24N6O10P4 [M − 2PF6]2+m/z = 1107.3739, found m/z = 1107. 4133.
:2) to give compound 11 (2.6 g, 87%). 1H NMR (500 MHz, CD2Cl2, TMS): δ = 1.22–1.24 (d, 6H, J = 8.0 Hz), 1.33 (s, 18H), 2.86–2.87 (m, 1H), 3.48–3.56 (m, 20H), 3.79–3.81 (m, 6H), 4.33–4.35 (m, 6H), 6.75–6.77 (d, 2H, J = 8.2 Hz), 6.82–6.83 (d, 2H, J = 7.8 Hz), 7.08–7.18 (m, 10H), 7.28–7.30 (d, 4H), 7.45–7.47 (d, 2H, J = 7.8 Hz), 7.88–7.90 ppm (d, 2H, J = 7.8 Hz). 13C NMR (125 MHz, CD2Cl2, TMS): δ = 23.6, 31.5, 33.9, 34.7, 61.7, 64.6, 69.4, 70.8, 108.2, 115.3, 116.1, 125.7, 126.8, 128.0, 128.9, 139.9, 145.2, 146.4, 149.5, 154.3, 156.5 ppm. MS: calcd for C62H81O10 [M + H]+m/z = 985.583, found m/z = 985.598.
:1 v/w) and the purple band was collected. Most of the solvent was removed in vacuo, followed by addition of H2O (15 mL). The resulting precipitate was collected by filtration, affording the dumbbell 13·2PF6 (39.1 mg, 41%). 1H NMR (500 MHz, (CD3)2CO, TMS): δ = 1.18–1.20 (d, 12H, J = 6.9 Hz), 1.31 (s, 36H), 2.83–2.85 (m, 2H), 3.58–3.66 (m, 12H), 3.70–3.72 (m, 4H), 3.73–3.80 (m, 16H), 3.82–3.84 (m, 4H), 3.93–3.96 (m, 8H), 4.03–4.04 (m, 4H), 4.09–4.12 (m, 4H), 4.18–4.21 (m, 8H), 4.76–4.78 (m, 4H), 6.73–6.78 (m, 8H), 7.05–7.12 (m, 20H), 7.29–7.31 (m, 8H), 7.41–7.43 (d, 4H, J = 8.4 Hz), 7.94–7.95 (d, 4H, J = 6.8 Hz), 8.13–8.15 (d, 4H, J = 8.4 Hz), 9.03–9.04 ppm (d, 4H, J = 6.8 Hz). 13C NMR (500 MHz, (CD3)2CO, TMS): δ = 23.3, 30.6, 33.2, 33.8, 53.1, 63.9, 69.2, 70.0, 70.4, 106.6, 113.8, 115.0, 121.6, 125.4, 126.1, 127.9, 128.7, 139.4, 144.6, 145.5, 145.9, 148.4, 150.6, 153.7, 156.7 ppm. HRMS: calcd for C134H166N2O18 [M − 2PF6]2+m/z = 1045.6068, found m/z = 1046.0909.
:1 v/w) and the purple band was collected. Most of the solvent was removed in vacuo, followed by addition of H2O (15 mL). The resulting precipitate was collected by filtration, affording the dumbbell 14·2PF6 (37.7 mg, 38%). 1H NMR (500 MHz, (CD3)2CO, TMS): δ = 1.18–1.21 (d, 12H), 1.65–1.73 (m, 6H), 1.32 (s, 36H), 2.82–2.84 (m, 2H), 3.58–3.65 (m, 12H), 3.70–3.73 (m, 4H), 3.74–3.81 (m, 16H), 3.82–3.85 (m, 4H), 3.92–3.96 (m, 8H), 4.03–4.07 (m, 8H), 4.10–4.13 (m, 4H), 4.18–4.22 (m, 8H), 4.75–4.78 (m, 4H), 6.74–6.78 (m, 8H), 7.06–7.14 (m, 24H), 7.28–7.32 (m, 8H), 7.42–7.44 (d, 4H, J = 8.4 Hz), 8.13–8.15 (d, 4H, J = 8.4 Hz), 8.85–8.87 ppm (d, 4H, J = 7.0 Hz). 13C NMR (500 MHz, (CD3)2CO, TMS): δ = 22.7, 23.4, 30.3, 33.0, 33.7, 53.2, 63.9, 68.6, 69.3, 70.1, 70.6, 100.4, 106.8, 113.8, 115.5, 125.1, 126.3, 127.9, 128.9, 139.5, 144.8, 145.7, 148.6, 150.3, 153.4, 156.5, 161.5 ppm. HRMS: calcd for C139H176N2O20 [M − 2PF6]2+m/z = 1096.6408, found m/z = 1096.6402.
:1 v/w) and the purple band was collected. Most of the solvent was removed in vacuo, followed by addition of H2O (15 mL). The resulting precipitate was collected by filtration, affording the dumbbell 15·2PF6 (36.4 mg, 36%). 1H NMR (500 MHz, (CD3)2CO, TMS): δ = 1.19–1.21 (d, 12H, J = 6.8 Hz), 1.30 (s, 36H), 2.84–2.85 (m, 2H), 3.58–3.65 (m, 16H), 3.71–3.74 (m, 4H), 3.72–3.81 (m, 20H), 3.81–3.84 (m, 4H), 3.92–3.96 (m, 8H), 4.03–4.05 (m, 4H), 4.08–4.13 (m, 4H), 4.21–4.32 (m, 12H), 4.73–4.75 (m, 4H), 6.74–6.80 (m, 8H), 7.08–7.13 (m, 24H), 7.29–7.32 (m, 8H), 7.41–7.43 (d, 4H, J = 8.4 Hz), 8.15–8.17 (d, 4H, J = 8.4 Hz), 8.89–8.91 ppm (d, 4H, J = 7.0 Hz). 13C NMR (500 MHz, (CD3)2CO, TMS): δ = 23.0, 30.5, 33.7, 34.2, 53.4, 64.1, 69.5, 70.1, 70.8, 100.2, 106.4, 113.8, 115.3, 125.7, 126.2, 127.9, 128.8, 140.3, 144.4, 145.8, 148.7, 150.2, 153.8, 156.5, 161.6 ppm. HRMS: calcd for C140H178N2O22 [M − 2PF6]2+m/z = 1119.6436, found m/z = 1119.6588.
