FeCl3-promoted alkylation of indoles by enamides†
Tianmin
Niu
,
Lehao
Huang
,
Tianxing
Wu
and
Yuhong
Zhang
*
Department of Chemistry, Zhejiang University, Hangzhou 310027, P.R. China. E-mail: yhzhang@zju.edu.cn; Fax: 0086-571-87953244; Tel: 0086-571-87952723
First published on 17th November 2010
Abstract
An efficient iron-promoted alkylation of indoles with enamides has been accomplished under mild reaction conditions. The reaction proceeded with remarkable regioselectivity leading exclusively to substitution by indoles at α-position of enamides.
Introduction
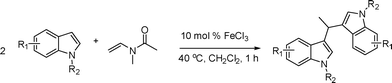
The hydroarylation of alkenes is one of the most important reactions for the functionalization of arenes and heteroarenes.1 One important application of this transformation is the alkylation of indoles, which are key structural motifs of numerous natural products and biologically active compounds.2 Although Friedel–Crafts reactions are well-known transformations for alkylation, the methods often suffer from drastic reaction conditions (high reaction temperature, strong acidic/base conditions) and regioselectivity problems.3 In recent years, the transition-metal catalyzed process has emerged as an attractive alternative to the conventional Friedel–Crafts reaction, and Cu(OTf)2,4 CeCl3,5 Zn(OTf)2,6 InBr3,7 SmI3,8 Sc(OTf)3,9 PtCl2,10 or AuCl311 catalytic systems have been developed. These processes have been proved remarkably effective under mild reaction conditions and enjoy a broad application in the synthesis of alkylated indoles. However, most of these investigations focus on the addition of indoles to electron-neutral alkenes or electron-deficient alkenes, which are activated by a conjugated electron-withdrawing group. There has been only scant attention in developing a general methodology for the addition of indoles to electron-rich alkenes such as enamides.12 To the best of our knowledge, the metal-catalyzed addition of indoles to enamides has not been explored. The development of an efficient procedure for the alkylation of indoles with electron-rich olefins such as enamides under mild conditions is highly desired.Recently, iron has been increasingly explored in organic transformations as an inexpensive and environmentally benign catalyst.13 There have been a series of reports concerning novel iron-catalyzed reactions, which lead to efficient C(sp2)–C(sp3),14 C(sp2)–C(sp2),15 C(sp2)–C(sp),16 and C–N17 bond formations. Herein, we report a highly efficient method for the addition of indoles to enamides in the presence of an iron catalyst under mild reaction conditions (Scheme 1). This facile catalytic system was also applicable to indolizines.
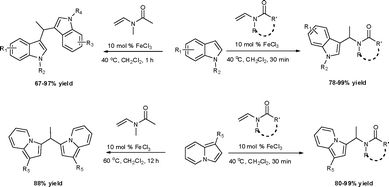 | ||
| Scheme 1 Iron-catalyzed alkylation of indoles and indolizines with enamides. | ||
Results and discussion
In our initial studies, we tested the effect of various metals on the addition of indole (1a) toward enamide (2a) in CH2Cl2. As shown in Table 1, no reaction was observed in the absence of metal catalyst (Table 1, entry 1). Treatment of 1a and 2a with ZnCl2, RuCl3 or InBr3 failed to give any product at 40 °C for 30 min (Table 1, entries 2–4). The desired addition product 1-(1-(1H-indol-3-yl)ethyl)pyrrolidin-2-one (3a) was isolated when NiCl2·6H2O or SnCl4 were applied as catalysts, but with very low yields (Table 1, entries 5–6). A more promising result was obtained by the use of 10 mol% CuCl2 (Table 1, entry 7). When BF3·Et2O was applied, 89% yield was isolated (Table 1, entry 8). Further exploration led to a discovery that a 99% isolated yield was obtained by employing FeCl3 as