Chiral separation by enantioselective liquid–liquid extraction
Boelo
Schuur†
bc,
Bastiaan J. V.
Verkuijl†
a,
Adriaan J.
Minnaard
a,
Johannes G.
de Vries
*ad,
Hero J.
Heeres
*b and
Ben L.
Feringa
*a
aUniversity of Groningen, Stratingh Institute for Chemisty, Nijenborgh 4, 9747 AG, Groningen, The Netherlands. E-mail: B.L.Feringa@rug.nl; Fax: +31 50 363 4278; Tel: +31 50 363 4296
bUniversity of Groningen, Chemical Engineering Department, Nijenborgh 4, 9747 AG, Groningen, The Netherlands. E-mail: H.J.Heeres@rug.nl; Fax: +31 50 363 4479; Tel: +31 50 363 4484
cEindhoven University of Technology, Chemical Engineering and Chemistry Department, P.O. Box 513, 5600 MB, Eindhoven, The Netherlands. E-mail: B.Schuur@tue.nl; Fax: +31 40 246 3966; Tel: +31 40 247 4716
dDSM Innovative Synthesis BV – A unit of DSM Pharma Chemicals, P.O. Box 16, 6160 MD, Geleen, The Netherlands. E-mail: Hans-JG.Vries-de@DSM.com; Fax: +3146 476 7604; Tel: +31 46 476 1572
First published on 25th November 2010
Abstract
The literature on enantioselective liquid–liquid extraction (ELLE) spans more than half a century of research. Nonetheless, a comprehensive overview has not appeared during the past few decades. Enantioselective liquid–liquid extraction is a technology of interest for a wide range of chemists and chemical engineers in the fields of fine chemicals, pharmaceuticals, agrochemicals, fragrances and foods. In this review the principles and advances of resolution through enantioselective liquid–liquid extraction are discussed, starting with an introduction on the principles of enantioselective liquid–liquid extraction including host–guest chemistry, extraction and phase transfer mechanisms, and multistage liquid–liquid extraction processing. Then the literature on enantioselective liquid–liquid extraction systems is reviewed, structured on extractant classes. The following extractant classes are considered: crown ether based extractants, metal complexes and metalloids, extractants based on tartrates, and a final section with all other types of chiral extractants.
Introduction
The availability of enantiopure compounds is of prime importance for the pharmaceutical1 and also to some extent for the agrochemical, flavour and fragrances industries.2 They may be obtained either via a synthetic approach,3 or via the separation of racemates (Fig. 1).4,5 The synthetic routes can be based on natural compounds, fermentation or asymmetric synthesis.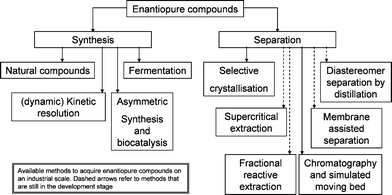 | ||
| Fig. 1 Available methods for acquiring enantiopure compounds. | ||
The chiral pool strategy uses chiral compounds from nature or products derived thereof (e.g. from fermentation processes6). Examples of industrial fermentations are the production of various acids (e.g.lactic acid, citric acid),7penicillin,8 and α-amino acids.9 Usually the production using this strategy is relatively cheap. Unfortunately, not all enantiopure precursors can be obtained from natural sources. When the chiral pool is not an option, conversion of prochiral compounds into chiral compounds by asymmetric synthesis10 is an attractive alternative. Although asymmetric catalysis is intrinsically very powerful, its use in practice may be hampered by high catalyst costs and limited development time caused by time-to-market pressure.11,12
Enantiopure compounds may also be obtained by racemic synthesis followed by enantiomer separation. The time-to-market for this strategy is potentially shorter than in asymmetric catalysis, provided the separation of enantiomers can be done with a technique that is broadly applicable to structurally diverse substrates. A facile synthesis of racemic compounds followed by a racemate separation using a broadly applicable technology that is easily developed is possibly very beneficial. Separation of racemates on industrial scale is usually based on resolution by crystallization.13–17 Although resolution by crystallization is the most frequently used route to enantiopure compounds, the main drawbacks are the low versatility and excessive solids handling and a maximum yield of 50%,18 unless racemization of the undesired enantiomer can be applied.19
Numerous laboratory techniques are available for enantioseparation.20–24 Typically, for scaling up of successful and very broadly applicable laboratory techniques such as chromatography25 and capillary electrophoresis,26 high capital investments are required.27 Nevertheless, it has been argued that for small volumes of single enantiomers needed in early development stages, the high separation costs by chromatographic techniques are still small compared to the total drug development costs.28 Some impressive preparative chiral separations have been demonstrated using centrifugal partition chromatography,29 and simulated moving bed chromatography.30,31 Another strategy for chiral separations is to use membrane-based approaches.12,32–35 When applying immobilized selectors in (liquid) membranes, the amount of selector needed can be reduced greatly. Limitations of this technology are the relatively low transport rates through the membranes requiring high membrane areas, delicate pressure control, and the risk of fouling.
In liquid–liquid extraction, diffusion and convection are the transport mechanisms, and high transport rates should be achievable.36 Liquid–liquid extraction is a mature technology that can easily be operated in a continuous countercurrent mode to fractionate the racemate into its enantiomers,37 which is advantageous when scaling-up. This possibility to operate at all scales, from laboratory separations to bulk processes in the chemical industry, makes the use of liquid–liquid extraction for enantioseparation especially interesting. The use of liquid–liquid extraction for enantioseparation has been known since 1959,38 and the first articles in the English literature appeared in the late 1960's.39–42 Enantioselective liquid–liquid extraction (ELLE) is an attractive alternative for chromatography and resolution by crystallization that, based on the scientific literature, still needs development towards commercialization. Studies cited in the literature deal mainly with the introduction of novel extraction systems. Only a few engineering studies have been reported.36,43–47 In the past 15 years, two reviews on aspects of chiral resolution by extraction have appeared, the first one (1994) with a strong emphasis on extraction chemistry,48 and the second one (2001) on extraction technology.37 In the current overview, the reader is first introduced to the principles of ELLE and the development strategy for successful application of ELLE in (continuous) industrial processes. Ample literature is available on liquid–liquid extraction equipment.49 However, the equipment that has typically been used by researchers in the field is different from traditionally and industrially applied equipment. Therefore, a concise overview of extraction equipment is provided. Next, an overview of ELLE chemistry, categorized according to chiral extractant classes, is presented as well as an overview of hosts. Finally, conclusions and an outlook on the field are given.
Principles and application of enantioselective liquid–liquid extraction
ELLE is closely affiliated to the large field of host–guest chemistry50 and chiral recognition can be regarded as a mere practical application of the developments in this wide field. Other closely related applications are the optical resolution of racemic compounds by inclusion complexation.51–54 and resolution by distillation with inclusion compounds.55ELLE combines the concepts of enantiomeric recognition56 and solvent extraction57–59 in a single technique. In this section, the principles underlying ELLE and the approaches for application of ELLE in multistage extraction processes will be introduced.Host–guest chemistry and chiral recognition
The principle of enantiomeric recognition is essential for enantioselective complexation and thus for ELLE. Without enantiomeric recognition, enantioselective processes are not possible. Encounter complexes of the extractant with the enantiomers are formed as a result of intermolecular interactions.60 These intermolecular interactions may include ion pairing, hydrogen bonding, π–π interactions, dipole and Van der Waals interactions.56 The complexation of a chiral host with a chiral guest bearing one stereocenter can be schematically represented as the guest binding with groups B, C and D to the groups B′, C′ and D′ of the host (Fig. 2, left). The opposite enantiomer will bind to two out of three sites (Fig. 2, right). The three point attachment (TPA) model states that the complex in Fig. 2 (left) is the preferred one.61 If the interactions, which are presented as dotted lines, represent not just attracting interactions, but also repulsive interactions, the TPA model is modified to the three point interaction (TPI) model.62,63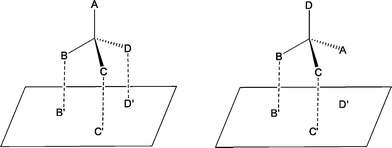 | ||
| Fig. 2 The three point model for the complexation of both substrate enantiomers to a chiral host. | ||
The field of enantiomeric recognition was boosted in the 1970's by Lehn and Cram50,64 and has led to a broad spectrum of host–guest chemistry. A wide variety of discriminative interactions has been reported. The field has been extensively reviewed and various approaches have been discussed: host–guest chemistry in general,65 anion recognition,66–68peptide and protein recognition,69carbohydrate recognition70 and recognition of carboxylic acids,71 neutral molecules72 and amino compounds.73
Extraction and phase transfer mechanisms
In addition to the chiral recognition principle, it is also essential for ELLE that there should be two (at least partially) immiscible phases, normally an aqueous and an organic phase. A typical ELLE system initially has the substrate predominantly confined in the aqueous phase (Fig. 3). After adding a lipophilic host or extractant, which prefers to be confined in the organic phase, a host-mediated phase transfer of the substrate occurs. In the example illustrated in Fig. 3, interaction of the host with the (S)-enantiomer is favored over interaction with the (R)-enantiomer. As a result, the organic phase will be enriched with the (S)-enantiomer and the aqueous phase enriched with the (R)-enantiomer.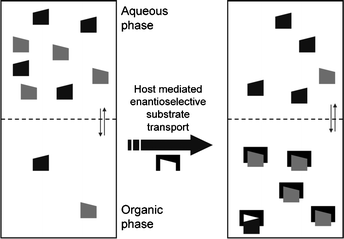 | ||
Fig. 3
Extraction and enantiomeric recognition in a biphasic system. Symbols:  : (S)-enantiomer, : (S)-enantiomer,  : (R)-enantiomer, : (R)-enantiomer,  : host. Taken from ref. 74 with permission, © 2009, ACS. : host. Taken from ref. 74 with permission, © 2009, ACS. | ||
Without going into mechanistic detail, the two main mechanisms given in the extraction literature46 are introduced here briefly. The two different types of extraction mechanism that have been proposed for ELLE are the homogeneous ligand addition mechanism, involving a homogeneous reaction in either one of the liquid phases between host and guest, and the interfacial ligand exchange mechanism.46,75 The two mechanisms are depicted in Fig. 4.