:1 v/w) as the mobile phase. The product was recovered from the silica gel by washing with an excess of eluent. The solution was concentrated to a minimum volume and the product was precipitated from the solution by addition of H2O (12 mL). The resulting precipitate was collected by filtration, affording the pure [2]rotaxane 2·6PF6 (50.1 mg, 36%). 1H NMR (500 MHz, CD3CN, TMS): δ = 1.10–1.11 (d, 6H, J = 7.0 Hz), 1.18–1.20 (d, 6H, J = 7.0 Hz), 1.28 (s, 18H), 1.30 (s, 18H), 2.57–2.59 (m, 2H), 2.86–2.89 (m, 2H), 3.60–3.67 (m, 12H), 3.71–3.78 (m, 20H), 3.89–3.95 (m, 12H), 4.04–4.06 (m, 4H), 4.20–4.25 (m, 8H), 4.36–4.38 (m, 4H), 4.73–4.76 (m, 4H), 5.69–5.74 (q, 8H, J = 8.0 Hz), 5.95–5.97 (m, 2H), 6.31–6.34 (m, 2H), 6.69–6.72 (d, 2H, J = 8.6 Hz), 6.83–6.86 (m, 4H), 7.05–7.19 (m, 28H), 7.32–7.36 (m, 10H), 7.73–7.75 (d, 2H, J = 8.6 Hz), 7.99–8.02 (m, 8H), 8.11–8.15 (m, 4H), 8.89–9.01 ppm (m, 12H). 13C NMR (500 MHz, CD3CN, TMS): δ = 23.6, 30.4, 33.3, 33.9, 53.2, 62.2, 64.0, 69.2, 70.1, 70.5, 106.6, 113.7, 115.3, 120.9, 121.6, 125.5, 126.3, 127.3, 128.0, 128.8, 134.6, 139.6, 144.1, 144.7, 145.3, 145.8, 148.6, 150.1, 150.7, 153.5, 156.8 ppm. HRMS: calcd for C170H198F24N6O18P4 [M − 2PF6]2+m/z = 1595.6665, found m/z = 1596.2242; calcd for C170H198F18N6O18P3 [M − 3PF6]3+m/z = 1015.4563, found m/z = 1015.9544.
:1 v/w) as the mobile phase. The product was recovered from the silica gel by washing with an excess of eluent. The solution was concentrated to a minimum volume and the product was precipitated from the solution by addition of H2O (12 mL). The resulting precipitate was collected by filtration, affording the pure [2]rotaxane 4·6PF6 (50.2 mg, 35%). 1H NMR (500 MHz, CD3CN, TMS): δ = 1.12–1.13 (d, 6H, J = 4.6 Hz), 1.19–1.21 (d, 6H, J = 4.6 Hz), 1.27 (s, 18H), 1.31 (s, 18H), 1.64–1.76 (m, 6H), 2.58–2.60 (m, 2H), 2.85–2.88 (m, 2H), 3.61–3.67 (m, 12H), 3.72–3.79 (m, 20H), 3.88–3.95 (m, 12H), 4.04–4.08 (m, 8H), 4.21–4.25 (m, 8H), 4.36–4.39 (m, 4H), 4.74–4.76 (m, 4H), 5.69–5.75 (q, 8H, J = 8.6 Hz), 5.95–5.97 (m, 2H), 6.32–6.34 (m, 2H), 6.70–6.73 (d, 2H, J = 8.2 Hz), 6.83–6.86 (m, 4H), 7.03–7.10 (m, 12H), 7.12–7.19 (m, 20H), 7.33–7.37 (m, 10H), 7.74–7.76 (d, 2H, J = 8.2 Hz), 7.98–8.02 (m, 8H), 8.89–9.01 ppm (m, 12H). 13C NMR (500 MHz, CD3CN, TMS): δ = 22.5, 23.7, 30.1, 33.4, 33.8, 53.1, 62.3, 63.8, 68.8, 69.5, 70.4, 70.9, 100.3, 106.6, 113.7, 115.3, 121.7, 125.4, 126.6, 127.4, 127.9, 128.7, 134.7, 139.2, 144.2, 144.9, 145.5, 148.3, 150.7, 153.6, 156.3, 161.8 ppm. HRMS: calcd for C175H208F24N6O20P4 [M − 2PF6]2+m/z = 1646.7006, found m/z = 1646.7394; calcd for C175H208F18N6O20P3 [M − 3PF6]3+m/z = 1049.4790, found m/z = 1049.5258.
:1 v/w) as the mobile phase. The product was recovered from the silica gel by washing with an excess of eluent. The solution was concentrated to a minimum volume and the product was precipitated from the solution by addition of H2O (12 mL). The resulting precipitate was collected by filtration, affording the pure [2]rotaxane 5·6PF6 (45.0 mg, 31%). 1H NMR (500 MHz, CD3CN, TMS): δ = 1.12–1.14 (d, 6H, J = 4.2 Hz), 1.18–1.20 (d, 6H, J = 4.2 Hz), 1.27 (s, 18H), 1.32 (s, 18H), 2.58–2.60 (m, 2H), 2.86–2.88 (m, 2H), 3.58–3.66 (m, 16H), 3.74–3.81 (m, 24H), 3.87–3.95 (m, 12H), 4.04–4.08 (m, 4H), 4.22–4.25 (m, 8H), 4.30–4.38 (m, 8H), 4.74–4.77 (m, 4H), 5.70–5.75 (q, 8H, J = 8.4 Hz), 5.96–5.98 (m, 2H), 6.32–6.34 (m, 2H), 6.71–6.73 (d, 2H, J = 8.6 Hz), 6.84–6.86 (m, 4H), 7.03–7.11 (m, 12H), 7.13–7.20 (m, 20H), 7.34–7.38 (m, 10H), 7.74–7.76 (d, 2H, J = 8.6 Hz), 7.97–8.02 (m, 8H), 8.89–9.02 ppm (m, 12H). 13C NMR (500 MHz, CD3CN, TMS): δ = 23.1, 30.6, 33.5, 34.7, 53.3, 61.8, 64.4, 69.9, 70.3, 71.1, 100.5, 106.1, 113.6, 115.7, 121.6, 125.4, 126.6, 127.1, 127.8, 128.4, 134.7, 140.6, 144.5, 145.3, 148.2, 150.4, 153.6, 156.8, 161.2 ppm. HRMS: calcd for C176H210F24N6O22P4 [M − 2PF6]2+m/z = 1669.7033, found m/z = 1669.7159; calcd for C176H210F18N6O22P3 [M − 3PF6]3+m/z = 1064.8141, found m/z = 1064.8356.
