Tubulin-binding dibenz[c,e]oxepines as colchinol analogues for targeting tumour vasculature†
David J.
Edwards
ab,
John A.
Hadfield
bc,
Timothy W.
Wallace
*a and
Sylvie
Ducki‡
c
aSchool of Chemistry, The University of Manchester, Oxford Road, Manchester, UK M13 9PL. E-mail: tim.wallace@manchester.ac.uk; Fax: +44 (0) 161 275 4939
bDrug Development Group, Paterson Institute for Cancer Research, Christie Hospital NHS Trust, Manchester, UK M20 4BX
cKidscan Laboratories, Centre for Molecular Drug Design, School of Environment and Life Sciences, University of Salford, Salford, UK M5 4WT
First published on 17th November 2010
Abstract
Various methoxy- and hydroxy-substituted dibenz[c,e]oxepines were prepared via the copper(I)-induced coupling of ether-tethered arylstannanes or the dehydrative cyclisation of 1,1′-biphenyl-2,2′-dimethanols, assembled using the Ullmann cross-coupling of ortho-bromoaryl carbonyl compounds. The dibenzoxepines were screened for their ability to inhibit tubulin polymerisation and the in vitrogrowth of K562 human chronic myelogenous leukemia cells. The most active was 5,7-dihydro-3,9,10,11-tetramethoxydibenz[c,e]oxepin-4-ol, whose tubulin inhibitory and cytotoxicity (IC50) values were 1 μM and 40 nM, respectively.
Introduction
Tumour growth requires the support of an associated blood supply, making tumour vasculature a potential target for anticancer therapy. This principle has inspired decades of research into the pathways of angiogenesis (the formation of new blood vessels), leading to the identification of a family of vascular endothelial growth factors (VEGFs) that stimulate this process.1 The subsequent search for VEGF inhibitors yielded inter alia the monoclonal antibody bevacizumab (Avastin®), which in 2004 became the first angiogenesis inhibitor to be approved in the USA for clinical use in the treatment of cancer.2 While the underlying principles and clinical practices of anti-angiogenic therapy continue to evolve,3 an alternative antivascular strategy is emerging. Tumour vasculature is structurally and functionally abnormal, being disorganised and prone to inefficiencies caused by branching, uneven diameter, shunts, etc., rendering it susceptible to the effects of vascular disrupting agents (VDAs).4 Various small molecules have come under scrutiny in this context,5 the most prominent being combretastatin A-4 (CA-4) 1, which was isolated by Pettit and coworkers from the African bush willow Combretum caffrum6 and shown to induce shutdown of tumour vasculature within minutes, while leaving normal vasculature intact.7 Clinical trials of the water-soluble prodrug CA-4P 2 (Zybrestat) were initiated in 1998, and the congener combretastatin A-1 3 is also under development as an antitumour agent in the form of the phosphate prodrug 4 (OXi4503).8 Other structures undergoing evaluation as VDAs include the combretastatin analogue 5 (AVE8062), which abruptly and irreversibly stops tumour blood flow in a range of cancer cell lines,9 the analogue 6 of the marine natural product dolastatin,10 the xanthenylacetic acid 7,11 the substituted heterocycles 8,129,1310,1411,151216 and 13,17 and the phosphate prodrug 14 of N-acetylcolchinol.18 With the notable exception of 7, which appears to act both through the host immune system, e.g. by stimulating the production of tumour necrosis factor (TNF),19 and directly by inducing vascular endothelial cell apoptosis,20 the biological target of these potential VDAs is tubulin.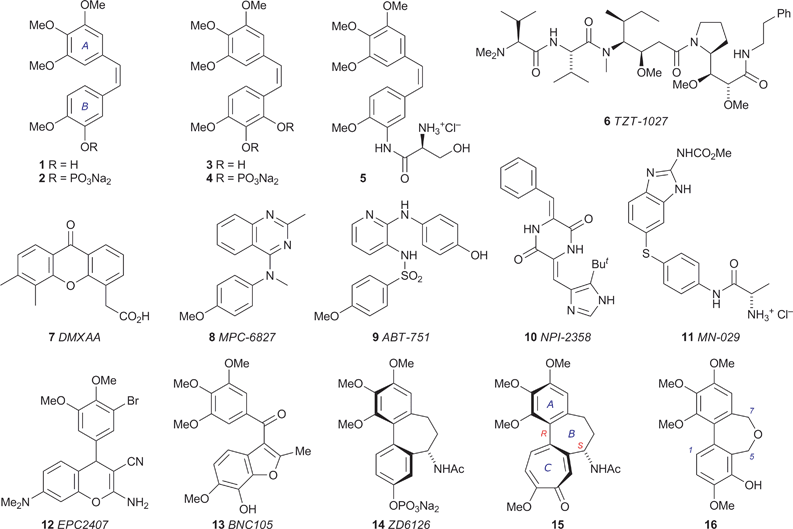