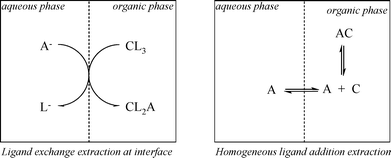 | ||
| Fig. 4 Interfacial ligand exchange mechanism of solute A with extractant C (left) and homogeneous organic phase ligand addition mechanism (right). | ||
The main difference between the two models is the locus of the complexation reaction. The interfacial reaction model obviously applies when the reaction takes place at the interface. This situation is likely to be valid when the guest is insoluble in the organic phase and the host is insoluble in the water phase. This type of mechanism is frequently observed in the field of metal-ion extractions,76,77 and is characteristic for ligand exchange extraction systems. Ligand exchange systems may also be appropriate for ELLE of ionic guests. When either the host or guest is soluble in the other phase, homogeneous reactions are also possible. The homogeneous reaction model is appropriate for ELLE systems with a substrate addition mechanism, and the solutes are at least slightly soluble in the extract phase (which is usually the case, as the solutes are typically organic compounds). This type of mechanism has been reported, for instance, for extractions of organic acids from aqueous fermentation broths.78,79
Understanding of the complexation mechanism is important when aiming at a process optimization. Typically, the substrates (α-amino acids, amino alcohols, and carboxylic acids) can exist in a neutral form, and in a (de)protonated form, depending on the actual pH in the aqueous phase. When the extraction proceeds through an interfacial anion exchange mechanism, the highest distributions are found at a pH where the guests prevail in the deprotonated form. When instead a homogeneous organic phase ligand addition mechanism is operative, the highest distributions may be found at a pH where the neutral form of the guest prevails.
Approaches for ELLE application in multistage processes
A key element in developing the technology of ELLE from the principles discussed above into a satisfactory separation process is the multistage countercurrent process approach. With this approach, it is not necessary to achieve a complete separation of enantiomers in a single stage and therefore also the selectivity requirements are not as extreme as in, for example, asymmetric catalysis. As a result, it is more likely that a single extractant can be applied to separate a broader range of racemates, which is desired.27 With versatile extractants for racemate separations, the development times for new synthetic routes of chiral compounds can be shortened. Thus, with the multistage approach, highly enantiopure compounds are obtainable in high yields using selectors that display only moderate selectivity. Several configurations may be applied, such as cocurrent multistage extraction,80 countercurrent multistage extraction,81 or countercurrent multistage fractional extraction,47 which may be considered as the most efficient configuration. Fig. 5 displays a scheme of a fractional extraction setup with a back-extraction section to recover the host to enable recycling. In fractional extraction, the feed enters the cascade of extractors or the extraction column at an intermediate stage. The stages from the feed stage F to stage N are called the strip section, the stages 1 to F form the wash section. This configuration allows efficient extraction of the desired enantiomer in the strip section, whereas the co-extracted enantiomer that is not desired is washed out of the extract stream in the wash section. The extract stream with the desired enantiomer leaves the wash section from stage 1, and enters the back-extraction section where the host is recovered to re-enter the strip section in stage N. The recovered enantiomer leaves the process in the back-extraction stream. If the enantiopurity in this stream is very high, part of the stream may be refluxed to join the wash stream and re-enter in stage 1 of the wash section. As a consequence, the increased concentration of the preferred enantiomer in the aqueous stream at the start of the cascade prevents the preferred enantiomer already complexed to the host in the organic phase from back-extracting into the aqueous phase. In this way the enantiomeric excess increases, so that a reduction of the number of extraction stages, and of the required amount of solvent is possible.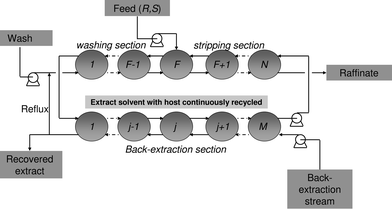 | ||
| Fig. 5 Multistage fractional extraction scheme with back-extraction section to recover the host. | ||
Several stages may be identified in the development of a multistage fractional extraction process. The first and probably the most essential step is the selection of a good host. Host selection is not easy and although over the years numerous hosts have been identified for the extraction of several substrate classes such as α-amino acids, amino alcohols and carboxylic acids, most hosts show only reasonable selectivity for a limited number of racemates. Besides being selective, the host–substrate complexation needs to be reversible and the host needs to be confined in a single phase as much as possible. These properties are vital for the application in a fractional extraction cascade. The hosts that have been reported thus far are discussed further on.
After a suitable host has been selected, single stage batch experiments are performed with the aim of the elucidation of the extraction mechanism and single stage extraction modeling. With an appropriate single stage model, the distribution of the enantiomers may be determined as function of the process conditions. Also, the single stage model parameters may be used in modeling multistage processes in order to determine the minimum wash, extract, and back-extraction flows to separate the enantiomers in the feed stream, and to determine the minimum number of stages for this separation. Heeres et al. demonstrated that ELLE is possible in continuous centrifugal contactor separator equipment.82 These continuous centrifugal contactor separators (CCS) seem highly suitable for ELLE, because the liquid hold-up is limited, and only a small amount of the precious host needs to be available in order to separate larger amounts of enantiomers in a continuous flow mode. Batch experimentation techniques were applied to develop an equilibrium stage model for the ELLE of racemic 3,5-dinitrobenzoyl-leucine using a Cinchona alkaloid type of host.75 The applicability of equilibrium stage modeling in this continuous process was also demonstrated. A single extraction stage corresponded to an equilibrium stage.
The relationship between the minimal number of required fractional extraction steps (Nmin) for full separation of both enantiomers of the substrate and the desired ee (or fraction equivalents, defined by xR and xS) of the substrate and the operational selectivity (αop) of a single extraction, is given by the Fenske equation (eqn (1)).
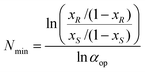 | (1) |
The relationship between Nmin and αop is depicted in Fig. 6 (left). At an αop of 1.5, Nmin is still 25. Nmin drops exponentially as αop increases. This graph illustrates that full separation is already possible at moderate levels of αop by using a fractional extraction CCS cascade with a reasonable number of fractional steps. Furthermore, it is evident that increasing αop beyond 7 provides only a slight decrease in Nmin. This illustrates the strength of the technology, as it is evident that with a limited number of stages a racemate can be separated, provided that there is a modest selectivity.
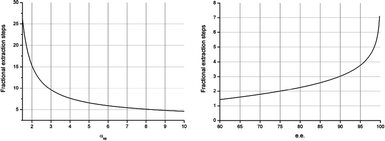 | ||
| Fig. 6 The minimal number of fractional extraction stages (Nmin) required for ee = 99% as a function of αop (left) and the minimal number of stages required for a system with αop = 7.0 as a function of ee (right). | ||
In Fig. 6 (right), the relationship between the desired ee and Nmin is depicted for an αop = 7.0. It is clear that only 4 stages are required to obtain 95% ee, and with 7 stages an ee as high as 99.8% ee is obtained.
The above mentioned examples of continuous extraction modeling with equilibrium stage models both involve discrete stages (like those displayed in Fig. 5). In more traditional extraction columns, no discrete stages can be identified, but a certain height corresponds with one theoretical equilibrium stage. For extraction columns, the number of stages calculated with an equilibrium model can thus be translated into the required height of the column using the height equivalent of a theoretical stage (HETS)49 approach. The HETS should be determined experimentally.
Single stage equilibria
The most common way to describe the performance of an ELLE process is using the ee. However, the ee alone does not fully describe the performance. This subsection lists the definitions required for adequate performance determination.27 The ee is a typical example of an operational definition, and not an intrinsic one. Operational definitions describe experimental observations. Another example of an operational definition is the operational selectivity of an ELLE system. The operational selectivity is defined as the ratio of the distributions of the enantiomers, when the (R) enantiomer is preferentially extracted: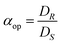 | (2) |
In eqn (2), the distributions are defined as:
 | (3) |
Both the aqueous phase concentration of the enantiomers and the organic phase concentration is the sum of the concentrations of all forms present in that phase, e.g. for the aqueous phase concentration of a weak Brønsted acid both the acid and its conjugated base should be taken into account. For example, the N-protected dinitrobenzoyl α-amino acids behave as weak acids, and depending on the pH either the neutral form or the deprotonated form is the predominant species.75 Similarly, in case of the homogeneous complexation model, the total concentration of the enantiomers in the organic phase is the sum of the host–guest complex concentrations and the concentrations of the free enantiomers in the organic phase (the interfacial ligand exchange model is most likely with almost zero solubility of the free enantiomers in the organic phase, hence only host–guest complexes are observed in the organic phase and no free enantiomers). The operational definitions are important to determine the actual performance of the extractive system and to establish the product purity, but do not give information on the intrinsic enantioselectivity of the extractant. The intrinsic selectivity of an ELLE system is defined as the ratio of the equilibrium constants, e.g. for preferential complexation of the host with the (R) enantiomer:
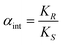 | (4) |
| [R]org = m[R]aq | (5) |
Thus, using the aqueous phase dissociation/protonation relations, the partition coefficient and the complexation equilibrium relations, a single stage can be described fully intrinsically. An example of such an intrinsically defined single stage extraction is given in Fig. 7.