:1 v/w) and the purple fractions were collected. The solvent was removed in vacuo to a minimal volume, followed by the addition of H2O (15 mL). The resulting precipitate was collected by filtration, affording the dumbbell 18·2PF6 (180 mg, 61%). 1H NMR (500 MHz, CD3CN): δ = 3.35 (t, 4H, J = 4.5 Hz), 3.69 (t, 4H, J = 5.0 Hz), 3.79 (t, 4H, J = 4.5 Hz), 3.97 (t, 4H, J = 4.0 Hz), 4.05 (t, 4H, J = 5.0 Hz), 4.13 (t, 4H, J = 5.0 Hz), 4.16 (t, 4H, J = 4.0 Hz), 4.75 (t, 4H, J = 5.0 Hz), 6.71 (d, 2H, J = 8.0 Hz), 6.82 (d, 2H, J = 7.5 Hz), 7.26 (t, 2H, J = 8.0 Hz), 7.30 (t, 2H, J = 7.5 Hz), 7.52 (d, 2H, J = 8.5 Hz), 7.54 (s, 4H), 7.66 (d, 2H, J = 8.5 Hz), 7.93 (d, 4H, J = 7.0 Hz), 8.73 ppm(d, 4H, J = 7.0 Hz). 13C NMR (125 MHz, CD3CN): δ = 50.1, 60.5, 67.2, 67.5, 68.6, 68.9, 69.0, 69.5, 105.2, 105.4, 113.7, 113.8, 124.2, 125.0, 125.1, 125.7, 126.1, 128.3, 135.8, 144.7, 153.6, 153.8, 154.0 ppm. HRMS: calcd for C52H56F6N8O8P [M − PF6]+m/z = 1065.3862, found m/z = 1065.3870.
:7:1)], then MeOH, Me2CO and 2% NH4PF6/Me2CO, respectively]. The purple fraction in Me2CO were collected, and concentrated to a minimum volume before the crude product was precipitated by the addition of H2O. The resulting solid was collected by filtration to afford 3·6PF6 (20 mg, 27%) as a purple powder. 1H NMR (500 MHz, CD3CN): δ = 1.17 (d, 12H, J = 5.5 Hz), 1.18 (d, 12H, J = 5.5 Hz), 2.55 (d, 1H, J = 6.0 Hz), 2.59 (d, 1H, J = 6.0 Hz), 3.35–3.40 (m, 4H), 3.90 (t, 2H, J = 3.5 Hz), 3.98 (t, 2H, J = 3.5 Hz), 4.03 (t, 2H, J = 4.5 Hz), 4.14 (t, 2H, J = 4.0 Hz), 4.20–4.23 (m, 4H), 4.27 (t, 2H, J = 3.0 Hz), 4.30 (t, 2H, J = 4.0 Hz), 4.36–4.41 (m, 6H), 4.49 (t, 2H, J = 4.3 Hz), 4.58 (t, 2H, J = 5.0 Hz), 4.73 (t, 2H, J = 4.0 Hz), 4.81 (s, 2H), 4.87 (t, 2H, J = 4.5 Hz), 4.88 (s, 2H), 5.05 (t, 2H, J = 4.3 Hz), 5.78 (d, 4H, J = 11.5 Hz), 5.86 (d, 4H, J = 11.5 Hz), 6.01 (t, 1H, J = 7.0 Hz), 6.05 (t, 1H, J = 7.0 Hz), 6.31 (d, 1H, J = 6.5 Hz), 6.38 (d, 1H, J = 6.5 Hz), 6.86 (t, 2H, J = 6.5 Hz), 7.09–7.15 (m, 6H), 7.31 (t, 2H, J = 7.0 Hz), 7.37 (d, 8H, J = 5.0 Hz), 7.64 (d, 1H, J = 6.5 Hz), 7.70 (d, 1H, J = 7.0 Hz), 7.84 (d, 2H, J = 7.5 Hz), 7.87 (s, 2H), 7.99 (s, 8H), 8.01 (d, 1H, J = 6.5 Hz), 8.11 (d, 2H, J = 6.5 Hz), 8.12 (s, 1H), 8.49 (d, 2H, J = 6.0 Hz), 8.68 (d, 2H, J = 5.5 Hz), 8.74 (b, 8H), 8.99 (d, 2H, J = 6.0 Hz). 13C NMR (125 MHz, CD3CN): δ = 23.3, 23.3, 26.5, 26.5, 28.7, 29.9, 50.1, 54.2, 61.0, 67.2, 67.5, 67.7, 67.9, 69.0, 69.3, 69.3, 69.4, 69.5, 69.8, 70.0, 104.5, 105.6, 105.9, 114.1, 124.1, 124.2, 124.3, 124.4, 124.7, 125.0, 125.2, 125.4, 125.6, 126.5, 129.1, 129.1, 131.3, 136.6, 141.6, 141.9, 143.7, 144.6, 145.0, 145.3, 150.9, 152.7, 152.8, 154.0. HRMS: calcd for C118H128F24N12O10P4 [M − 2PF6]2+m/z = 1226.9238, found m/z = 1226.9281.
:1 v/w) and the purple band was collected. Most of the solvent was removed in vacuo, followed by addition of H2O (15 mL). The resulting precipitate was collected by filtration, affording the dumbbell 7·2PF6 (24.2 mg, 42%). 1H NMR (500 MHz, (CD3)2CO, TMS): δ = 1.14–1.15 (d, 24H, J = 7.0 Hz), 3.35–3.37 (m, 4H), 3.66–3.79 (m, 20H), 4.47–4.51 (m, 12H), 6.89–6.91 (d, 4H, J = 7.6 Hz), 7.07–7.10 (d, 6H), 7.43–7.45 (d, 4H, J = 7.6 Hz), 7.89–7.90 (d, 4H, J = 7.6 Hz), 8.12–8.14 (d, 4H, J = 7.8 Hz), 9.09–9.11 ppm (d, 4H, J = 7.8 Hz). 13C NMR (125 MHz, (CD3)2CO, TMS): δ = 23.5, 28.4, 68.4, 69.9, 70.5, 108.1, 115.8, 121.7, 123.6, 124.3, 126.2, 128.6, 142.0, 144.8, 148.7, 150.8, 154.3 ppm. HRMS: calcd for C70H86F6N2O10P [M − PF6]+m/z = 1259.5924, found m/z = 1259.5914; calcd for C70H86N2O10 [M − 2PF6]2+m/z = 557.3141, found m/z = 557.3179.
:1 v/w) as the mobile phase. The product was recovered from the silica gel by washing with an excess of eluent. The solution was concentrated to a minimum volume and the product was precipitated from the solution by addition of H2O (12 mL). The resulting precipitate was collected by filtration, affording the pure [2]rotaxane 1·6PF6 (37.1 mg, 37%). 1H NMR (500 MHz, CD3CN, TMS): δ = 1.07–1.08 (d, 12H, J = 6.8 Hz), 1.16–1.17 (d, 12H, J = 6.8 Hz), 2.31–2.33 (m, 2H), 3.21–3.23 (m, 2H), 3.32–3.34 (m, 2H), 3.59–3.89 (m, 20H), 4.32–4.47 (m, 12H), 5.68–5.74 (m, 8H), 5.86–5.88 (m, 2H), 6.21–6.23 (m, 2H), 6.86–6.88 (d, 2H, J = 8.0 Hz), 7.03–7.16 (m, 14H), 7.44–7.46 (d, 2H, J = 8.0 Hz), 7.77–7.79 (d, 2H, J = 8.0 Hz), 7.88–7.92 (m, 8H), 8.08–8.14 (m, 4H), 8.89–8.91 (d, 2H, J = 7.0 Hz), 8.94–8.96 (d, 2H, J = 7.0 Hz), 9.06–9.11 ppm (m, 8H). 13C NMR (125 MHz, CD3CN, TMS): δ = 23.4, 28.6, 62.4, 68.7, 69.5, 70.7, 108.2, 115.7, 121.2, 121.7, 123.4, 124.6, 126.2, 127.5, 128.8, 134.7, 142.1, 144.9, 148.6, 150.8, 154.4 ppm. HRMS: calcd for C106H118F30N6O10P5 [M − PF6]+m/z = 2359.7118, found m/z = 2359.7467; calcd for C106H118F24N6O10P4 [M − 2PF6]2+m/z = 1107.3739, found m/z = 1107. 4133.