Tubulin, in the form of various isotypes, is an abundant component of the cytoplasm of animal cells. Two GTP-binding monomers, α- and β-tubulin, form a heterodimer whose polymerisation generates microtubules, which occupy a pivotal role in a range of intracellular processes involving structure, shape, signalling and transport, including chromosome segregation and positioning during mitosis. The dynamics of microtubule assembly and disassembly are responsive to the identity of the nucleotide bound to the β-tubulin of its terminal heterodimer unit, the microtubule being viable when this is GTP but prone to rapid shrinkage when it is exchanged for GDP.21 The dynamics of tubulin–microtubule interconversion are delicately poised and can be disrupted by coordinating species: some of the tubulin binders in clinical use (taxanes, epothilones) stabilise the GDP-bound tubulin in the microtubule, thereby inhibiting its disassembly,22 while others (vincristine, etoposide) bind to the αβ-tubulin heterodimer and inhibit microtubule assembly.23 The origin and subtleties of the latter type of inhibition have become more apparent since the acquisition of high-resolution crystallographic structures of tubulin-drug complexes by Knossow and coworkers.24,25Tubulin binding agents can exert a direct cytotoxic effect by perturbing microtubule dynamics, thereby undermining mitosis (which requires the rapid turnover of microtubules at all stages) and bringing about cell death. Microtubules also play a prominent role in maintaining the physical structure of the endothelial cells lining new tumour vasculature, which lack the well-defined actin cytoskeleton and other strengthening features of mature endothelial cells, and it has been established that some tubulin-binding VDAs induce morphological changes in the endothelial cells of immature tumour vasculature, e.g. rounding and detachment, leading to reduced blood flow and tumour necrosis.4,18b,26 Significantly, vascular shutdown by CA-4 1 is achieved using substantially less than the maximum tolerated dose (MTD), illustrating one of the potential advantages of targeting tumour vasculature rather than the mitotic apparatus, which generally involves less favourable therapeutic margins.
In common with CA-4 1,27 the structures 8–14 bind to tubulin at or close to the same site as colchicine 15, the best known tubulin-binding agent and ‘spindle toxin’.28 The toxicity of colchicine 15 precludes its clinical use as an antimitotic agent, but it is clear that the colchicine binding site of tubulin can accommodate a diverse range of structures and so offers considerable scope for the design of binding agents with minimised pharmacokinetic half-lives and cardiostimulatory effects,29 the latter being a potentially generic problem with tubulin-targeting VDAs.18d,30 Our interest in the axial chirality of colchicine 15 led us to analyse the variation of the interaryl dihedral angle in a series of heterocyclic variants of the bridged biaryl core,31 and we observed that the degree of helicity in dibenz[c,e]oxepines closely matches that found in colchicine 15. In pursuing this line we synthesised a series of new dibenz[c,e]oxepines and assessed their ability to inhibit tubulin polymerisation, which led to the identification of 5,7-dihydro-3,9,10,11-tetramethoxydibenz[c,e]oxepin-4-ol 16 as a new lead in the search for effective VDAs.32 Our results, herein described in detail, suggest that a dibenz[c,e]oxepine unit may be capable of providing the helical core of a new series of tubulin-binding small molecules.33
Synthesis of materials
Initially we assessed the methods available for biaryl synthesis, seeking to identify those suited for use in approaches to polysubstituted dibenz[c,e]oxepines. Although not ideal from an environmental viewpoint, the copper(I)-mediated intramolecular coupling of tethered arylstannane units had been shown by Piers et al.34 to be an effective source of the target heterocyclic system, and we therefore chose to apply this method to our first targets. To prepare the required series of doubly-stannylated dibenzyl ethers, we first acquired stocks of the stannylated trimethoxybenzyl bromides 2034 and 21 (Scheme 1) and then used analogous lithiation–transmetallation sequences to convert benzyl alcohol 22 and veratryl alcohol 24 into the respective tributyltin derivatives 23 and 25 (Scheme 2).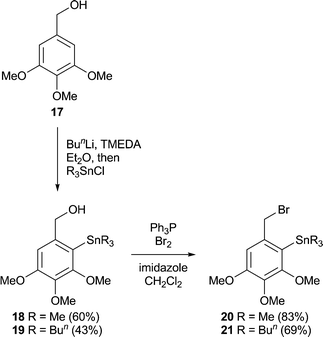 | ||