Enantioselective liquid–liquid extraction equipment
Traditional equipment for liquid–liquid extraction used in the chemical industry covers a wide range, both in types and in sizes.49 Selection of the most suitable type is dependent on the process conditions and constraints and may be supported by selection charts.83 There are two basic types of extractors, the mixer–settlers that form discrete stages and are used for the bulkiest of all separation processes, and the extraction columns that are most broadly applied in all kinds of extraction processes. Extraction columns are considered as differential equipment, as no discrete stages can be distinguished. Therefore, the number of stages in a column is determined by expressing the height equivalent of a theoretical stage (HETS). Many types of columns are available with a broad range of internals used to enhance the mass transport facilitating the separation.57 For special needs, such as short contact times to avoid product degradation, numerous types of centrifugal extractors have been developed,84 some resembling the discrete stages in traditional mixer–settlers (e.g. the centrifugal contactor separators) and others resembling the differential contacting equipment (e.g. the Podbielniak centrifugal extractor).85Bulk liquid membranes
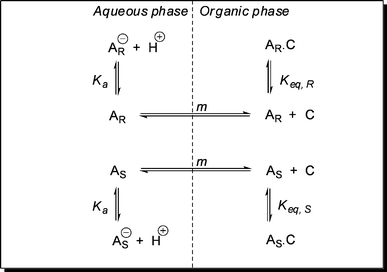
Industrial equipment is less suitable for fundamental research on extraction mechanisms. Therefore, researchers have come up with innovative types of equipment that allow for demonstration of enantioselectivity or elucidation of intrinsic parameters. Fundamental research on extraction kinetics is commonly performed using Lewis type of stirred cells.76,86 Next to the traditional Lewis cell, novel devices have been developed for ELLE characterization. The first novel device that was especially designed for ELLE purposes was the chiral resolution machine developed by Cram and coworkers87 (Fig. 8).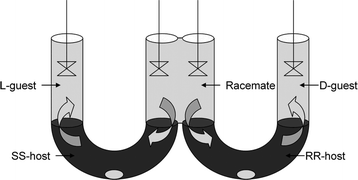 | ||
| Fig. 8 Cram's catalytic resolving device, reproduced from ref. 87 with permission. © 1979, ACS. | ||
This simple resolution device is fed with aqueous racemate in the middle compartment, and the two U-tubes filled with heavier chlorinated organic liquids that contain each one of the enantiomers of the host. The individual enantiomers from the racemate travel preferentially through one of the chiral host phases, depending on the enantiomer of the host applied and end up in the outer aqueous receiving compartments.
The groups of de Mendoza88 and De Vries89 have applied single U-tubes with only one enantiomer of the hosts to examine the resolving process. With the U-tube type of resolving equipment, having exactly determined interfacial areas, mass transfer characteristics could be studied, however, they are mostly applied without a stirrer in the feed and receive phase. The use of a device as in Fig. 8 could ensure well mixed aqueous phases in order to determine mass transfer characteristics properly.
Supported liquid membranes
Abe et al. reported the continuous separation of mandelic acid enantiomers with their liquid particle extractor, which may be considered as a continuous chromatographic device.90 Although the step from batch to continuous separation is worth mentioning, the device has never matured to preparative scale, most likely due to the large solvent flows required. More recently, supported liquid membrane technology (see Fig. 9) was reported as another type of continuous enantioseparation process.34 With this technology very high purities may be obtained with only limited amounts of precious hosts; however the practical use is limited by the risk of fouling, loss of extract phase, and the slow transport rates.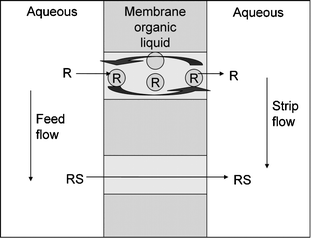 | ||
| Fig. 9 Supported liquid membrane flow. | ||
Centrifugal contactor separators (CCS)
Until recently, the state of the art in ELLE was limited to a demonstration of the proof of principle for ELLE using the above discussed types of equipment for batch-wise operation, or applying chromatographic or membrane approaches. The research on continuous ELLE processes in liquid–liquid biphasic systems to design multistage countercurrent fractional extraction processes for separation of racemates into single enantiomers with only moderate extractant selectivity had not been reported. This is possibly due to the large volumes required in the traditional types of continuous extraction equipment. Making use of centrifugal contactor separators (CCS), only small amounts of the precious chiral extractants are required for multistage continuous separation of enantiomers by extraction, as demonstrated by Heeres et al.91,92In this CCS (Fig. 10), the organic and aqueous liquids enter the device separately in the annular mixing zone, where they move down by means of gravity and become intensely mixed due to the high shear stress between the fast rotating centrifuge and the static wall of the device, allowing interfacial mass transfer and enantioselective partitioning when a chiral, enantioselective extractant is used. It was demonstrated that because of the intensive mixing and the fast reaction, one experimental stage corresponded with an equilibrium stage. Next, the dispersed liquids are moved to the centrifuge through a hole in the bottom. The liquids are efficiently separated by the large centrifugal forces. The device is thus a centrifugal version of a mixer–settler with the advantage that through use of centrifugal force only small amounts of liquids are needed.
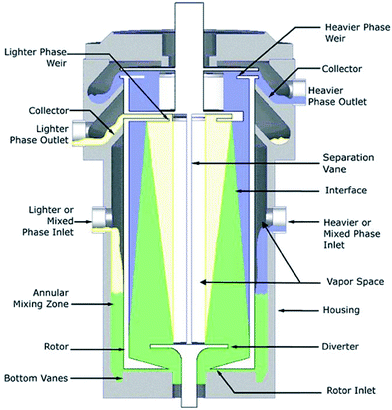 | ||
| Fig. 10 Centrifugal contactor separator, courtesy of CINC Solutions, Doetinchem, The Netherlands. | ||
Extractant classes
Crown ether based reactive extractants
The crown ether based systems are among the most enantioselective reactive extractants known. The discovery, synthesis and application of crown ethers were pioneered by Pedersen.93,94 The crown ethers were provided with chiral moieties and applied as chiral hosts by the groups of Lehn95 and Cram.96 The versatility of crown ether based extractants in ELLE may be limited to ammonium salts, but their operational selectivities up to 31 make it the most important extractants in the field.Cram's BINOL crown ether system
Cram's dilocular host contains a macrocyclic uncharged crown ether carrier sided by two BINOL moieties of which one BINOL has two methyl directing side-groups at the 3,3′-positions (Fig. 11).96 It gives selectivities up to 12 for amine salts,87 and up to 31 for α-amino acid ester salts.97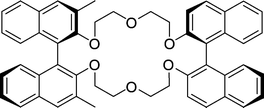 | ||
| Fig. 11 Cram's dilocular crown ether host. | ||
The enantioselective recognition is restricted to primary amine salts, since the ammonium cation complexes in a tripodal configuration to the crown ether. Steric hindrance by side groups connected to the 3,3′-positions of one BINOL backbone direct the bulky groups into a favorable position. When the D-enantiomer of an α-amino acid ester complexes with a (R,R)-host, the R substituents in the α-amino acid ester occupy the most spacious cavity, and the NCH hydrogen rests against the chiral barrier of the host formed by the directing methyl group of the host (Fig. 12).
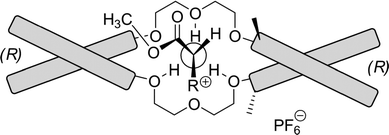 | ||
| Fig. 12 The complexation of an α-amino ester salt with the dilocular crown ether host. Schematic bars represent binaphthyl moieties. | ||
In subsequent years, Cram et al. managed to design a chiral host with only one chiral element (Fig. 13).98 The directing side-groups (A) turned out to be vital for chiral recognition, giving low selectivity for A = H. The highest selectivity was observed for A = Ph, ranging from 3.9 up to 19.5 for the ammonium salts of α-amino acids and α-amino acid methyl esters as substrates. The selectivity towards phenylglycine was increased to 23.4 using a mixture of acetonitrile and chloroform as the organic phase.
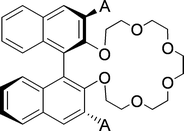 | ||
| Fig. 13 Chiral crown ether based host with only one chiral element, A = directing side group. | ||
Guanidinium-crown ether tethered systems
The guanidinium-crown ether tethered system published by de Mendoza et al.99 is a beautiful example of the use of multiple functionalities to achieve chiral recognition of α-amino acids (Fig. 14, top). The molecule was designed with four possible interaction sites, of which one proved not to be essential. One important issue was that the functional groups interacting with the ammonium and carboxylate, respectively, need to be non-self-complementary. Therefore, a crown ether and a guanidinium function were used to prevent the host from internal collapse. The naphthalene ring was introduced for extra π–π interaction with the side chains of aromatic α-amino acids. Finally, the structure was made homochiral to achieve enantioselective recognition. | ||
| Fig. 14 De Mendoza's guanidinium–crown ether tethered host (top), and proposed complexation of the host complexed with L-tryptophan (bottom). | ||
The structure proposed in 1992 shows a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex between the (S,S) structure of the host (depicted in Fig. 14, bottom) and L-Trp. However, structural and molecular dynamics studies on the same complex showed that in the case of L-tryptophan, the indole moiety is folded over the aza-crown ether (Fig. 14, bottom), thereby excluding some additional polar surface from interactions with the apolar solvent (chloroform).88 In the case of D-tryptophan the indolyl ring dangles into the solvent.
:1 complex between the (S,S) structure of the host (depicted in Fig. 14, bottom) and L-Trp. However, structural and molecular dynamics studies on the same complex showed that in the case of L-tryptophan, the indole moiety is folded over the aza-crown ether (Fig. 14, bottom), thereby excluding some additional polar surface from interactions with the apolar solvent (chloroform).88 In the case of D-tryptophan the indolyl ring dangles into the solvent.