catalyst (Table 1, entry 9). Importantly, the reaction was carried out in open air without exclusion of oxygen from the reaction flask, and FeCl3·6H2O showed almost the same reactivity with FeCl3 (Table 1, entry 10). In contrast, iron reagents such as Fe2O3, FeCl2 and Fe(acac)3 were inactive (Table 1, entries 11–13), showing that the source of iron species influenced the reaction significantly. Catalytic amounts (10 mol%) of Brønsted acids delivered no or relatively lower product yields (Table 1, entries 14–16). The reaction could be performed at room temperature in lower yield (Table 1, entry 17). The solvents are crucial to the reaction. No reaction was observed in DMF (Table 1, entry 18). The comparable results were obtained by the use of CH3CN, toluene, acetone, and THF as solvent (Table 1, entries 19–22). It should be noted that the reaction could carried out in water to give a 78% yield in 40 °C for 30 min (Table 1, entry 23).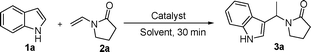
| Entry | Catalyst | Solvent | T/°C | Yield (%)b |
|---|---|---|---|---|
| a Reaction conditions: indole (0.5 mmol), 1-vinyl-2-pyrrolidinone (0.6 mmol), catalyst (0.05 mmol), solvent (5 ml), 40 °C, 30 min. b Isolated yields based on indole. c The isolated yield in anhydrous CH2Cl2 under nitrogen atmosphere is given in parentheses. d Concentrated hydrochloric acid (wt%: 36.5%) was used. | ||||
| 1 | none | CH2Cl2 | 40 | n.r |
| 2 | ZnCl2 | CH2Cl2 | 40 | n.r |
| 3 | RuCl3 | CH2Cl2 | 40 | trace |
| 4 | InBr3 | CH2Cl2 | 40 | n.r |
| 5 | NiCl2·6H2O | CH2Cl2 | 40 | 42 |
| 6 | SnCl4 | CH2Cl2 | 40 | 41 |
| 7 | CuCl2 | CH2Cl2 | 40 | 90 |
| 8 | BF3·Et2O | CH2Cl2 | 40 | 89 |
| 9 | FeCl3 | CH2Cl2 | 40 | 99 (98)c |
| 10 | FeCl3·6H2O | CH2Cl2 | 40 | 97 |
| 11 | Fe2O3 | CH2Cl2 | 40 | 32 |
| 12 | FeCl2 | CH2Cl2 | 40 | trace |
| 13 | Fe(acac)3 | CH2Cl2 | 40 | n.r |
| 14d | HCl | CH2Cl2 | 40 | 77 |
| 15 | TMSCl | CH2Cl2 | 40 | 74 |
| 16 | HOAc | CH2Cl2 | 40 | n.r |
| 17 | FeCl3 | CH2Cl2 | r.t. | 53 |
| 18 | FeCl3 | DMF | 40 | n.r |
| 19 | FeCl3 | CH3CN | 40 | 86 |
| 20 | FeCl3 | THF | 40 | 78 |
| 21 | FeCl3 | toluene | 40 | 70 |
| 22 | FeCl3 | acetone | 40 | 80 |
| 23 | FeCl3 | H2O | 40 | 78 |
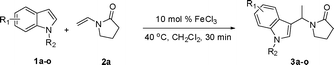
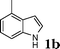
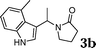
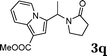
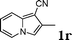
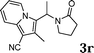
Under the optimized reaction conditions, we examined the reactivity of various indoles as summarized in Table 2. In general, indoles with both electron-rich and electron-deficient substituents are active to give the adducts in high yields. Indoles with an electron-donating group were highly active to afford the alkylated indoles in excellent yields at 40 °C within 30 min (Table 2, entries 1–6). The indoles with C2 substituents delivered the corresponding alkylated indoles in high yields (Table 2, entries 7–8), illustrating that steric hindrance played a poor role to the reaction. Indole with moderate electron-withdrawing bromide group was active also to afford the corresponding alkylated indole in 98% yield (Table 2, entry 9). However, the strong electron-withdrawing substituents in indoles led to the decrease of the reaction rate, and the prolongation of the reaction time was needed to access the high yields (Table 2, entries 10–12). N-Substituted indoles presented equally high efficiency in respect to that of free indoles to give the adducts in high yields (Table 2, entries 13–15). Furthermore, this facile catalytic system was also applicable to various indolizines (Table 2, entries 16–18).