:2) to give compound 11 (2.6 g, 87%). 1H NMR (500 MHz, CD2Cl2, TMS): δ = 1.22–1.24 (d, 6H, J = 8.0 Hz), 1.33 (s, 18H), 2.86–2.87 (m, 1H), 3.48–3.56 (m, 20H), 3.79–3.81 (m, 6H), 4.33–4.35 (m, 6H), 6.75–6.77 (d, 2H, J = 8.2 Hz), 6.82–6.83 (d, 2H, J = 7.8 Hz), 7.08–7.18 (m, 10H), 7.28–7.30 (d, 4H), 7.45–7.47 (d, 2H, J = 7.8 Hz), 7.88–7.90 ppm (d, 2H, J = 7.8 Hz). 13C NMR (125 MHz, CD2Cl2, TMS): δ = 23.6, 31.5, 33.9, 34.7, 61.7, 64.6, 69.4, 70.8, 108.2, 115.3, 116.1, 125.7, 126.8, 128.0, 128.9, 139.9, 145.2, 146.4, 149.5, 154.3, 156.5 ppm. MS: calcd for C62H81O10 [M + H]+m/z = 985.583, found m/z = 985.598.
:1 v/w) and the purple band was collected. Most of the solvent was removed in vacuo, followed by addition of H2O (15 mL). The resulting precipitate was collected by filtration, affording the dumbbell 13·2PF6 (39.1 mg, 41%). 1H NMR (500 MHz, (CD3)2CO, TMS): δ = 1.18–1.20 (d, 12H, J = 6.9 Hz), 1.31 (s, 36H), 2.83–2.85 (m, 2H), 3.58–3.66 (m, 12H), 3.70–3.72 (m, 4H), 3.73–3.80 (m, 16H), 3.82–3.84 (m, 4H), 3.93–3.96 (m, 8H), 4.03–4.04 (m, 4H), 4.09–4.12 (m, 4H), 4.18–4.21 (m, 8H), 4.76–4.78 (m, 4H), 6.73–6.78 (m, 8H), 7.05–7.12 (m, 20H), 7.29–7.31 (m, 8H), 7.41–7.43 (d, 4H, J = 8.4 Hz), 7.94–7.95 (d, 4H, J = 6.8 Hz), 8.13–8.15 (d, 4H, J = 8.4 Hz), 9.03–9.04 ppm (d, 4H, J = 6.8 Hz). 13C NMR (500 MHz, (CD3)2CO, TMS): δ = 23.3, 30.6, 33.2, 33.8, 53.1, 63.9, 69.2, 70.0, 70.4, 106.6, 113.8, 115.0, 121.6, 125.4, 126.1, 127.9, 128.7, 139.4, 144.6, 145.5, 145.9, 148.4, 150.6, 153.7, 156.7 ppm. HRMS: calcd for C134H166N2O18 [M − 2PF6]2+m/z = 1045.6068, found m/z = 1046.0909.
:1 v/w) and the purple band was collected. Most of the solvent was removed in vacuo, followed by addition of H2O (15 mL). The resulting precipitate was collected by filtration, affording the dumbbell 14·2PF6 (37.7 mg, 38%). 1H NMR (500 MHz, (CD3)2CO, TMS): δ = 1.18–1.21 (d, 12H), 1.65–1.73 (m, 6H), 1.32 (s, 36H), 2.82–2.84 (m, 2H), 3.58–3.65 (m, 12H), 3.70–3.73 (m, 4H), 3.74–3.81 (m, 16H), 3.82–3.85 (m, 4H), 3.92–3.96 (m, 8H), 4.03–4.07 (m, 8H), 4.10–4.13 (m, 4H), 4.18–4.22 (m, 8H), 4.75–4.78 (m, 4H), 6.74–6.78 (m, 8H), 7.06–7.14 (m, 24H), 7.28–7.32 (m, 8H), 7.42–7.44 (d, 4H, J = 8.4 Hz), 8.13–8.15 (d, 4H, J = 8.4 Hz), 8.85–8.87 ppm (d, 4H, J = 7.0 Hz). 13C NMR (500 MHz, (CD3)2CO, TMS): δ = 22.7, 23.4, 30.3, 33.0, 33.7, 53.2, 63.9, 68.6, 69.3, 70.1, 70.6, 100.4, 106.8, 113.8, 115.5, 125.1, 126.3, 127.9, 128.9, 139.5, 144.8, 145.7, 148.6, 150.3, 153.4, 156.5, 161.5 ppm. HRMS: calcd for C139H176N2O20 [M − 2PF6]2+m/z = 1096.6408, found m/z = 1096.6402.
:1 v/w) and the purple band was collected. Most of the solvent was removed in vacuo, followed by addition of H2O (15 mL). The resulting precipitate was collected by filtration, affording the dumbbell 15·2PF6 (36.4 mg, 36%). 1H NMR (500 MHz, (CD3)2CO, TMS): δ = 1.19–1.21 (d, 12H, J = 6.8 Hz), 1.30 (s, 36H), 2.84–2.85 (m, 2H), 3.58–3.65 (m, 16H), 3.71–3.74 (m, 4H), 3.72–3.81 (m, 20H), 3.81–3.84 (m, 4H), 3.92–3.96 (m, 8H), 4.03–4.05 (m, 4H), 4.08–4.13 (m, 4H), 4.21–4.32 (m, 12H), 4.73–4.75 (m, 4H), 6.74–6.80 (m, 8H), 7.08–7.13 (m, 24H), 7.29–7.32 (m, 8H), 7.41–7.43 (d, 4H, J = 8.4 Hz), 8.15–8.17 (d, 4H, J = 8.4 Hz), 8.89–8.91 ppm (d, 4H, J = 7.0 Hz). 13C NMR (500 MHz, (CD3)2CO, TMS): δ = 23.0, 30.5, 33.7, 34.2, 53.4, 64.1, 69.5, 70.1, 70.8, 100.2, 106.4, 113.8, 115.3, 125.7, 126.2, 127.9, 128.8, 140.3, 144.4, 145.8, 148.7, 150.2, 153.8, 156.5, 161.6 ppm. HRMS: calcd for C140H178N2O22 [M − 2PF6]2+m/z = 1119.6436, found m/z = 1119.6588.