Lipophilic crown ethers
The ELLE of amine picrates using lipophilic crown ethers (Fig. 15, upper panel) has been reported by Costero et al.100 and Nazarenko et al.101 with selectivities up to 3. Picrates (Fig. 15, lower panel) are salts or esters of picric acid and known to be soluble in both the aqueous and organic phase.102 | ||
| Fig. 15 Crown ethers (upper panel) for extraction of picrates (lower panel). | ||
Azophenolic crown ethers
An important member among the family of chiral crown ether hosts is the azophenolic crown ether shown in Fig. 16 (left). Azophenolic crown ethers were synthesized by Naemura et al.103 to study complexation in homogeneous solutions of chiral amines and amino alcohols.104 The crown ether is able to form a tripod-like complex with a neutral amine (Fig. 16 (right). The acidic proton of the phenol forms a hydrogen bond with the amine. The acidity of the phenol is increased by the electron withdrawing nitro groups. This increased acidity is presumed to enhance the complexation of the azophenolic crown ether with a primary amine. | ||
| Fig. 16 Azophenolic crown ether host (left) and the proposed complexation structure of an azophenolic crown ether with a neutral primary amine (right). | ||
This system has been extensively studied by De Haan et al.45 With phenylglycinol, an intrinsic selectivity of up to 12 was achieved, although other amines show lower selectivity (1.5–3.2).
Metal complexes and metalloids as reactive extractants
 | ||
| Fig. 17 Proposed complex of the chiral stationary phase (left) and the diastereomeric complex between copper(II)(L-proline) as the stationary phase and D-phenylalanine (right). | ||
Takeuchi et al.109 were the first to use this system for enantioselective solvent extraction of neutral α-amino acids. N-Alkylated-L-proline derivatives were used as ligands for copper, to keep the host confined into the organic phase. The use of L-4-hydroxyproline gave an increase in operational selectivity (Fig. 18). The complexing constants of the α-amino acid enantiomers were determined by variation of the cupric-ion concentration with respect to the pure enantiomers. The resulting intrinsic selectivities ranged from 1.8 to 4.5 for non-aromatic α-amino acids.
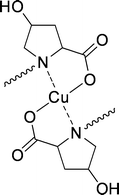 | ||
| Fig. 18 Copper(II)(N-alkyl-4-hydroxyproline)2. | ||
Pickering and Chaudhuri44,110 demonstrated the use of two different extractants Cu(II)(N-dodecyl-(L)-hydroxyproline)2, Cu(II)(C-12Hyp)2 and (S)-bis(phenylnaphtho)-20-crown-6) for the ELLE of phenylalanine. The selectivity using the Cu(II)(C-12Hyp)2 complex was in the range reported earlier by Takeuchi et al.,109 establishing the versatility of the system. De Haan et al. studied the Cu(II)(N-(2-hydroxydodecyl)-L-hydroxyproline) complex as an extractant for a series of primary amines.27 They found dependency between distribution and operational selectivity as well as the pH. Similar results of these correlations were also shown by Pickering111 and Ding.112 The Cu(II)(N-(2-hydroxydodecyl)-L-hydroxyproline) complex has a selectivity of 1.8 for norephedrine while for other amines an αop below 1.2 was obtained. Dimitrova and Bart have demonstrated the application of microemulsions for the enantioselective extraction of phenylalanine and tryptophan with copper hydroxyprolines.113
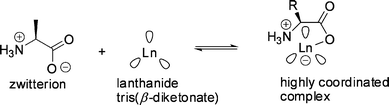 | ||
Fig. 19 The proposed complexation of a zwitterionic amino acid with a lanthanide tris(β-diketonate) ( represents β-diketonate ligand). represents β-diketonate ligand). | ||
Upon variation of the lanthanide cation, opposing trends were observed for distribution and enantioselectivity.117 The correlation between the ion size of the central metal cation (Pr(III) > Eu(III), Eu(II) > Yb(III)) and the extractability was negative, whereas the enantioselectivity increased. This may be due to the smaller cation providing closer contact with the α-amino acid to enhance the αop up to 2.2 in the case of the ytterbium complex 2e (Fig. 20) with a (+)-camphor-derived ligand for the extraction of tryptophan.118
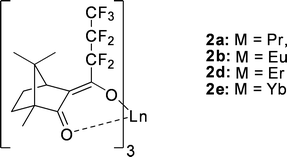 | ||
| Fig. 20 Lanthanide complexes with (+)-camphor derived ligand. | ||
:4 in the organic phase.
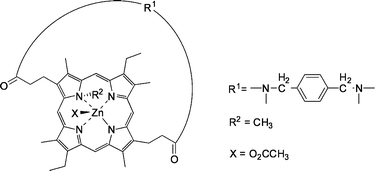 | ||
| Fig. 21 Strapped N-alkylporphyrin zinc host. | ||
The unstrapped zinc-porphyrin host face is blocked by the N-substituent (R2 in Fig. 21). In the strapped cavity, the complexation of N-Cbz protected amino acids is presumed to take place. The proposed structure of the N-Cbz α-amino acid with the zinc porphyrin involves the complexation of the carboxylate to the zinc ion, which is considered to be the primary interaction. Hydrogen bonding of the benzoyl functionality of the substrate with the amide groups on the strap was confirmed by NMR and IR studies.
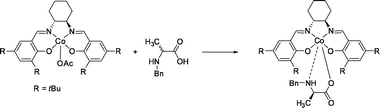 | ||
| Fig. 22 Enantioselective complexation of the chiral cobalt(III)salen host with N-benzyl α-amino acids. | ||
In the back-extraction procedure, the free substrate was obtained via reductive cleavage using 10 eq. sodium dithionite. The formed Co(II) complex can be air-oxidized and re-used without loss of activity. Further studies were performed on the dependence of the protecting group122 and the extraction of β3-amino acids.123 The reductive back-extraction using sodium dithionite is a major drawback with respect to future application.
 | ||
| Fig. 23 Proposed extraction model for amino acid extraction by palladium BINAP complexes.74 © 2009 ACS. | ||
The use of xyl-BINAP gave even better results, with the αop up to 7.0 for phenylalanine analogues, which is the highest reported selectivity for these class of substrates.124 A bidentate binding of the amino acid substrate with the palladium complex was proposed. The extraction of the product of an enzyme catalyzed reaction with a chiral palladium complex even facilitated the isolation of enantiopure β-phenylalanine in high yields.125
Tartrate assisted extractants
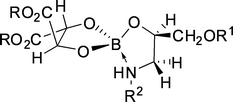 | ||
| Fig. 24 Proposed structure of the complex formed by boric acid, dialkyl tartrate and amino alcohol substrate. | ||
Viegas et al.36 modeled this system based on propanolol/boric acid and di-n-dodecyl tartrates in chloroform. The mathematical approach developed involves the use of a hybrid model, with an experimentally determined component, describing the material balance equations of the reactions involved, and a neural network, which allows accounting for the variable non-selective partitioning of propranolol. The modeled intrinsic selectivity was 2.77, indicating that the operational selectivity achieved experimentally by Abe et al. (2.71)127 was close to the intrinsic optimum of the system.
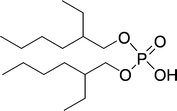 | ||
| Fig. 25 Structure of di(2-ethylhexyl)phosphoric acid (D2EHPA). | ||
Luo et al. developed a synergetic system comparable to the dialkyl tartrate–boric acid system as described above,131 although this system is different, as the tartaric acid component is the free acid, instead of an ester. The D2EHPA is able to extract the tryptophan, albeit without any enantioselectivity. On addition of the tartaric acid, the extraction becomes enantioselective, which makes the system synergetic. The authors propose that the extracting species is a complex formed between D2EHPA and the tartaric acid derivative (O,O′-dibenzoyl-(2R,3R)-tartaric acid, (−)-DBTA). The complex was employed in the extraction of Trp and the operational selectivity achieved a maximum of 5.3.132 Ee's up to 57% in the aqueous phase were measured. However, this includes partial diastereomeric salt formation, in other words classic resolution. Luo et al. also published the synergistic extraction using tartrates with Aliquat 336 (trialkyl methyl ammonium chloride, containing mixtures of C8–C10 alkyl chains) with comparable results.133
Diphosphonium salt bearing binaphthyl hosts
A study was reported by Ohki et al.134 on diphosphonium salts that form a complex with (−)-O-dibenzoyltartrate. The structure of this complex is shown in Fig. 26.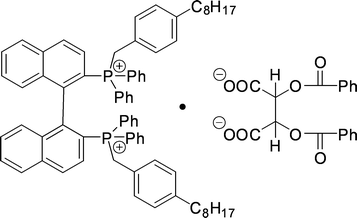 | ||
| Fig. 26 Structure of the diphosphonium salt complex with (−)-O-dibenzoyltartrate. | ||
A reasonable operational selectivity of 1.36 was obtained. Experiments with a monophosphonium salt (one phosphonium salt at the 2- position of the binaphthyl backbone was replaced by a phosphate) showed almost no selectivity, demonstrating the necessity of the bifunctionality of the host.