| Entry | Indole/indolizine | Product | Yield (%)b |
|---|---|---|---|
| a Reaction conditions: indoles (0.5 mmol), 1-vinyl-2-pyrrolidinone (0.6 mmol), FeCl3 (8 mg, 0.05 mmol), in CH2Cl2 (5 ml), 40 °C, 30 min. b Isolated yields based on indole. c 6-Methyl-1H-indole (0.75 mmol), 1-vinyl-2-pyrrolidinone (0.5 mmol), isolated yields based on N-vinylformamide, 3 h was used. d The reaction time was 6 h. | |||
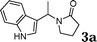
| 1 |
|
|
99 |
| 2 |
|
|
82 |
| 3 |
|
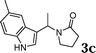
|
78 |
| 4 |
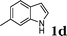
|
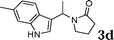
|
96c |
| 5 |
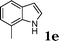
|
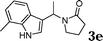
|
95 |
| 6 |
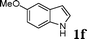
|
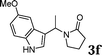
|
88 |
| 7 |
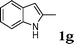
|
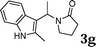
|
96 |
| 8 |
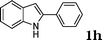
|
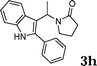
|
90 |
| 9 |
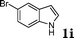
|
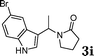
|
98 |
| 10 |
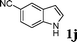
|
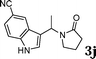
|
99d |
| 11 |
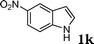
|
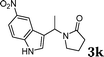
|
83d |
| 12 |
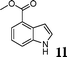
|
|
86 |
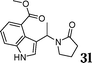
| 13 |
|
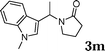
|
98 |
| 14 |
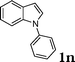
|
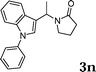
|
97 |
| 15 |
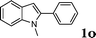
|
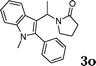
|
88 |
| 16 |
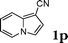
|
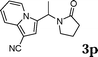
|
95 |
| 17 |
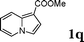
|
|
99 |
| 18 |
|
|
92 |
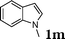
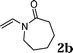
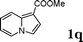
The reactivity of various enamindes was examined and the results are summarized in Table 3. Satisfying results were obtained when 1-vinylpyrrolidin-2-one 2a was replaced with 1-vinylazepan-2-one 2b (Table 3, entries 1–3). However, N-vinylformamide 2c gave the corresponding products in low yields under the reaction conditions. To our delight, when the ratio of the substrates was modified from indole![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :enamide = 1:1.2 to enamide:indole = 1:1.5, the reaction was dramatically improved to afford the products in excellent yields (Table 3, entries 4–6). N-Methyl-N-vinylacetamide 2d afforded low yield in the reaction with N-free indole 1a possibly due to the decomposition of the product, and a 79% yield was obtained when the reaction was performed at room temperature (Table 3, entry 7). On the contrary, N-methyl indole 1m participated in the reaction smoothly to afford 6b in 81% yield at 40 °C within 30 min (Table 3, entry 8). In the case of indolizine 1q, longer reaction time and higher temperature were required (Table 3, entries 3 and 9).
:enamide = 1:1.2 to enamide:indole = 1:1.5, the reaction was dramatically improved to afford the products in excellent yields (Table 3, entries 4–6). N-Methyl-N-vinylacetamide 2d afforded low yield in the reaction with N-free indole 1a possibly due to the decomposition of the product, and a 79% yield was obtained when the reaction was performed at room temperature (Table 3, entry 7). On the contrary, N-methyl indole 1m participated in the reaction smoothly to afford 6b in 81% yield at 40 °C within 30 min (Table 3, entry 8). In the case of indolizine 1q, longer reaction time and higher temperature were required (Table 3, entries 3 and 9).