:1 v/w) as the mobile phase. The product was recovered from the silica gel by washing with an excess of eluent. The solution was concentrated to a minimum volume and the product was precipitated from the solution by addition of H2O (12 mL). The resulting precipitate was collected by filtration, affording the pure [2]rotaxane 2·6PF6 (50.1 mg, 36%). 1H NMR (500 MHz, CD3CN, TMS): δ = 1.10–1.11 (d, 6H, J = 7.0 Hz), 1.18–1.20 (d, 6H, J = 7.0 Hz), 1.28 (s, 18H), 1.30 (s, 18H), 2.57–2.59 (m, 2H), 2.86–2.89 (m, 2H), 3.60–3.67 (m, 12H), 3.71–3.78 (m, 20H), 3.89–3.95 (m, 12H), 4.04–4.06 (m, 4H), 4.20–4.25 (m, 8H), 4.36–4.38 (m, 4H), 4.73–4.76 (m, 4H), 5.69–5.74 (q, 8H, J = 8.0 Hz), 5.95–5.97 (m, 2H), 6.31–6.34 (m, 2H), 6.69–6.72 (d, 2H, J = 8.6 Hz), 6.83–6.86 (m, 4H), 7.05–7.19 (m, 28H), 7.32–7.36 (m, 10H), 7.73–7.75 (d, 2H, J = 8.6 Hz), 7.99–8.02 (m, 8H), 8.11–8.15 (m, 4H), 8.89–9.01 ppm (m, 12H). 13C NMR (500 MHz, CD3CN, TMS): δ = 23.6, 30.4, 33.3, 33.9, 53.2, 62.2, 64.0, 69.2, 70.1, 70.5, 106.6, 113.7, 115.3, 120.9, 121.6, 125.5, 126.3, 127.3, 128.0, 128.8, 134.6, 139.6, 144.1, 144.7, 145.3, 145.8, 148.6, 150.1, 150.7, 153.5, 156.8 ppm. HRMS: calcd for C170H198F24N6O18P4 [M − 2PF6]2+m/z = 1595.6665, found m/z = 1596.2242; calcd for C170H198F18N6O18P3 [M − 3PF6]3+m/z = 1015.4563, found m/z = 1015.9544.
:1 v/w) as the mobile phase. The product was recovered from the silica gel by washing with an excess of eluent. The solution was concentrated to a minimum volume and the product was precipitated from the solution by addition of H2O (12 mL). The resulting precipitate was collected by filtration, affording the pure [2]rotaxane 4·6PF6 (50.2 mg, 35%). 1H NMR (500 MHz, CD3CN, TMS): δ = 1.12–1.13 (d, 6H, J = 4.6 Hz), 1.19–1.21 (d, 6H, J = 4.6 Hz), 1.27 (s, 18H), 1.31 (s, 18H), 1.64–1.76 (m, 6H), 2.58–2.60 (m, 2H), 2.85–2.88 (m, 2H), 3.61–3.67 (m, 12H), 3.72–3.79 (m, 20H), 3.88–3.95 (m, 12H), 4.04–4.08 (m, 8H), 4.21–4.25 (m, 8H), 4.36–4.39 (m, 4H), 4.74–4.76 (m, 4H), 5.69–5.75 (q, 8H, J = 8.6 Hz), 5.95–5.97 (m, 2H), 6.32–6.34 (m, 2H), 6.70–6.73 (d, 2H, J = 8.2 Hz), 6.83–6.86 (m, 4H), 7.03–7.10 (m, 12H), 7.12–7.19 (m, 20H), 7.33–7.37 (m, 10H), 7.74–7.76 (d, 2H, J = 8.2 Hz), 7.98–8.02 (m, 8H), 8.89–9.01 ppm (m, 12H). 13C NMR (500 MHz, CD3CN, TMS): δ = 22.5, 23.7, 30.1, 33.4, 33.8, 53.1, 62.3, 63.8, 68.8, 69.5, 70.4, 70.9, 100.3, 106.6, 113.7, 115.3, 121.7, 125.4, 126.6, 127.4, 127.9, 128.7, 134.7, 139.2, 144.2, 144.9, 145.5, 148.3, 150.7, 153.6, 156.3, 161.8 ppm. HRMS: calcd for C175H208F24N6O20P4 [M − 2PF6]2+m/z = 1646.7006, found m/z = 1646.7394; calcd for C175H208F18N6O20P3 [M − 3PF6]3+m/z = 1049.4790, found m/z = 1049.5258.
:1 v/w) as the mobile phase. The product was recovered from the silica gel by washing with an excess of eluent. The solution was concentrated to a minimum volume and the product was precipitated from the solution by addition of H2O (12 mL). The resulting precipitate was collected by filtration, affording the pure [2]rotaxane 5·6PF6 (45.0 mg, 31%). 1H NMR (500 MHz, CD3CN, TMS): δ = 1.12–1.14 (d, 6H, J = 4.2 Hz), 1.18–1.20 (d, 6H, J = 4.2 Hz), 1.27 (s, 18H), 1.32 (s, 18H), 2.58–2.60 (m, 2H), 2.86–2.88 (m, 2H), 3.58–3.66 (m, 16H), 3.74–3.81 (m, 24H), 3.87–3.95 (m, 12H), 4.04–4.08 (m, 4H), 4.22–4.25 (m, 8H), 4.30–4.38 (m, 8H), 4.74–4.77 (m, 4H), 5.70–5.75 (q, 8H, J = 8.4 Hz), 5.96–5.98 (m, 2H), 6.32–6.34 (m, 2H), 6.71–6.73 (d, 2H, J = 8.6 Hz), 6.84–6.86 (m, 4H), 7.03–7.11 (m, 12H), 7.13–7.20 (m, 20H), 7.34–7.38 (m, 10H), 7.74–7.76 (d, 2H, J = 8.6 Hz), 7.97–8.02 (m, 8H), 8.89–9.02 ppm (m, 12H). 13C NMR (500 MHz, CD3CN, TMS): δ = 23.1, 30.6, 33.5, 34.7, 53.3, 61.8, 64.4, 69.9, 70.3, 71.1, 100.5, 106.1, 113.6, 115.7, 121.6, 125.4, 126.6, 127.1, 127.8, 128.4, 134.7, 140.6, 144.5, 145.3, 148.2, 150.4, 153.6, 156.8, 161.2 ppm. HRMS: calcd for C176H210F24N6O22P4 [M − 2PF6]2+m/z = 1669.7033, found m/z = 1669.7159; calcd for C176H210F18N6O22P3 [M − 3PF6]3+m/z = 1064.8141, found m/z = 1064.8356.