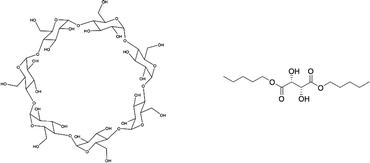 | ||
| Fig. 27 β-Cyclodextrin (a) and L-dipentyl tartrate (b), applied as chiral hosts in biphasic recognition. | ||
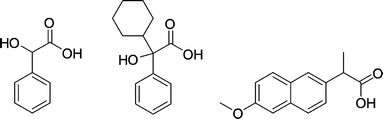 | ||
| Fig. 28 Mandelic acid (a), α-cyclohexyl mandelic acid (b) and naproxen (c). | ||
Other chiral extractants
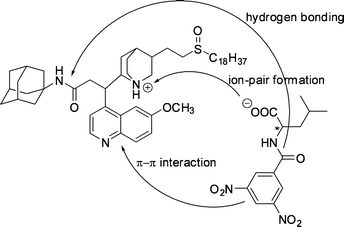 | ||
| Fig. 29 The interactions proposed to be responsible for the chiral discrimination in extraction of DNB-leucine by a modified Cinchona alkaloid extractant. | ||
De Vries et al. studied a number of Cinchona alkaloids using two continuous centrifugal separators (CCS, see Fig. 30).89 The use of continuous equipment was thus demonstrated in a configuration resembling the classical resolving machine of Cram.87 Heeres et al. have applied a continuous single stage extraction approach using the same type of equipment,82 and also showed that the ligand addition mechanism with homogeneous organic phase complexation applies to extractions of DNB substituted α-amino acids using Cinchona alkaloid extractants.75 It was demonstrated that equilibrium modeling is applicable to describe ELLE in a continuous operation mode.82
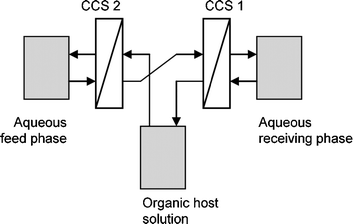 | ||
| Fig. 30 The setup used by De Vries et al.89 incorporating continuous centrifugal extractors (CCS) to facilitate phase transfer. | ||
Heeres et al. have also developed a multistage countercurrent cascade process for the separation of DNB-leucine enantiomers.91,92 This was the first time that ELLE was applied in a continuous countercurrent mode, enabling the complete separation of racemates into single enantiomers. A scheme of the setup that was applied is displayed in Fig. 31.
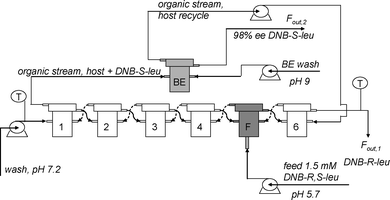 | ||
| Fig. 31 Fractional six stage countercurrent ELLE process setup with host recycle in back-extraction stage (BE) and feed (F) at stage five as used by Schuur et al.92 © 2009 ACS. | ||
A single back-extraction stage was proven sufficient for complete recovery of the extracted DNB-(S)-Leu, and the recycling of the host to the extraction section. The host was recycled up to 50 times without loss of enantiomeric excess in the process. With the thus validated multistage equilibrium modeling approach, it was determined that up to 17.7 kg racemate may be separated completely into its enantiomers using 12 units of the bench scale CCS type of equipment. In this process, both enantiomers are obtained with an ee exceeding 99%.91,92
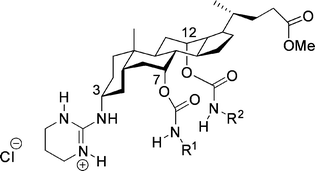 | ||
| Fig. 32 Steroidal guanidinium host. | ||
The phenyl-substituted carbamates (R1 and R2, Fig. 32) gave high enantioselectivities in extraction. These hosts were capable of extracting N-acyl protected α-amino acids into the organic phase.149,150 The extraction is thought to take place via a mechanism involving exchange of the chloride for the carboxylate. The diastereomeric ratio was determined with 1H-NMR spectroscopy. The ratio of the extracted substrate enantiomers was up to 10:1 (L:D). The most sterically hindered substrate (N-Ac-tert-leucine) gave the lowest selectivity. Enantioselective transport was performed with these type of hosts, using a U-tube and a hollow-fiber set-up.151 The host showed multiple turnovers during the transport and the transported substrate present in the receiving phase had an ee of around 60%.
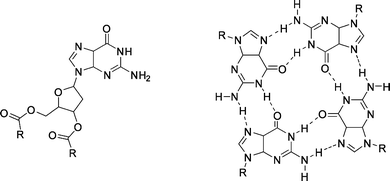 | ||
| Fig. 33 Deoxyguanosine derivative (left) and quartet structure formed by self-assembly (right). R = p-(n-C12H25O)C6H4. | ||
:1 (L-Phe:D-Phe) ratio.
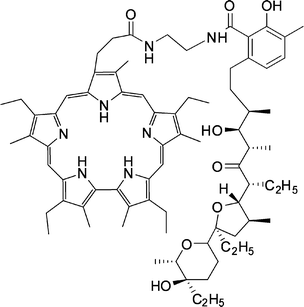 | ||
| Fig. 34 Sapphyrin–lasalocid host. | ||
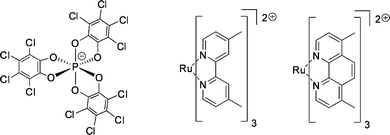 | ||
| Fig. 35 TRISPHAT anion (left) and tris(diimine)ruthenium(II) complexes (right). | ||
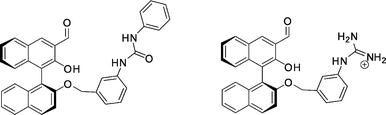 | ||
| Fig. 36 BINOL–aldehyde hosts with a urea (left) and a guanidinium (right) functionality. | ||
The preferred conformations of both diastereomers of the host–guest complexes shown in Fig. 37 provide an explanation for the large difference in selectivity. The steric repulsion between hydrogen and alkyl/aryl around the imine bond of the host and the (S)-amino-alcohol (Fig. 37, left) is larger than that in the case of the (R)-amino-alcohol when the only hydrogen atoms are eclipsed (Fig. 37, right).
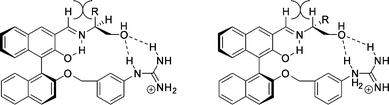 | ||
| Fig. 37 Proposed complexes of the guanidinium BINOL–aldehyde host with (S)-amino-alcohols (left) and (R)-amino-alcohols (right). | ||
 | ||
| Fig. 38 Diaryl-BINOL phosphoric acid extractant (left) and primary amine substrate (right). | ||
Operational selectivities as high as 2.2 were achieved in the extraction of phenylglycinol. Titration studies by NMR and FTIR provided evidence for the formation of a 1:1 host–guest complex. U-tube experiments proved the reversibility of the system.
Summary for hosts
The hosts discussed in this paper are listed in Table 1.| Extractant | Substrate | Scope (αop) (−) or [ee](%) | References |
|---|---|---|---|
| a Intrinsic selectivity. | |||
| Cram dilocular host | α-Amino acid and α-amino acid ester salts | 5 examples (1.5–12)a, 18 examples (1.0–31)a | 87, 96, 97 |
| BINOL-crown ether | α-Amino acids and ester salts | 6 examples (1.1–19.5)a | 98 |
| Guanidium-crown ether | α-Amino acids | 1 example [up to 30%] | 99 |
| Lipophilic crown ethers | Picrate ammonium salts | 2 examples (2.2–3.0) | 100, 101 |
| Azophenolic crown ethers | Primary amines | 6 examples (1–12)a | 45–47 |
| Proline derivative Cu(II) complexes | α-Amino acids | 5 examples (1.0–4.5) | 44, 109–113 |
| Lanthanide(III) tris(β-diketonate) complexes | α-Amino acids and amino alcohols | 6 examples (up to 2.2) | 114, 117 |
| Metalloporphyrins | N-Cbz α-amino acids | 15 examples [50:50–96:4] |
119 |
| Palladium–BINAP complexes | α-Amino acids | 22 examples (1.1–7.0) | 74, 124 |
| Salen–cobalt(III) complexes | N-Acyl α- and β-amino acids | 9 examples [90–96%] | 120, 122, 123 |
| Dialkyl tartrates and boric acid | β-Amino alcohols | 4 examples (2.20–2.54) | 36, 126, 127 |
| D2EHPA and tartaric acid derivative | Amino acids | 4 examples (1.36–5.3) | 131, 132 |
| Diphosphonium salt | Tartaric acid derivative | 1 example (1.36) | 134 |
| Alkyl tartrate with β-cyclodextrin | Carboxylic acids, cyclopyrrolone derivate | 7 examples (1.20–2.8) | 135–140 |
| Carbamoylated quinine Derivatives | N-Protected α-amino acids | Various examples (3–5) | 75, 82, 89, 91, 92, 143 |
| Steroidal guanidinium | N-Acyl amino acids | 8 examples [1:1–10:1] |
149–151 |
| Deoxyguanosine derivatives | DNB-N-protected α-amino acids | 6 examples (1.15–3.03) | 153 |
| Sapphyrin–lasalocid | α-Amino acids | Low selectivity | 157 |
| TRISPHAT | Tris(diimine)ruthenium(II) complexes | 2 examples [12:1–49:1] |
163, 164 |
| 3-Aldehyde substitued BINOL | Amino alcohols | 7 examples (1.0–15)a | 166, 167 |
| Diaryl-BINOL phosphoric acid | Primary amines | 1.0–2.2 | 168 |
Conclusions and outlook
The field of enantioselective extraction is undergoing a revival. For decades, satisfying operational selectivities could only be achieved with chiral hosts containing crown ether-like structures. Despite excellent selectivities in several cases the drawback of these systems is the limited scope restricted to primary amines or their ammonium salts. Furthermore, the complex multi-step synthesis restricts access to sufficient amounts of host. With the lanthanide(III) tris(β-diketonate) complexes and the carbamoylated quinine derivatives introduced about ten years ago and the salen–cobalt(III) complexes reported in 2006, it has been shown that reactive extraction does not need to be limited to crown ether systems to achieve good operational selectivities. Furthermore, the experimental verification of the separation of a racemate in a multistage countercurrent cascade demonstrated the proof of principle of the separation of a racemate using a host with intermediate selectivity.The scalability, recyclability and easy handling of the process makes ELLE in combination with multistage countercurrent cascade technology a fierce rival in the field of new chiral separation technologies. However, there are still major challenges left in the field. Most systems show moderate operational selectivities and a limited substrate scope. For industrial applications, a synthetically acceptably accessible host with a high selectivity towards a wide substrate scope is important to reduce the development time for the separation processes.