| Entry | Enamide | Indole/indolizine | Product | Yield (%)b |
|---|---|---|---|---|
| a Reaction conditions: indoles (0.5 mmol), enamides (0.6 mmol), FeCl3 (8 mg, 0.05 mmol), in CH2Cl2 (5 ml), 40 °C, 30 min. b Isolated yields based on indole. c 3 h, 60 °C, were used. d Indole (0.75 mmol), N-vinylformamide enamide (0.5 mmol), isolated yields based on N-vinylformamide. e Isolated yield when concentrated HCl (10 mol %) was used as catalyst. f Isolated yield when TMSCl (10 mol%) used as catalyst. g The reaction was at room temperature. | ||||
| 1 |
|
1a |
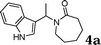
|
90 |
| 2 | 2b | 1m |
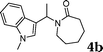
|
91 |
| 3 | 2b | 1q |
|
99c |
| 4d |
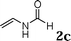
|
1a |
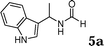
|
97
67e 81f |
| 5d | 2c | 1m |
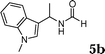
|
99 |
| 6d | 2c | 1q |
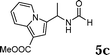
|
99 |
| 7 |
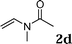
|
1a |
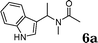
|
79g |
| 8 | 2d | 1m |
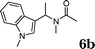
|
81 |
| 9 | 2d | 1q |
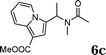
|
80c |
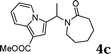
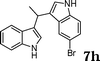
During the study of the reaction of indoles with N-methyl-N-vinylformamide 2d, we found that the ratio of indoles to enamides influenced the final products. An excess of indole led to the substitution of indoles to 2d to give a mixture of alkylated indole 6a and a second alkylation product of bis-indolylmethane 7a. Since bis-indolylmethane and its derivatives are key structural units in many natural drugs used as cancer growth inhibitors,18 we investigated the reaction conditions for selective formation of bis-indolylmethane products as shown in Table 4. It was found that the amount of indole played a key role for the second alkylation. When 3 equivalents of indole were used, only bis-indolylmethane 7a was obtained in 91% yield (Table 4, entry 1). This transformation was compatible with a broad of functional groups, including –CH3, –Br, –CN, and N–CH3 (Table 4, entries 2–6). Importantly, asymmetric bis-indolylmethanes could be prepared by the use of 6a as initial substrate (Table 4, entry 7–8). The reaction was also applicable to indolizine (Table 4, entry 9). The enamides 2a, 2b, and 2c failed to furnish the bis-indolylmethane under the reaction conditions.
| Entry | Indole/indolizine | Product | Yield (%)b |
|---|---|---|---|
| a Reaction conditions: indoles (1.5 mmol), N-methyl-N-vinylacetamide (0.5 mmol), FeCl3 (8 mg, 0.05 mmol), in CH2Cl2 (5 ml), 40 °C, 1 h. b Isolated yields based on N-methyl-N-vinylacetamide. c The reaction time prolongs to 5 h. d 12 h was used. e The reaction was in 60 °C for 12 h. | |||
| 1 |
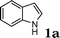
|
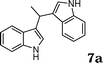
|
91 |
| 2 |
|
|
89 |
| 3 |
|
|
88c |
| 4 |
|
|
67d |
| 5 |
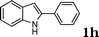
|
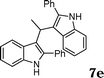
|
75 |
| 6 |
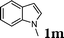
|
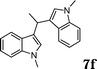
|
90 |
| 7 |
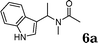
|
|
97 |
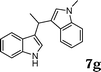
| 8 | 6a |
|
96 |
| 9 |
|
|
88e |
There are two possible pathways for the intermolecular C-3 indole alkylation as shown in Scheme 2. (1) With the aid of Lewis acid FeCl3, enamide 2a transforms to the iminium species I, which reacts with indole 1avia a Friedel–Crafts-type process to afford the alkylation product 3a (Scheme 1, path A).19 (2) The protonation of enamide 2a by the Brønsted acid hydrolyzed from FeCl3 generates the iminium II, which undergoes nucleophilic addition with indoles to give the final product (Scheme 1, path B).12,20 We performed the reaction in anhydrous CH2Cl2 under nitrogen atmosphere, and it was found that a high yield of desired product 1-(1-(1H-indol-3-yl)ethyl)pyrrolidin-2-one 3a was obtained (Table 1, entry 9). Furthermore, typical Lewis acid BF3·Et2O also worked well in this transformation (Table 1, entry 8). On the other hand, the employment of hydrochloric acid (10 mol%) and trimethyl chlorosilane (TMSCl) (10 mol%) as catalysts resulted in the desired products with enamide 2a and 2c in moderate yields (Table 1, entries 14 and 15; Table 4, entry 4), and the reaction with enamides 2b and 2d failed. Thus, we postulate the reaction takes place mainly through path A.