| Scheme 1 | ||
 | ||
| Scheme 2 Reagents: i, Montmorillonite K10, CH2Cl2, RT, 3 h. | ||
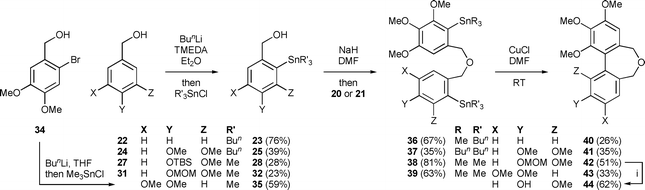
To extend the series, the protected vanillin 26 was reduced to 27 for use in the lithiation–stannylation sequence. However, in this case the yield of the desired alcohol 28 was poor (28%) and the by-products included the doubly-stannylated species 29 (11%). The formation of the latter, which was characterised inter alia by the 1H NMR signals from its diastereotopic SnCH2Si group [δH (400 MHz, CDCl3) 0.00 (1 H, d, J 19.4 Hz), −0.07 (1 H, d, J 19.4 Hz)], serves as a reminder35 of the increased susceptibility of a t-butyldimethylsilyloxy group towards deprotonation by an alkyllithium when a second lithium coordination site, in this case the methoxy group, is located nearby. Repeating the sequence with the MOM-protected vanillin 31 also proved troublesome, with poor conversion to the desired alcohol 32 (23%) and the formation of a comparable amount of the regioisomeric product 33 (19%). Using an alternative stannylation procedure based on lithium–bromine exchange, the bromo alcohol 34 provided a fair yield of 35 in straightforward fashion.
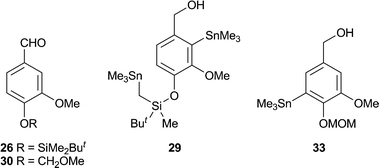
The alcohols 23, 25, 32 and 35 were converted into the respective dibenzyl ethers 36–39 using the appropriate aryl bromide 20 or 21. These etherifications were more efficient when using the trimethylstannyl bromide 20, presumably for steric reasons. Subjecting the ethers 36–39 to the conditions of the cyclisation process34 gave the desired dibenz[c,e]oxepines 40–43 in yields that were, at best, modest, but allowed the isolation of sufficient material for testing purposes and they therefore remain to be optimised. A sample of the MOM-protected variant 42 was transformed into the corresponding phenol 44 using a mild procedure.36
Other dibenz[c,e]oxepines were synthesised using the Ullmann reaction37 to effect the coupling of appropriate substituted haloarenes. Although conventional Ullmann cross-couplings tend to give mixtures containing homocoupled products, they offer rapid access to certain types of target structure and can be optimised by modifying the reaction conditions and stoichiometry so as to inhibit homocoupling,38,39 or by the quantitative formation of the intermediate arylcopper species from one of the reaction partners prior to its exposure to the other.40 In our first Ullmann approach (Scheme 3), we found that combining mole equivalents of the bromoaldehyde 45 and the bromoester 46 gave an acceptable yield of 47, although chromatography was required to isolate it from the homocoupled product 48 (12%). Reduction of the carbonyl functions of 47 provided, successively, the ester-alcohol 49 and the diol 50. The latter was transformed into the oxepine 51 upon treatment with aqueous acid.41
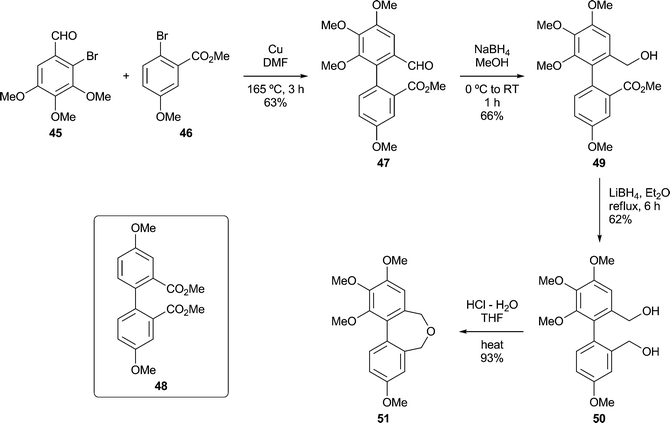 | ||
| Scheme 3 | ||
In a second Ullmann cross-coupling (Scheme 4), reacting 45 with 6-bromopiperonal 52 gave the dialdehyde 53 (22%), the low isolated yield in this case being partly due to the use of crystallisations to remove the contaminating dialdehyde 54. The dialdehyde 5338 and related structures are useful lignan precursors that can also be prepared via Suzuki–Miyaura coupling protocols.42Reduction of 53 to the diol 55, followed by ring closure as before, gave the dibenzoxepine 56 as a crystalline solid.