The search for new and better host systems is challenging. De Haan et al. made an excellent survey on various chiral separator systems used in chromatography and electrophoresis.27 It turns out to be difficult to transfer the existing systems used in these separation technologies to application in ELLE, mainly because most intrinsic selectivities in these technologies are below 1.2, whereas for ELLE a minimum selectivity of 1.5 is desired to limit the number of extraction stages. Moreover, chromatography usually relies on interactions at the solid–liquid interface. The solid support has a large influence on the mechanism of complexation, which causes the selectivity to be different from the selectivity in a liquid–liquid environment.
New opportunities may come from the field of enantioselective catalysis. Homogeneous asymmetrically catalyzed reactions take place in a fully liquid environment by definition and show unprecedented high enantioselectivities using a wide range of chiral catalysts. These catalysts can be the inspiration for the design of new chiral hosts applied in ELLE. Also, little attention has been paid to systematic variation of functional groups of a specific host. The variation mainly is focussed on the solubility in the organic phase of the host, but seldom to enhance enantiospecific interactions. It is this synthetic approach that deserves more attention.
Another point to stress is the clarification of the mechanism of various systems and the relationship between intrinsic and operational selectivity. Most systems seem to be clear examples of fully reversible systems. Operational selectivity shows dependence on for instance pH, initial concentrations of both substrates and hosts, type of buffer, organic solvents and temperature. It is rarely clear if this dependency between operation selectivity and the various parameters can be adequately explained with either the ligand exchange mechanism or the homogeneous ligand addition mechanism. Both models would imply a strong relation between the intrinsic selectivity and the operational selectivity. This relation is frequently seen in various extraction systems and can open doors towards large scale substrate scanning by examining the intrinsic selectivity with spectroscopic methods and the subsequent optimization of extraction conditions. Little effort has been made towards this goal.
In conclusion, chiral separation by reactive liquid–liquid extraction is an exciting research field, which already has shown proof of principle. Major challenges remain, especially the development of new hosts, which are easily accessible and show a high selectivity for a wide substrate scope, the study of the extraction mechanisms, the examination of the intrinsic selectivities which would enable easy substrate scanning and the application of new systems in a fractional countercurrent multistage cascade to achieve full separation of the substrate enantiomers in a continuous fashion.
Acknowledgements
This work was financially supported by the Netherlands Organization for Scientific Research (NWO-CW) through the Separation Technology program (project nr. 700.54.654) in cooperation with DSM and Schering-Plough.Notes and references
- D. J. Ager, Handbook of Chiral Chemicals, Marcel Dekker, New York, 2005 Search PubMed.
- G. M. Ramos Tombo and D. Bellus, Angew. Chem., Int. Ed. Engl., 1991, 30, 1193–1215 CrossRef.
- R. Noyori, Asymmetric Catalysis in Organic Synthesis, Wiley, New York, 1994 Search PubMed.
- R. A. Sheldon, Chirotechnology, Marcel Dekker, New York, 1993 Search PubMed.
- A. N. Collins, G. N. Sheldrake, and J. Crosby, Chirality in Industry II, New York, Wiley and Sons, 1997, p. 411 Search PubMed.
- P. K. Ajikumar, K. Tyo, S. Carlsen, O. Mucha, T. H. Phon and G. Stephanopoulos, Mol. Pharmaceutics, 2008, 5, 167–190 CrossRef CAS.
- M. J. Waites, Industrial Microbiology, Blackwell Science, Oxford, 2001 Search PubMed.
- M. Reschke and K. Schugerl, Chem. Ing. Tech., 1984, 56, 141 CrossRef CAS.
- D. Cascaval, C. Oniscu and A. I. Galaction, Biochem. Eng. J., 2001, 7, 171–176 CrossRef CAS.
- E. N. Jacobsen, A. Pfalz, and H. Yamamoto, Comprehensive Asymmetric Catalysis, Springer, Berlin, 1999 Search PubMed.
- J. G. de Vries and A. H. M. de Vries, Eur. J. Org. Chem., 2003, 799–811 CrossRef CAS.
- N. M. Maier, P. Franco and W. Lindner, J. Chromatogr., A, 2001, 906, 3–33 CrossRef CAS.
- A. Bruggink, Rational Design in Resolutions, in Chirality in Industry II, ed. A. N. Collins, G. N. Sheldrake and J. Crosby, John Wiley & sons Ltd., Chichester, 1997, pp. 81–98 Search PubMed.
- F. Faigl, E. Fogassy, M. Nogradi, E. Palovics and J. Schindler, Tetrahedron: Asymmetry, 2008, 19, 519–536 CrossRef CAS.
- E. Fogassy, M. Nogradi, D. Kozma, G. Egri, E. Palovics and V. Kiss, Org. Biomol. Chem., 2006, 4, 3011–3030 RSC.
- D. Kozma, CRC Handbook of Optical Resolutions Via Diastereomeric Salt Formation, CRC Press LLC, Boca Raton, 2002 Search PubMed.
- M. Leeman, G. Brasile, E. Gelens, T. Vries, B. Kaptein and R. Kellogg, Angew. Chem., Int. Ed., 2008, 47, 1287–1290 CrossRef CAS.
- S. Houlton, Manuf. Chem., 2001, 72, 16–17 Search PubMed.
- Y. Fujima, M. Ikunaka, T. Inoue and J. Matsumoto, Org. Process Res. Dev., 2006, 10, 905–913 Search PubMed.
- S. Ahuja, Chiral Separations: Application and Technology, ACS, Washington, 1997 Search PubMed.
- G. Subramanian, Chiral Separation Techniques: A Practical Approach, Wiley-VCH, Weinheim, 2001 Search PubMed.
- F. Toda, Enantiomer Separation: Fundamentals and Practical Methods, Kluwer Academic Publishers, Dordrecht, 2004 Search PubMed.
- G. Guebitz and M. G. Schmid, Chiral Separations, Humana Press, Totowa (NJ), 2004 Search PubMed.
- K. W. Busch and M. A. Busch, Chiral Analysis, Elsevier, Amsterdam, 2004 Search PubMed.
- V. A. Davankov, J. Chromatogr., A, 1994, 666, 55–76 CrossRef CAS.
- T. Jira, A. Bunke, M. G. Schmid and G. Gubitz, J. Chromatogr., A, 1997, 761, 269–275 CrossRef.
- M. Steensma, N. J. M. Kuipers, A. B. de Haan and G. Kwant, Chirality, 2006, 18, 314–328 CrossRef CAS.
- G. B. Cox, Preparative Enantioselective Chromatography, Blackwell Publishing, Oxford, 2005 Search PubMed.
- E. Gavioli, N. M. Maier, C. Minguillon and W. Lindner, Anal. Chem., 2004, 76, 5837–5848 CrossRef.
- E. Francotte, T. Leutert, L. La Vecchia, F. Ossola, P. Richert and A. Schmidt, Chirality, 2002, 14, 313–317 CrossRef CAS.
- G. Zenoni, F. Quattrini, M. Mazzotti, C. Fuganti and M. Morbidelli, Flavour Fragrance J., 2002, 17, 195–202 CrossRef CAS.
- C. A. M. Afonso and J. G. Crespo, Angew. Chem., Int. Ed., 2004, 43, 5293–5295 CrossRef CAS.
- J. T. F. Keurentjes, L. J. W. M. Nabuurs and E. A. Vegter, J. Membr. Sci., 1996, 113, 351–360 CrossRef CAS.
- A. Maximini, H. Chmiel, H. Holdik and N. W. Maier, J. Membr. Sci., 2006, 276, 221–231 CrossRef CAS.
- R. Xie, L. Y. Chu and J. G. Deng, Chem. Soc. Rev., 2008, 37, 1243–1263 RSC.
- R. M. C. Viegas, C. A. M. Afonso, J. G. Crespo and I. M. Coelhoso, Sep. Purif. Technol., 2007, 53, 224–234 CrossRef CAS.
- A. B. De Haan and B. Simandi, Extraction Technology for the Separation of Optical Isomers, in Ion Exchange and Solvent Extraction, ed. Y. Marcus, M. M. Sharma and J. A. Marinsky, Marcel Dekker, Inc, New York, 2001, pp. 255–294 Search PubMed.
- L. B. Dashkevic, Tr. Leningr. Khim.-Farm. Inst., 1959, 6, 29 Search PubMed.
- K. Bauer, H. Falk and K. Schlogl, Monatsh. Chem., 1968, 99, 2186–2194 CrossRef CAS.
- N. S. Bowman, G. T. Mccloud and G. K. Schweitz, J. Am. Chem. Soc., 1968, 90, 3848–3852 CrossRef CAS.
- S. J. Romano, K. H. Wells, H. L. Rothbart and W. Rieman, Talanta, 1969, 16, 581–590 CrossRef CAS.
- G. K. Schweitz, I. R. Supernaw and N. S. Bowman, J. Inorg. Nucl. Chem., 1968, 30, 1885–1890 CrossRef.
- J. Koska and C. A. Haynes, Chem. Eng. Sci., 2001, 56, 5853–5864 CrossRef CAS.
- P. J. Pickering and J. B. Chaudhuri, Chem. Eng. Sci., 1997, 52, 377–386 CrossRef CAS.
- M. Steensma, N. J. Kuipers, A. B. de Haan and G. Kwant, J. Chem. Technol. Biotechnol., 2006, 81, 588–597 CrossRef CAS.
- M. Steensma, N. J. M. Kuipers, A. B. de Haan and G. Kwant, Chem. Eng. Sci., 2007, 62, 1395–1407 CrossRef CAS.
- M. Steensma, N. J. M. Kuipers, A. B. de Haan and G. Kwant, Chem. Eng. Proc., 2007, 46, 996–1005 CrossRef CAS.
- E. Eliel, S. Wilen and L. Mander, Miscellaneous Separation Methods, in Stereochemistry of organic compounds, Wiley, New York, NY, 1994, pp. 416–424 Search PubMed.