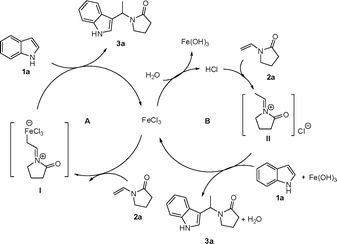 | ||
| Scheme 2 Proposed mechanism for alkylation of indoles from enamide. | ||
The production of bis-indolylmethanes in Table 4 might take place through the elimination of amine moiety of N-(1-(1H-indol-3-yl)ethyl)-N-methylacetamide 6a with the assistance of iron catalyst to form the intermediate III, which undergoes a Friedel–Crafts-type process with indoles or indolizine to deliver the symmetrical or unsymmetrical bis-indolylmethanes (Scheme 3).18a,21
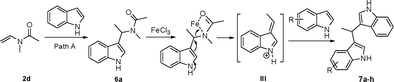 | ||
| Scheme 3 Proposed mechanism for bis-indolylmethanes. | ||
Conclusion
In summary, we have developed a highly efficient iron-catalyzed process for the addition of indoles to enamides under mild reaction conditions. The simple catalytic system worked well with a broad range of indoles and allowed the facile synthesis of alkylated indoles as well as symmetric and unsymmetric bis-indolylmethanes.Experimental section
General
Unless otherwise stated, all reactions were carried out in an oven-dried flask in air. 1H NMR spectra were recorded at 400 or 500 MHz and the chemical shifts are reported in parts per million (δ) relative to internal standard TMS (0 ppm) for CDCl3 or DMSO-d6. The peak patterns are indicated as follows: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; dd, doublet of doublet; dt, doublet of triplet. The coupling constants, J, are reported in Hertz (Hz). 13C NMR spectra were recorded at 100 or 125 MHz and referenced to the internal solvent signals (center peak is 77.00 ppm in CDCl3 or 39.90 ppm in DMSO-d6). Mass spectroscopy data were collected on an HRMS-EI instrument. Melting points were measured on a Yanaco MP-500 apparatus and uncorrected. FT-IR spectra were recorded on a Nicolet Nexus 470 FT-IR spectrophotometer and the data were reported in reciprocal centimetres (cm−1). Indoles and enamide materials were purchased from common commercial sources and used without additional purification.Representative procedure for the preparation of 3-alkylindole
To a mixture of indole (59 mg, 0.5 mmol), FeCl3 (8 mg, 0.05 mmol) and CH2Cl2 (5 mL), 1-vinylpyrrolidin-2-one (67 mg, 0.6 mmol) was added dropwise at room temperature. The resulting mixture was stirred at 40 °C for 30 min. After the reaction, the reaction solution was filtered through a pad of celite, and the solvent was removed under reduced pressure. Purification by flash chromatography over silica gel, eluting with acetate–dichloromethane (1:10), provided the desired compound as a white solid (113 mg, 99%): 1H NMR (400 MHz, CDCl3, TMS) δ 8.89 (s, 1 H), 7.62 (d, J = 8.0 Hz, 1 H), 7.38 (d, J = 7.6 Hz, 1 H), 7.19 (t, J = 7.2 Hz, 1 H), 7.13 (s, 1 H), 7.08 (t, J = 7.2 Hz, 1 H), 5.80 (q, J = 6.8 Hz, 1 H), 3.26 (dt, J = 8.6, 5.4 Hz, 1 H), 2.86 (dt, J = 9.0, 5.6 Hz, 1 H), 2.51–2.38 (m, 2 H), 1.95–1.85 (m, 1 H), 1.82–1.71 (m, 1 H), 1.58 (d, J = 7.2 Hz, 3 H); 13C NMR (100 MHz, CDCl3) δ 174.4, 136.6, 126.4, 122.3, 122.2, 119.7, 119.3, 115.7, 111.3, 42.7, 42.2, 31.8, 17.7, 16.7. HRMS (EI) Calcd for C14H16N2O: [M]+ 228.1263; Found, 228.1260; IR ν (KBr) 3244, 3058, 2973, 2932, 1659, 1493, 1455, 1428, 1342, 1287, 1248, 1198, 1116, 771, 745, 659. cm−1; mp: 147–149 °C.