:1 v/w) and the purple fractions were collected. The solvent was removed in vacuo to a minimal volume, followed by the addition of H2O (15 mL). The resulting precipitate was collected by filtration, affording the dumbbell 18·2PF6 (180 mg, 61%). 1H NMR (500 MHz, CD3CN): δ = 3.35 (t, 4H, J = 4.5 Hz), 3.69 (t, 4H, J = 5.0 Hz), 3.79 (t, 4H, J = 4.5 Hz), 3.97 (t, 4H, J = 4.0 Hz), 4.05 (t, 4H, J = 5.0 Hz), 4.13 (t, 4H, J = 5.0 Hz), 4.16 (t, 4H, J = 4.0 Hz), 4.75 (t, 4H, J = 5.0 Hz), 6.71 (d, 2H, J = 8.0 Hz), 6.82 (d, 2H, J = 7.5 Hz), 7.26 (t, 2H, J = 8.0 Hz), 7.30 (t, 2H, J = 7.5 Hz), 7.52 (d, 2H, J = 8.5 Hz), 7.54 (s, 4H), 7.66 (d, 2H, J = 8.5 Hz), 7.93 (d, 4H, J = 7.0 Hz), 8.73 ppm(d, 4H, J = 7.0 Hz). 13C NMR (125 MHz, CD3CN): δ = 50.1, 60.5, 67.2, 67.5, 68.6, 68.9, 69.0, 69.5, 105.2, 105.4, 113.7, 113.8, 124.2, 125.0, 125.1, 125.7, 126.1, 128.3, 135.8, 144.7, 153.6, 153.8, 154.0 ppm. HRMS: calcd for C52H56F6N8O8P [M − PF6]+m/z = 1065.3862, found m/z = 1065.3870.
:7:1)], then MeOH, Me2CO and 2% NH4PF6/Me2CO, respectively]. The purple fraction in Me2CO were collected, and concentrated to a minimum volume before the crude product was precipitated by the addition of H2O. The resulting solid was collected by filtration to afford 3·6PF6 (20 mg, 27%) as a purple powder. 1H NMR (500 MHz, CD3CN): δ = 1.17 (d, 12H, J = 5.5 Hz), 1.18 (d, 12H, J = 5.5 Hz), 2.55 (d, 1H, J = 6.0 Hz), 2.59 (d, 1H, J = 6.0 Hz), 3.35–3.40 (m, 4H), 3.90 (t, 2H, J = 3.5 Hz), 3.98 (t, 2H, J = 3.5 Hz), 4.03 (t, 2H, J = 4.5 Hz), 4.14 (t, 2H, J = 4.0 Hz), 4.20–4.23 (m, 4H), 4.27 (t, 2H, J = 3.0 Hz), 4.30 (t, 2H, J = 4.0 Hz), 4.36–4.41 (m, 6H), 4.49 (t, 2H, J = 4.3 Hz), 4.58 (t, 2H, J = 5.0 Hz), 4.73 (t, 2H, J = 4.0 Hz), 4.81 (s, 2H), 4.87 (t, 2H, J = 4.5 Hz), 4.88 (s, 2H), 5.05 (t, 2H, J = 4.3 Hz), 5.78 (d, 4H, J = 11.5 Hz), 5.86 (d, 4H, J = 11.5 Hz), 6.01 (t, 1H, J = 7.0 Hz), 6.05 (t, 1H, J = 7.0 Hz), 6.31 (d, 1H, J = 6.5 Hz), 6.38 (d, 1H, J = 6.5 Hz), 6.86 (t, 2H, J = 6.5 Hz), 7.09–7.15 (m, 6H), 7.31 (t, 2H, J = 7.0 Hz), 7.37 (d, 8H, J = 5.0 Hz), 7.64 (d, 1H, J = 6.5 Hz), 7.70 (d, 1H, J = 7.0 Hz), 7.84 (d, 2H, J = 7.5 Hz), 7.87 (s, 2H), 7.99 (s, 8H), 8.01 (d, 1H, J = 6.5 Hz), 8.11 (d, 2H, J = 6.5 Hz), 8.12 (s, 1H), 8.49 (d, 2H, J = 6.0 Hz), 8.68 (d, 2H, J = 5.5 Hz), 8.74 (b, 8H), 8.99 (d, 2H, J = 6.0 Hz). 13C NMR (125 MHz, CD3CN): δ = 23.3, 23.3, 26.5, 26.5, 28.7, 29.9, 50.1, 54.2, 61.0, 67.2, 67.5, 67.7, 67.9, 69.0, 69.3, 69.3, 69.4, 69.5, 69.8, 70.0, 104.5, 105.6, 105.9, 114.1, 124.1, 124.2, 124.3, 124.4, 124.7, 125.0, 125.2, 125.4, 125.6, 126.5, 129.1, 129.1, 131.3, 136.6, 141.6, 141.9, 143.7, 144.6, 145.0, 145.3, 150.9, 152.7, 152.8, 154.0. HRMS: calcd for C118H128F24N12O10P4 [M − 2PF6]2+m/z = 1226.9238, found m/z = 1226.9281.
Acknowledgements
The research was supported by the US National Science Foundation (NSF) under grant number CHE-0924620. We thank the NSF for the award of a graduate research fellowship to A. C. F.References
- (a) V. Balzani, A. Credi, F. M. Raymo and J. F. Stoddart, Angew. Chem., Int. Ed., 2000, 39, 3348–3391 CrossRef CAS; (b) A. R. Pease, J. O. Jeppesen, J. F. Stoddart, Y. Luo, C. P. Collier and J. R. Heath, Acc. Chem. Res., 2001, 34, 433–444 CrossRef CAS; (c) R. Ballardini, V. Balzani, A. Credi, M. T. Gandolfi and M. Venturi, Acc. Chem. Res., 2001, 34, 445–455 CrossRef CAS; (d) C. A. Schalley, K. Beizai and F. Vögtle, Acc. Chem. Res., 2001, 34, 465–476 CrossRef CAS; (e) J.-R. Collin, C. Dietrich-Buchecker, P. Gaviña, M. C. Jimenez-Molero and J.-P. Sauvage, Acc. Chem. Res., 2001, 34, 477–487 CrossRef CAS; (f) B. L. Feringa, Acc. Chem. Res., 2001, 34, 504–513 CrossRef CAS; (g) T. R. Kelly, Acc. Chem. Res., 2001, 34, 514–522 CrossRef CAS; (h) V. Balzani, A. Credi and M. Venturi, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4814–4817 CrossRef CAS; (i) Y. Shirai, J.-F. Morin, T. Sasaki, J. M. Guerrero and J. M. Tour, Chem. Soc. Rev., 2006, 35, 1043–1055 RSC; (j) V. Balzani, A. Credi, S. Silvi and M. Venturi, Chem. Soc. Rev., 2006, 35, 1135–1149 RSC; (k) W. R. Browne and B. L. Feringa, Nat. Nanotechnol., 2006, 1, 25–35 CrossRef CAS; (l) E. R. Kay, D. A. Leigh and F. Zerbetto, Angew. Chem., Int. Ed., 2007, 46, 72–191 CrossRef CAS; (m) S. Saha, K. C.-F. Leung, T. D. Nguyen, J. F. Stoddart and J. I. Zink, Adv. Funct. Mater., 2007, 17, 685–693 CrossRef CAS; (n) K. Patel, S. Angelos, W. R. Dichtel, A. Coskun, Y.-W. Yang, J. I. Zink and J. F. Stoddart, J. Am. Chem. Soc., 2008, 130, 2382–2383 CrossRef CAS; (o) J. F. Stoddart, Chem. Soc. Rev., 2009, 38, 1802–1820 RSC; (p) J. F. Stoddart, Nat. Chem., 2009, 1, 14–15 CrossRef CAS; (q) K. K. Cotí, M. E. Belowich, M. Liong, M. W. Ambrogio, Y. A. Lau, H. A. Khatib, J. I. Zink, N. M. Khashab and J. F. Stoddart, Nanoscale, 2009, 1, 16–39 RSC; (r) M. Liong, S. Angelos, E. Choi, K. Patel, J. F. Stoddart and J. I. Zink, J. Mater. Chem., 2009, 19, 6251–6257 RSC.