- J. C. Godfrey and M. J. Slater, Liquid–Liquid Extraction Equipment, John Wiley & Sons, New York, 1994 Search PubMed.
- D. J. Cram and J. M. Cram, Container Molecules and Their Guests, Royal Society of Chemistry, Cambridge, 1994 Search PubMed.
- F. Toda, K. Tanaka, C. W. Leung, A. Meetsma and B. L. Feringa, J. Chem. Soc., Chem. Commun., 1994, 2371–2372 RSC.
- K. Yoshizawa, S. Toyota and F. Toda, Tetrahedron, 2004, 60, 7767–7774 CrossRef CAS.
- F. Toda, K. Yoshizawa, S. Hyoda, S. Toyota, S. Chatziefthimiou and I. M. Mavridis, Org. Biomol. Chem., 2004, 2, 449–451 RSC.
- J. Liao, X. X. Sun, X. Cui, K. B. Yu, J. Zhu and J. G. Deng, Chem.–Eur. J., 2003, 9, 2611–2615 CrossRef CAS.
- G. Kaupp, Angew. Chem., Int. Ed. Engl., 1994, 33, 728–729 CrossRef.
- W. H. Pirkle and T. C. Pochapsky, Chem. Rev., 1989, 89, 347–362 CrossRef CAS.
- T. C. Lo, M. H. I. Baird, and C. Hanson, Handbook of Solvent Extraction, Krieger Publishing Company, Malabar, USA, 1991 Search PubMed.
- J. D. Thornton, Science and Practice of Liquid–Liquid Extraction, Clarendon, Oxford, 1992 Search PubMed.
- R. E. Treybal, Liquid Extraction, 2nd Ed., McGraw-Hill, New York, 1963 Search PubMed.
- N. M. Maier, S. Schefzick, G. M. Lombardo, M. Feliz, K. Rissanen, W. Lindner and K. B. Lipkowitz, J. Am. Chem. Soc., 2002, 124, 8611–8629 CrossRef CAS.
- V. A. Davankov, Chirality, 1997, 9, 99–102 CrossRef CAS.
- V. Sundaresan and R. Abrol, Chirality, 2005, 17, S30–S39 CrossRef CAS.
- E. L. Eliel and S. H. Wilen, Stereochemistry of Organic Compounds, Wiley-Interscience, New York, 1994 Search PubMed.
- J. M. Lehn, Supramolecular Chemistry: Concepts and Perspectives, Weinheim, VCH, 1995 Search PubMed.
- J. H. Hartley, T. D. James and C. J. Ward, J. Chem. Soc., Perkin Trans. 1, 2000, 3155–3184 RSC.
- P. D. Beer and P. A. Gale, Angew. Chem., Int. Ed., 2001, 40, 486–516 CrossRef.
- I. Stibor and P. Zlatuskova, Anion Sensing, 2005, 255, 31–63 Search PubMed.
- C. Seel, A. Galan and J. deMendoza, Supramolecular Chemistry II - Host Design and Molecular Recognition, 1995, 175, 101–132 Search PubMed.
- M. W. Peczuh and A. D. Hamilton, Chem. Rev., 2000, 100, 2479–2493 CrossRef CAS.
- A. P. Davis and R. S. Wareham, Angew. Chem., Int. Ed., 1999, 38, 2978–2996 CrossRef.
- R. J. Fitzmaurice, G. M. Kyne, D. Douheret and J. D. Kilburn, J. Chem. Soc., Perkin Trans. 1, 2002, 841–864 RSC.
- T. H. Webb and C. S. Wilcox, Chem. Soc. Rev., 1993, 22, 383–395 RSC.
- X. X. Zhang, J. S. Bradshaw and R. M. Izatt, Chem. Rev., 1997, 97, 3313–3361 CrossRef CAS.
- B. J. V. Verkuijl, A. J. Minnaard, J. G. de Vries and B. L. Feringa, J. Org. Chem., 2009, 74, 6526–6533 CrossRef CAS.
- B. Schuur, J. G. M. Winkelman and H. J. Heeres, Ind. Eng. Chem. Res., 2008, 47, 10027–10033 CrossRef CAS.
- H. J. Bart, Reactive Extraction, Springer Verlag, Berlin, 2001 Search PubMed.
- M. Cox, Liquid–Liquid Extraction in Hydrometallurgy, in Science and Practice of Liquid–Liquid Extraction, ed. J. D. Thornton, Clarendon, Oxford, 1992, pp. 1–101 Search PubMed.
- C. J. King, Chemtech, 1992, 22, 285–291 CAS.
- K. L. Wasewar, A. A. Yawalkar, J. A. Moulijn and V. G. Pangarkar, Ind. Eng. Chem. Res., 2004, 43, 5969–5982 CrossRef CAS.
- G. F. Cegla, W. W. Meinke and K. F. Mattil, J. Food Sci., 1977, 42, 816–820 CrossRef CAS.
- M. Tanaka, K. Koyama and J. Shibata, Ind. Eng. Chem. Res., 1997, 36, 4353–4357 CrossRef CAS.
- B. Schuur, J. Floure, A. J. Hallett, J. G. M. Winkelman, J. G. de Vries and H. J. Heeres, Org. Process Res. Dev., 2008, 12, 950–955 Search PubMed.
- G. S. Laddha and T. E. Degaleesan, Transport Phenomena in Liquid Extraction, Tata McGraw-Hill, New Delhi, 1978 Search PubMed.
- M. Hafez, Centrifugal Extractors, in Handbook of Solvent Extraction, ed. T. C. Lo, M. H. C. Baird, and C. Hanson, Krieger Publishing Company, Malabar, 1991, pp. 459–474 Search PubMed.
- L. S. Ettre, J. Chromatogr. Sci., 1999, 37, 2A–8A CAS.
- G. J. Hanna and R. D. Noble, Chem. Rev., 1985, 85, 583–598 CrossRef CAS.
- M. Newcomb, J. L. Toner, R. C. Helgeson and D. J. Cram, J. Am. Chem. Soc., 1979, 101, 4941–4947 CrossRef CAS.
- P. Breccia, M. Van Gool, R. Perez-Fernandez, S. Martin-Santamaria, F. Gago, P. Prados and J. C. de Mendoza, J. Am. Chem. Soc., 2003, 125, 8270–8284 CrossRef CAS.
- A. J. Hallett, G. J. Kwant and J. G. de Vries, Chem.–Eur. J., 2009, 15, 2111–2120 CrossRef CAS.
- H. Nishizawa, K. Tahara, A. Hayashida and Y. Abe, Anal. Sci., 1993, 9, 611–615 CAS.
- B. Schuur, J. G. M. Winkelman, J. G. de Vries and H. J. Heeres, Chem. Eng. Sci., 2010, 65, 4682–4690 CrossRef CAS.
- B. Schuur, A. J. Hallett, J. G. M. Winkelman, J. G. de Vries and H. J. Heeres, Org. Process Res. Dev., 2009, 13, 911–914 Search PubMed.
- C. J. Pedersen, J. Am. Chem. Soc., 1967, 89, 7017–7019 CrossRef CAS.
- C. J. Pedersen, J. Am. Chem. Soc., 1967, 89, 2495–2497 CrossRef CAS.
- J. M. Lehn and A. Moradpour, Helv. Chim. Acta, 1978, 61, 2407–2418 CrossRef CAS.
- E. P. Kyba, J. M. Timko, L. J. Kaplan, F. Dejong, G. W. Gokel and D. J. Cram, J. Am. Chem. Soc., 1978, 100, 4555–4568 CrossRef CAS.
- S. C. Peacock, L. A. Domeier, F. C. A. Gaeta, R. C. Helgeson, J. M. Timko and D. J. Cram, J. Am. Chem. Soc., 1978, 100, 8190–8202 CrossRef CAS.
- D. S. Lingenfelter, R. C. Helgeson and D. J. Cram, J. Org. Chem., 1981, 46, 393–406 CrossRef CAS.
- A. Galan, D. Andreu, A. Echavarren, P. Prados and J. De Mendoza, J. Am. Chem. Soc., 1992, 114, 1511–1512 CrossRef CAS.
- M. Colera, A. M. Costero, P. Gavina and S. Gil, Tetrahedron: Asymmetry, 2005, 16, 2673–2679 CrossRef CAS.
- A. Y. Nazarenko, P. Huszthy, J. S. Bradshaw, J. D. Lamb and R. M. Izatt, J. Inclusion Phenom. Mol. Recognit. Chem., 1994, 20, 13–22 CrossRef CAS.
- S. S. Moore, T. L. Tarnowski, M. Newcomb and D. J. Cram, J. Am. Chem. Soc., 1977, 99, 6398–6405 CrossRef CAS.
- K. Naemura, K. Nishioka, K. Ogasahara, Y. Nishikawa, K. Hirose and Y. Tobe, Tetrahedron: Asymmetry, 1998, 9, 563–574 CrossRef CAS.
- K. Naemura, K. Matsunaga, J. Fuji, K. Ogasahara, Y. Nishikawa, K. Hirose and Y. Tobe, Anal. Sci., 1998, 14, 175–182 CrossRef CAS.
- P. E. Hare and E. Gil-Av, Science, 1979, 204, 1226–1228 CAS.
- G. Gübitz, W. Jellenz and W. Santi, J. Liq. Chromatogr. Relat. Technol., 1981, 4, 701–712.
- V. A. Davankov and S. V. Rogozhin, J. Chromatogr., A, 1971, 60, 280–283 CrossRef CAS.
- G. Gubitz, W. Jellenz and W. Santi, J. Chromatogr., A, 1981, 203, 377–384 CrossRef.
- T. Takeuchi, R. Horikawa and T. Tanimura, Anal. Chem., 1984, 56, 1152–1155 CrossRef CAS.