Representative procedure for the preparation of bis-indolylmethanes
N-Methyl-N-vinylacetamide (50 mg, 0.5 mmol), indole (176 mg, 1.5 mmol), FeCl3 (8 mg, 0.05 mmol) and CH2Cl2 (5 mL) were introduced into the reaction vessel at room temperature. The resulting mixture was stirred at 40 °C for 1 h. After the reaction, the reaction solution was filtered through a pad of celite, and the solvent was removed under reduced pressure. Purification by flash chromatography over silica gel, eluting with acetate–petrol ether (1:10), provided the desired compound as a white solid (118 mg, 91%): 1H NMR (400 MHz, CDCl3, TMS) δ 7.75 (s, 2 H), 7.63 (d, J = 7.2 Hz, 2 H), 7.34 (d, J = 8.0 Hz, 2 H), 7.22 (t, J = 7.4 Hz, 2 H), 7.10 (t, J = 7.6 Hz, 2 H), 6.84 (s, 2 H), 4.72 (q, J = 7.2 Hz, 1 H), 1.85 (d, J = 7.2 Hz, 3 H); 13C NMR (100 MHz, CDCl3) δ 136.6, 126.9, 121.8, 121.6, 121.3, 119.7, 119.0, 111.5, 28.2, 21.8.
Acknowledgements
Funding from Natural Science Foundation of China (No. 20872126) and Zhejiang Provincial Natural Science Foundation of China (R407106) is acknowledged.Notes and references
- (a) S. Cacchi and G. Fabrizi, Chem. Rev., 2005, 105, 2873 CrossRef CAS; (b) M. Bandini and A. Eichholzer, Angew. Chem., Int. Ed., 2009, 48, 9608 CAS; (c) T. Arai and N. Yokoyama, Angew. Chem., Int. Ed., 2008, 47, 4989 CrossRef CAS.
- (a) N. K. Garg, R. Sarpong and B. M. Stoltz, J. Am. Chem. Soc., 2002, 124, 13179 CrossRef CAS; (b) H. Mizoguchi, H. Oguri, K. Tsuge and H. Oikawa, Org. Lett., 2009, 11, 3016 CrossRef CAS; (c) H. Ueda, H. Satoh, K. Matsumoto, K. Sugimoto, T. Fukuyama and H. Tokuyama, Angew. Chem., Int. Ed., 2009, 48, 7600 CrossRef CAS; (d) B. Kenda, Y. Quesnel, A. Ates, P. Michel, L. Turet and J. Mercier, Chem. Abstr., 2006, 146, 45393 WO 2006128692; (e) M. Lehr, Arch. Pharm., 1996, 329, 386 CrossRef CAS; (f) T. D. Aicher, B. Balkan, P. A. Bell, L. J. Brand, S. H. Cheon, R. O. Deems, J. B. Fell, W. S. Fillers, J. D. Fraser, J. Gao, D. C. Knorr, G. G. Kahle, C. L. Leone, J. Nadelson, R. Simpson and H. C. Smith, J. Med. Chem., 1998, 41, 4556 CrossRef CAS; (g) Z. Guo, M. Xian, W. Zhang, A. McGill and P. G. Wang, Bioorg. Med. Chem., 2001, 9, 99 CrossRef CAS; (h) K. Oh, W. Mar, S. Kim, J. Kim, M. Oh, J. Kim, D. Shin, C. J. Sim and J. Shinc, Bioorg. Med. Chem. Lett., 2005, 15, 4927 CrossRef CAS; (i) B. Bao, Q. Sun, X. Yao, J. Hong, C.-O. Lee, H. Y. Cho and J. H. Jung, J. Nat. Prod., 2007, 70, 2 CrossRef CAS; (j) Y. Wu and R. P. J. Ryan, Chem. Abstr., 1972, 77, 164476 US 3679702 19720725; (k) H. Kohn, K. N. Sawhney, P. LeGall, J. D. Conley, D. W. Robertson and J. D. Leander, J. Med. Chem., 1990, 33, 919 CrossRef CAS.