- (a) T. Hugel, N. B. Holland, A. Cattani, L. Moroder, M. Seitz and H. E. Gaub, Science, 2002, 296, 1103–1106 CrossRef; (b) D. A. Leigh, J. K. Y. Wong, F. Dehez and F. Zerbetto, Nature, 2003, 424, 174–179 CrossRef CAS; (c) S. P. Fletcher, F. Dumur, M. M. Pollard and B. L. Feringa, Science, 2005, 310, 80–82 CrossRef CAS; (d) J. Berná, D. A. Leigh, M. Lubomska, S. M. Mendoza, E. M. Pérez, P. Rudolf, G. Teobaldi and F. Zerbetto, Nat. Mater., 2005, 4, 704–710 CrossRef CAS; (e) M. N. Chatterjee, E. R. Kay and D. A. Leigh, J. Am. Chem. Soc., 2006, 128, 4058–4073 CrossRef CAS; (f) V. Serreli, C.-F. Lee, E. R. Kay and D. A. Leigh, Nature, 2007, 445, 523–527 CrossRef CAS; (g) F. Chiaravalloti, L. Gross, K.-H. Rieder, S. M. Stojkovic, A. Gourdon, C. Joachim and F. Moresco, Nat. Mater., 2007, 6, 30–33 CrossRef CAS; (h) L. Grill, K.-H. Rieder, F. Moresco, G. Rapenne, S. M. Stojkovic, X. Bouju and C. Joachim, Nat. Nanotechnol., 2007, 2, 95–98 CrossRef CAS; (i) G. Fioravanti, N. Haraszkiewicz, E. R. Kay, S. M. Mendoza, C. Bruno, M. Marcaccio, P. G. Wiering, F. Paolucci, P. Rudolf, A. M. Brouwer and D. A. Leigh, J. Am. Chem. Soc., 2008, 130, 2593–2601 CrossRef CAS; (j) M. von Delius, E. M. Geertsema and D. A. Leigh, Nat. Chem., 2010, 2, 96–101 CrossRef CAS.
- (a) C. P. Collier, G. Mattersteig, E. W. Wong, Y. Luo, K. Beverly, J. Sampaio, F. M. Raymo, J. F. Stoddart and J. R. Heath, Science, 2000, 289, 1172–1175 CrossRef CAS; (b) D. W. Steuerman, H.-R. Tseng, A. J. Peters, A. H. Flood, J. O. Jeppesen, K. A. Nielsen, J. F. Stoddart and J. R. Heath, Angew. Chem., Int. Ed., 2004, 43, 6486–6491 CrossRef CAS; (c) J. D. Badjić, V. Balzani, A. Credi, S. Silvi and J. F. Stoddart, Science, 2004, 303, 1845–1849 CrossRef CAS; (d) V. Balzani, M. Clemente-León, A. Credi, B. Ferrer, M. Venturi, A. H. Flood and J. F. Stoddart, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 1178–1183 CrossRef CAS; (e) J. E. Green, J. W. Choi, A. Boukai, Y. Bunimovich, E. Johnston-Halperin, E. Delonno, Y. Luo, B. A. Sheriff, K. Xu, Y. S. Shin, H.-R. Tseng, J. F. Stoddart and J. R. Heath, Nature, 2007, 445, 414–417 CrossRef CAS; (f) J. R. Heath, Annu. Rev. Mater. Res., 2009, 39, 1–23 CrossRef CAS; (g) S. J. Van Der Molen and P. Liljeroth, J. Phys.: Condens. Mater., 2010, 22, 1–30; (h) W. Zhang, E. DeIonno, W. R. Dichtel, L. Fang, A. Trabolsi, J.-C. Olsen, D. Benítez, J. R. Heath and J. F. Stoddart, J. Mater. Chem., 2011, 21, 1487–1495 RSC.
- (a) J. O. Jeppesen, J. Perkins, J. Becher and J. F. Stoddart, Angew. Chem., Int. Ed., 2001, 40, 1216–1221 CrossRef CAS; (b) J. O. Jeppesen, K. A. Nielsen, J. Perkins, S. A. Vignon, A. di Fabio, R. Ballardini, M. T. Gandolfi, M. Venturi, V. Balzani, J. Becher and J. F. Stoddart, Chem.–Eur. J., 2003, 9, 2982–3007 CrossRef CAS; (c) S. Kang, S. A. Vignon, H.-R. Tseng and J. F. Stoddart, Chem.–Eur. J., 2004, 10, 2555–2564 CrossRef CAS; (d) I. Aprahamian, T. Yasuda, T. Ikeda, S. Saha, W. R. Dichtel, K. Isoda, T. Kato and J. F. Stoddart, Angew. Chem., Int. Ed., 2007, 46, 4675–4679 CrossRef CAS; (e) Y.-L. Zhao, I. Aprahamian, A. Trabolsi, N. Erina and J. F. Stoddart, J. Am. Chem. Soc., 2008, 130, 6348–6350 CrossRef CAS.
- (a) H.-R. Tseng, S. A. Vignon and J. F. Stoddart, Angew. Chem., Int. Ed., 2003, 42, 1491–1495 CrossRef CAS; (b) H.-R. Tseng, S. A. Vignon, P. C. Celestre, J. Perkins, J. O. Jeppesen, A. di Fabio, R. Ballardini, M. T. Gandolfi, M. Venturi, V. Balzani and J. F. Stoddart, Chem.–Eur. J., 2004, 10, 155–172 CrossRef CAS.