- P. J. Pickering and J. B. Chaudhuri, Chirality, 1997, 9, 261–267 CrossRef CAS.
- P. J. Pickering and J. B. Chaudhuri, J. Membr. Sci., 1997, 127, 115–130 CrossRef.
- H. B. Ding, P. W. Carr and E. L. Cussler, AIChE J., 1992, 38, 1493–1498 CrossRef CAS.
- P. Dimitrova and H. J. Bart, Chem. Eng. Technol., 2009, 32, 1527–1534 CrossRef CAS.
- H. Tsukube, J. Uenishi, T. Kanatani, H. Itoh and O. Yonemitsu, Chem. Commun., 1996, 477–478 RSC.
- G. R. Sullivan, D. Ciavarel and H. S. Mosher, J. Org. Chem., 1974, 39, 2411–2412 CrossRef CAS.
- C. Kremer, J. Torres, S. Dominguez and A. Mederos, Coord. Chem. Rev., 2005, 249, 567–590 CrossRef CAS.
- H. Tsukube, S. Shinoda, J. Uenishi, T. Kanatani, H. Itoh, M. Shiode, T. Iwachido and O. Yonemitsu, Inorg. Chem., 1998, 37, 1585–1591 CrossRef CAS.
- R. Rinaldi, Prog. Nucl. Magn. Reson. Spectrosc., 1982, 15, 291–352 CrossRef CAS.
- K. Konishi, K. Yahara, H. Toshishige, T. Aida and S. Inoue, J. Am. Chem. Soc., 1994, 116, 1337–1344 CrossRef CAS.
- T. B. Reeve, J. P. Cros, C. Gennari, U. Piarulli and J. G. de Vries, Angew. Chem., Int. Ed., 2006, 45, 2449–2453 CrossRef CAS.
- J. R. Larrow and E. N. Jacobsen, Top. Organomet. Chem., 2004, 7, 123–152.
- P. Dzygiel, T. B. Reeve, U. Piarulli, M. Krupicka, I. Tvaroska and C. Gennari, Eur. J. Org. Chem., 2008, 1253–1264 CrossRef CAS.
- P. Dzygiel, C. Monti, U. Piarulli and C. Gennari, Org. Biomol. Chem., 2007, 5, 3464–3471 RSC.
- B. J. V. Verkuijl, B. Schuur, A. J. Minnaard, J. G. de Vries and B. L. Feringa, Org. Biomol. Chem., 2010, 8, 3045–3054 RSC.
- B. J. V. Verkuijl, W. Szymanski, B. Wu, A. J. Minnaard, D. B. Janssen, J. G. de Vries and B. L. Feringa, Chem. Commun., 2010, 46, 901–903 RSC.
- Y. Abe, T. Shoji, M. Kobayashi, W. Qing, N. Asai and H. Nishizawa, Chem. Pharm. Bull., 1995, 43, 262–265 CAS.
- Y. Abe, T. Shoji, S. Fukui, M. Sasamoto and H. Nishizawa, Chem. Pharm. Bull., 1996, 44, 1521–1524 CAS.
- I. Van de Voorde, L. Pinoy, E. Courtijn and F. Verpoort, Solvent Extr. Ion Exch., 2006, 24, 893–914 CrossRef CAS.
- M. Teramoto, T. Yamashiro, A. Inoue, A. Yamamoto, H. Matsuyama and Y. Miyake, J. Membr. Sci., 1991, 58, 11–32 CrossRef CAS.
- Y. S. Liu, Y. Y. Dai and J. D. Wang, Sep. Sci. Technol., 2000, 35, 1439–1454 CrossRef CAS.
- B. Tan, G. S. Luo, X. Qi and J. D. Wang, Sep. Purif. Technol., 2006, 49, 186–191 CrossRef CAS.
- B. Tan, G. S. Luo and H. D. Wang, Tetrahedron: Asymmetry, 2006, 17, 883–891 CrossRef CAS.
- B. Tan, G. S. Luo and J. D. Wang, Sep. Purif. Technol., 2007, 53, 330–336 CrossRef CAS.
- A. Ohki, R. Miyashita, K. Naka and S. Maeda, Bull. Chem. Soc. Jpn., 1991, 64, 2714–2719 CrossRef CAS.
- F. P. Jiao, X. Q. Chen, W. G. Hu, F. R. Ning and K. L. Huang, Chem. Pap., 2007, 61, 326–328 Search PubMed.
- K. Tang, Y. Chen, K. Huang and J. Liu, Tetrahedron: Asymmetry, 2007, 18, 2399–2408 CrossRef CAS.
- K. W. Tang, J. M. Yi, Y. B. Liu, X. Y. Jiang and Y. Pan, Chem. Eng. Sci., 2009, 64, 4081–4088 CrossRef CAS.
- K. W. Tang, L. T. Song, Y. B. Liu, Y. Pan and X. Y. Jiang, Chem. Eng. J., 2010, 158, 411–417 CrossRef CAS.
- K. W. Tang, L. T. Song, Y. Pan, X. Y. Jiang and J. B. Miao, Chin. J. Chem., 2010, 28, 119–124 CrossRef CAS.
- K. W. Tang, Y. Y. Chen and J. J. Liu, Sep. Purif. Technol., 2008, 62, 681–686 CrossRef CAS.
- M. Lammerhofer and W. Lindner, J. Chromatogr., A, 1996, 741, 33–48 CrossRef.
- R. D. Mhaskar and M. M. Sharma, Chem. Eng. Sci., 1975, 30, 811–818 CrossRef CAS.
- K. H. Kellner, A. Blasch, H. Chmiel, M. Lammerhofer and W. Lindner, Chirality, 1997, 9, 268–273 CrossRef CAS.
- C. Czerwenka, M. M. Zhang, H. Kahlig, N. M. Maier, K. B. Lipkowitz and W. Lindner, J. Org. Chem., 2003, 68, 8315–8327 CrossRef CAS.
- H. Caner, P. U. Biedermann and I. Agranat, Chirality, 2003, 15, 637–645 CrossRef CAS.
- G. D. H. Dijkstra, R. M. Kellogg and H. Wynberg, J. Org. Chem., 1990, 55, 6121–6131 CrossRef CAS.
- G. D. H. Dijkstra, R. M. Kellogg, H. Wynberg, J. S. Svendsen, I. Marko and K. B. Sharpless, J. Am. Chem. Soc., 1989, 111, 8069–8076 CrossRef CAS.
- G. Uccello-Barretta, F. Balzano, C. Quintavalli and P. Salvadori, J. Org. Chem., 2000, 65, 3596–3602 CrossRef CAS.
- L. J. Lawless, A. G. Blackburn, A. J. Ayling, M. N. Perez-Payan and A. P. Davis, J. Chem. Soc., Perkin Trans. 1, 2001, 1329–1341 RSC.
- A. P. Davis and L. J. Lawless, Chem. Commun., 1999, 9–10 RSC.
- B. Baragana, A. G. Blackburn, P. Breccia, A. P. Davis, J. De Mendoza, J. M. Padron-Carrillo, P. Prados, J. Riedner and J. G. de Vries, Chem.–Eur. J., 2002, 8, 2931–2936 CrossRef CAS.
- J. T. Davis, Angew. Chem., Int. Ed., 2004, 43, 668–698 CrossRef CAS.
- V. Andrisano, G. Gottarelli, S. Masiero, E. H. Heijne, S. Pieraccini and G. P. Spada, Angew. Chem., Int. Ed., 1999, 38, 2386–2388 CrossRef CAS.
- I. Spasojevic and A. L. Crumbliss, J. Chem. Soc., Dalton Trans., 1998, 4021–4027 RSC.
- J. L. Sessler and A. Andrievsky, Chem. Commun., 1996, 1119–1120 RSC.
- A. Andrievsky and J. L. Sessler, Abstr. Pap. ACS, 1996, 211, 76–77 Search PubMed.
- J. L. Sessler and A. Andrievsky, Chem.–Eur. J., 1998, 4, 159–167 CrossRef CAS.
- A. Vonzelewsky, P. Belser, P. Hayoz, R. Dux, X. Hua, A. Suckling and H. Stoecklievans, Coord. Chem. Rev., 1994, 132, 75–85 CrossRef.
- J. K. Barton, Science, 1986, 233, 727–734 CAS.
- V. Balzani, A. Credi, and M. Venturi, Molecular Devices and Machines: Concepts and Perspectives for the Nanoworld, Wiley-VCH, Weinheim, 2008 Search PubMed.
- R. D. Gillard and R. E. E. Hill, J. Chem. Soc., Dalton Trans., 1974, 1217–1236 RSC.
- J. Lacour, S. Torche-Haldimann, J. J. Jodry, C. Ginglinger and F. Favarger, Chem. Commun., 1998, 1733–1734 RSC.
- J. Lacour, C. Goujon-Ginglinger, S. Torche-Haldimann and J. J. Jodry, Angew. Chem. Int. Ed., 2000, 39, 3695–3697 CrossRef CAS.
- J. J. Jodry and J. Lacour, Chem.–Eur. J., 2000, 6, 4297–4304 CrossRef CAS.
- H. Ratni, J. J. Jodry, J. Lacour and E. P. Kundig, Organometallics, 2000, 19, 3997–3999 CrossRef CAS.
- L. Tang, S. Choi, R. Nandhakumar, H. Park, H. Chung, J. Chin and K. M. Kim, J. Org. Chem., 2008, 73, 5996–5999 CrossRef CAS.
- K. M. Kim, H. Park, H. J. Kim, J. Chin and W. Nam, Org. Lett., 2005, 7, 3525–3527 CrossRef CAS.
- B. J. V. Verkuijl, J. G. de Vries and B. L. Feringa, Chirality, 2010 DOI:10.1002/chir.20834.
Footnote |
| † These two authors contributed equally to this manuscript. |
| This journal is © The Royal Society of Chemistry 2011 |