- (a) F. Andreanti, R. Andrisano, C. D. Case and M. Tramontini, J. Chem,. Soc., 1970, 1157 Search PubMed; (b) S. Nunomoto, Y. Kawakami, Y. Yamashita, H. Takeuchi and S. Eguchi, J. Chem. Soc., Perkin Trans. 1, 1990, 111 RSC; (c) Z. Zhang, X. Wang and R. A. Widenhoefer, Chem. Commun., 2006, 3717 RSC.
- (a) T. B. Poulsen and K. A. Jørgensen, Chem. Rev., 2008, 108, 2903 CrossRef CAS; (b) S. Yamazaki and Y. Iwata, J. Org. Chem., 2006, 71, 739 CrossRef CAS; (c) H. Yang, Y.-T. Hong and S. Kim, Org. Lett., 2007, 9, 2281 CrossRef CAS.
- G. Bartoli, M. Bartolacci, M. Bosco, G. Foglia, A. Giuliani, E. Marcantoni, L. Sambri and E. Torregiani, J. Org. Chem., 2003, 68, 4594 CrossRef CAS.
- Y.-X. Jia, S.-F. Zhu, Y. Yang and Q.-L. Zhou, J. Org. Chem., 2006, 71, 75 CrossRef CAS.
- (a) M. Bandini, P. G. Cozzi, P. Melchiorre and A. Umani-Ronchi, J. Org. Chem., 2002, 67, 5386 CrossRef CAS; (b) M. Yasuda, T. Somyo and A. Baba, Angew. Chem., Int. Ed., 2006, 45, 793 CrossRef CAS.
- H. Mao, X. Wang, W. Wang, L. He, L. Kong and J. Liu, Synthesis, 2008, 2582 CAS.
- (a) I. Komoto and S. Kobayashi, Org. Lett., 2002, 4, 1115 CrossRef CAS; (b) J. S. Yadav, B. V. S. Reddy, K. V. R. Rao and G. G. K. S. N. Kumar, Tetrahedron Lett., 2007, 48, 5573 CrossRef CAS.
- (a) C. Liu and R. A. Widenhoefer, J. Am. Chem. Soc., 2004, 126, 10250 CrossRef CAS; (b) C. Liu, X. Han, X. Wang and R. A. Widenhoefer, J. Am. Chem. Soc., 2004, 126, 3700 CrossRef CAS; (c) X. Han and R. A. Widenhoefer, Org. Lett., 2006, 8, 3801 CrossRef CAS.
- (a) M. M. Rozenman, M. W. Kanan and D. R. Liu, J. Am. Chem. Soc., 2007, 129, 14933 CrossRef CAS; (b) W. Rao and P. W. H. Chan, Org. Biomol. Chem., 2008, 6, 2426 RSC.
- (a) M. Terada and K. Sorimachi, J. Am. Chem. Soc., 2007, 129, 292 CrossRef CAS; (b) Y.-X. Jia, J. Zhong, S.-F. Zhu, C.-M. Zhang and Q.-L. Zhou, Angew. Chem., Int. Ed., 2007, 46, 5565 CrossRef CAS.
- (a) C. Bolm, J. Legros, J. L. Paih and L. Zani, Chem. Rev., 2004, 104, 6217 CrossRef CAS; (b) J. Bonnamour and C. Bolm, Org. Lett., 2008, 10, 2665 CrossRef CAS; (c) A. Correa, M. Carril and C. Bolm, Angew. Chem., Int. Ed., 2008, 47, 2880 CrossRef; (d) A. Fürstner, A. Leitner, M. Méndez and H. Krause, J. Am. Chem. Soc., 2002, 124, 13856 CrossRef; (e) B. Plietker, Angew. Chem., Int. Ed., 2006, 45, 6053 CrossRef CAS; (f) C. C. Kofink, B. Blank, S. Pagano, N. Götz and P. Knochel, Chem. Commun., 2007, 1954 RSC; (g) J. Kischel, I. Jovel, K. Mertins, A. Zapf and M. Beller, Org. Lett., 2006, 8, 19 CrossRef CAS.