- (a) Y. Liu, A. H. Flood, P. A. Bonvallet, S. A. Vignon, B. H. Northrop, H.-R. Tseng, J. O. Jeppesen, T. J. Huang, B. Brough, M. Baller, S. Magonov, S. D. Solares, W. A. Goddard III, C.-M. Ho and J. F. Stoddart, J. Am. Chem. Soc., 2005, 127, 9745–9759 CrossRef CAS; (b) S. Nygaard, K. C.-F. Leung, I. Aprahamian, T. Ikeda, S. Saha, B. W. Laursen, S.-Y. Kim, S. W. Hansen, P. C. Stein, A. H. Flood, J. F. Stoddart and J. O. Jeppesen, J. Am. Chem. Soc., 2007, 129, 960–970 CrossRef CAS; (c) I. Yoon, O. Š. Miljanić, D. Benítez, S. I. Khan and J. F. Stoddart, Chem. Commun., 2008, 4561–4563 RSC; (d) I. Yoon, D. Benítez, Y.-L. Zhao, O. Š. Miljanić, S.-Y. Kim, E. Tkatchouk, K. C.-F. Leung, S. I. Khan, W. A. Goddard III and J. F. Stoddart, Chem.–Eur. J., 2009, 15, 1115–1122 CAS.
- A. Coskun, D. C. Friedman, H. Li, K. Patel, H. A. Khatib and J. F. Stoddart, J. Am. Chem. Soc., 2009, 131, 2493–2495 CrossRef CAS.
- UV-Vis spectroelectrochemical experiments support firmly that the translational movement of the CBPQT4+ ring in the bistable [2]rotaxanes occurs after the first one-electron oxidation of the TTF unit on the dumbbell components.
- In this manuscript, “bipyridinium” refers to N,N′-dialkyl-4,4′-bipyridinium (BIPY2+) units in which two pyridinium moieties attached to each other directly without any other connection units (in compound 1·6PF6 and 2·6PF6). The term “bispyridinium” refers to units in which the two pyridinium moieties are connected indirectly in the 4,4′ positions by either a benzene ring (in compound 3·6PF6) or a saturated chain (in compound 4·6PF6 and 5·6PF6).
- (a) A. Trabolsi, N. Khashab, A. C. Fahrenbach, D. C. Friedman, M. T. Colvin, K. K. Cotí, D. Benítez, E. Tkatchouk, J.-C. Olsen, M. E. Belowich, R. Carmielli, H. A. Khatib, W. A. Goddard III, M. R. Wasielewski and J. F. Stoddart, Nat. Chem., 2010, 2, 42–49 CrossRef CAS; (b) A. Trabolsi, A. C. Fahrenbach, S. K. Dey, A. I. Share, D. C. Friedman, S. Basu, T. B. Gasa, N. M. Khashab, S. Saha, I. Aprahamian, H. A. Khatib, A. H. Flood and J. F. Stoddart, Chem. Commun., 2010, 46, 871–873 RSC.
- P.-L. Anelli, P. R. Ashton, R. Ballardini, V. Balzani, M. Delgado, M. T. Gandolfi, T. T. Goodnow, A. E. Kaifer, D. Philp, M. Pietraszkiewicz, L. Prodi, M. V. Reddington, A. M. Z. Slawin, N. Spencer, J. F. Stoddart, C. Vicent and D. J. Williams, J. Am. Chem. Soc., 1992, 114, 193–218 CrossRef CAS.
- (a) C. O. Dietrich-Buchecker, J.-P. Sauvage and J.-P. Kintzinger, Tetrahedron Lett., 1983, 24, 5095–5098 CrossRef CAS; (b) C. O. Dietrich-Buchecker and J.-P. Sauvage, Chem. Rev., 1987, 87, 795–810 CrossRef CAS; (c) B. Odell, M. V. Reddington, A. M. Z. Slawin, N. Spencer, J. F. Stoddart and D. J. Williams, Angew. Chem., Int. Ed. Engl., 1988, 27, 1547–1550 CrossRef; (d) M. Asakawa, P. R. Ashton, V. Balzani, A. Credi, C. Hamers, G. Mattersteig, M. Montalti, A. N. Shipway, N. Spencer, J. F. Stoddart, M. S. Tolley, M. Venturi, A. J. P. White and D. J. Williams, Angew. Chem., Int. Ed., 1998, 37, 333–337 CrossRef CAS; (e) K. E. Griffiths and J. F. Stoddart, Pure Appl. Chem., 2008, 80, 485–506 CrossRef CAS.
- (a) J. A. Bravo, F. M. Raymo, J. F. Stoddart, A. J. P. White and D. J. Williams, Eur. J. Org. Chem., 1998, 2565–2571 CrossRef CAS; (b) C. Seel and F. Vögtle, Chem.–Eur. J., 2000, 6, 21–24 CrossRef CAS; (c) A.-M. Fuller, D. A. Leigh, P. J. Lusby, I. D. H. Oswald, S. Parsons and D. B. Walker, Angew. Chem., Int. Ed., 2004, 43, 3914–3918 CrossRef CAS; (d) F. Aricó, J. D. Badjić, S. J. Cantrill, A. H. Flood, K. C.-F. Leung, Y. Liu and J. F. Stoddart, Top. Curr. Chem., 2005, 249, 203–259 CAS; (e) J. Wu, K. C.-F. Leung and J. F. Stoddart, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 17266–17271 CrossRef CAS.
- (a) A. Harada, J. Li and M. Kamachi, Nature, 1992, 356, 325–327 CrossRef CAS; (b) C. Gong, Q. Ji, C. Subramaniam and H. W. Gibson, Macromolecules, 1998, 31, 1814–1818 CrossRef CAS; (c) F. M. Raymo and J. F. Stoddart, Chem. Rev., 1999, 99, 1643–1663 CrossRef CAS.
- O. Š. Miljanić, W. R. Dichtel, S. I. Khan, S. Mortezaei, J. R. Heath and J. F. Stoddart, J. Am. Chem. Soc., 2007, 129, 8236–8246 CrossRef CAS.
- P. R. Ashton, R. Ballardini, V. Balzani, M. Bělohradský, M. T. Gandolfi, D. Philp, L. Prodi, F. M. Raymo, M. V. Reddington, N. Spencer, J. F. Stoddart, M. Venturi and D. J. Williams, J. Am. Chem. Soc., 1996, 118, 4931–4951 CrossRef CAS.
- (a) K. Biradha and M. Fujita, J. Chem. Soc., Dalton Trans., 2000, 3805–3810 RSC; (b) Y.-S. Su and C.-F. Chen, Org. Lett., 2010, 12, 1888–1891 CrossRef CAS.
- M. Asakawa, W. Dehaen, G. L'abbé, S. Menzer, J. Nouwen, F. M. Raymo, J. F. Stoddart and D. J. Williams, J. Org. Chem., 1996, 61, 9591–9595 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c0ob00937g |
| ‡ These authors contributed equally to this work. |
| § Current Address: Division of Semiconductor Business, Samsung Electronics Co. Ltd., San #16 Banwol-Dong, Hwasung-City, Gyeonggi-Do 445-701, Korea. |
| This journal is © The Royal Society of Chemistry 2011 |