- (a) Y.-Z. Li, B.-J. Li, X.-Y. Lu, S. Lin and Z.-J. Shi, Angew. Chem., Int. Ed., 2009, 48, 3817 CrossRef CAS; (b) W. M. Czaplik, M. Mayer and A. J. Wangelin, Angew. Chem., Int. Ed., 2009, 48, 607 CrossRef CAS; (c) C. M. R. Volla and P. Vogel, Angew. Chem., Int. Ed., 2008, 47, 1305 CrossRef CAS.
- (a) T. Hatakeyama, S. Hashimoto, K. Ishizuka and M. Nakamura, J. Am. Chem. Soc., 2009, 131, 11949 CrossRef CAS; (b) T. Hatakeyama and M. Nakamura, J. Am. Chem. Soc., 2007, 129, 9844 CrossRef CAS; (c) T. Nagano and T. Hayashi, Org. Lett., 2005, 7, 491 CrossRef CAS; (d) N. Yoshikai, A. Matsumoto, J. Norinder and E. Nakamura, Angew. Chem., Int. Ed., 2009, 48, 2925 CrossRef CAS; (e) J. Wen, J. Zhang, S.-Y. Chen, J. Li and X.-Q. Yu, Angew. Chem., Int. Ed., 2008, 47, 8897 CrossRef CAS.
- (a) M. Carril, A. Correa and C. Bolm, Angew. Chem., Int. Ed., 2008, 47, 4862 CrossRef CAS; (b) T. Hatakeyama, Y. Yoshimoto, T. Gabriel and M. Nakamura, Org. Lett., 2008, 10, 5341 CrossRef CAS.
- (a) B. Yao, Z. Liang, T. Niu and Y. Zhang, J. Org. Chem., 2009, 74, 4630 CrossRef CAS; (b) A. Correa and C. Bolm, Angew. Chem., Int. Ed., 2007, 46, 8862 CrossRef CAS; (c) D. Guo, H. Huang, J. Xu, H. Jiang and H. Liu, Org. Lett., 2008, 10, 4513 CrossRef CAS; (d) B. Anxionnat, A. Guérinot, S. Reymond and J. Cossy, Tetrahedron Lett., 2009, 50, 3470 CrossRef CAS; (e) Z. Zhan, J. Yu, H. Liu, Y. Cui, R. Yang, W. Yang and J. Li, J. Org. Chem., 2006, 71, 8298 CrossRef CAS.
- (a) M. Shiri, M. A. Zolfigol, H. G. Kruger and Z. Tanbakouchian, Chem. Rev., 2010, 110, 2250 CrossRef CAS; (b) R. Bell, S. Carmeli and N. Sar, J. Nat. Prod., 1994, 57, 1587 CrossRef CAS; (c) X. Guo, S. Pan, J. Liu and Z. Li, J. Org. Chem., 2009, 74, 8848 CrossRef CAS; (d) R. Veluri, I. Oka, I. Wagner-Döbler and H. Laatsch, J. Nat. Prod., 2003, 66, 1520 CrossRef CAS; (e) Q.-L. He, F.-L. Sun, X.-J. Zheng and S.-L. You, Synlett, 2009, 1111 CAS.
- (a) E. Angelini, C. Balsamini, F. Bartoccini, S. Lucarini and G. Piersanti, J. Org. Chem., 2008, 73, 5654 CrossRef CAS; (b) Y.-X. Jia, J.-H. Xie, H.-F. Duan, L.-X. Wang and Q.-L. Zhou, Org. Lett., 2006, 8, 1621 CrossRef CAS; (c) M. C. Kimber, Org. Lett., 2010, 12, 1128 CrossRef CAS.
- T. Akiyama, Chem. Rev., 2007, 107, 5744 CrossRef CAS.
- (a) G. Herrán, A. Segura and A. G. Csákÿ, Org. Lett., 2007, 9, 961 CrossRef CAS; (b) R. Ballini, A. Palmieri, M. Petrini and E. Torregiani, Org. Lett., 2006, 8, 4093 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data for all new compounds along with copies of 1H and 13C NMR spectral data. See DOI: 10.1039/c0ob00709a |
| This journal is © The Royal Society of Chemistry 2011 |