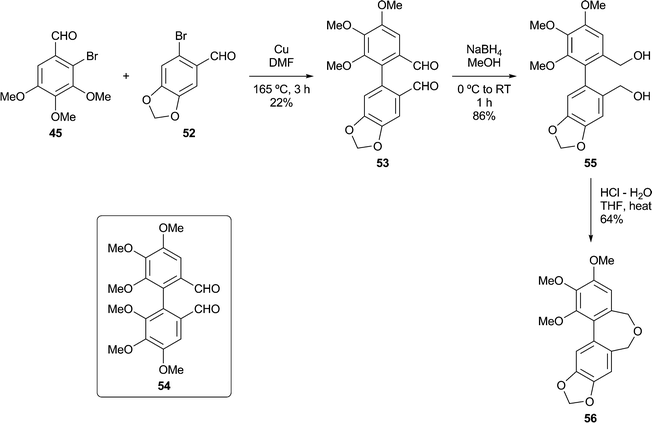 | ||
| Scheme 4 | ||
Based on our analysis of the structure–activity relationships in known colchicine, allocolchicine and combretastatin derivatives, we surmised that the substitution pattern present in the dibenzoxepine 16 was particularly worthy of study. The proposed route to this compound required a hydroxyl protecting group that would withstand the conditions of the Ullmann coupling reaction, and encouraged by a literature precedent43 we elected to use a methanesulfonyl (mesyl) group in this capacity. The route to the target therefore began with the mesylation of the commercially available bromoaldehyde 57 under standard conditions (Scheme 5). This provided a good yield of the mesylate 58, together with a by-product identified as the sultone 59 (22%) derived from 58via the base-induced condensation of the mesylate and aldehyde groups. We surmise that the formation of 59, whose heterocyclic core is rare, is assisted by the buttressing effect of the flanking OMe and Br functions. Ullmann coupling of the mesylate 58 with the bromoaldehyde 45 (3 equiv.), followed by chromatography, gave an acceptable yield of the dialdehyde 60, and subsequent reduction followed by acid-induced cyclisation gave the desired mesylate 62. Alkaline hydrolysis of 62 provided the target dibenzoxepine 16 as a white crystalline solid, m.p. 145–147 °C (MeOH). The 1H NMR spectrum of 16 (Fig. 1) illustrates the line broadening for the diastereotopic methylene signals that is typical of compounds of this type, in which the biaryl unit is non-planar and fluctional, undergoing axis inversion slowly on the NMR time scale.44
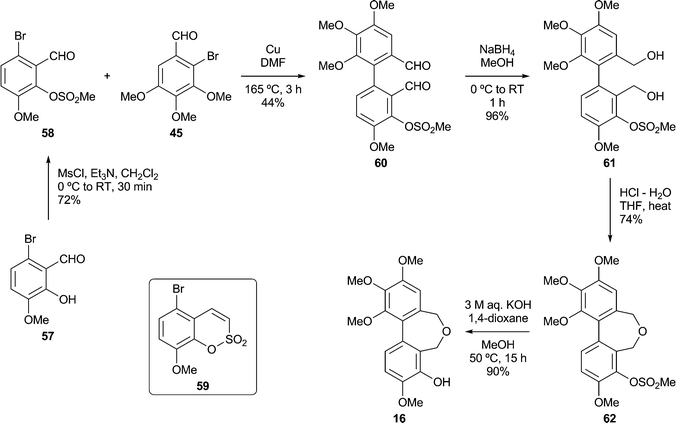 | ||
| Scheme 5 | ||
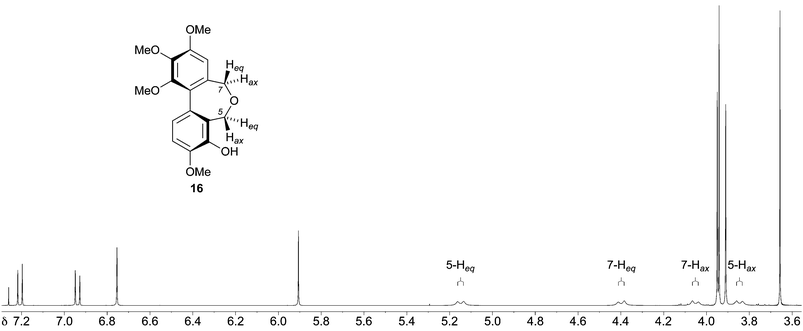 | ||
| Fig. 1 1H NMR spectrum (400 MHz, CDCl3, 25 °C) of the dibenzoxepine 16. | ||
Compound evaluation
New materials were screened for their ability to inhibit microtubule assembly45 and for growth inhibitory activity (IC50) against the K562 human chronic myelogenous leukemia cell-line.46 Both of these assays are routinely used for the evaluation of test compounds and provide a useful comparison with benchmarks such as CA-4 1 and colchicine 15. The results (Table 1) show that, as might be expected, biological activity is dependent on the aromatic substitution pattern, and reveal useful levels of activity in the dibenzoxepines 16, 51 and 56 (entries 3, 9 and 10). In particular, the phenol 16 inhibits tubulin assembly to the same extent as CA-4 1, whose substituent array is the same, and returned an IC50 value of 40 nM against the K562 cell-line. None of the other compounds inhibited tubulin polymerisation to a significant degree, although 43 and 44 (entries 7 and 8) displayed some in vitro cytotoxicity.| Entry | Compound | Tubulin assembly | K562 MTT assay |
|---|---|---|---|
| IC50/μMa | IC50/μMb | ||
| a Concentration required for 50% inhibition of tubulin assembly. b Concentration that inhibits the growth of the K562 cell line by 50% after incubation for 5 days. Each drug concentration was tested in triplicate, and the standard error of each value is <10%. c All entries in this column are normalised to this value for 1, which varied over the range 0.0010–0.0022 between batches. d Value taken from ref. 47. | |||
| 1 | 1 | 1.3 | 0.0010c |
| 2 | 15 | 3.9 | 0.0022d |
| 3 | 16 | 1.0 | 0.04 |
| 4 | 40 | >10 | >20 |
| 5 | 41 | >10 | >20 |
| 6 | 42 | >10 | >20 |
| 7 | 43 | >10 | 3 |
| 8 | 44 | >10 | 19 |
| 9 | 51 | 7.4 | 0.13 |
| 10 | 56 | >10 | 0.10 |
| 11 | 62 | >10 | >20 |
| 12 | 63 | >10 | >400 |
| 13 | 66 | >10 | 73 |
| 14 | 67 | >10 | 360 |
| 15 | 68 | >10 | >400 |
| 16 | 69 | >10 | >400 |
| 17 | 70 | >10 | >400 |
Discussion
The difference in potency of 40 and 51 as tubulin polymerisation inhibitors mirrors that of their respective carbocyclic analogues 71 (IC50 > 50 μM)48 and 72 (IC50 1.5 μM),49 and indicates that at least one oxygen substituent in the C-ring is a prerequisite for tubulin-binding activity. However, the hexamethoxy series comprising 63 and 66–70, which had been prepared from the diol 64 for our crystallographic study,31 was almost devoid of activity. The cytotoxicity of the dibenzazepine 66 (entry 13) is intriguing, given that its C-ring oxygenation pattern cannot be viewed as optimal, although it is recognised that the link between cytotoxicity and the ability to inhibit tubulin polymerisation is potentially complex.The properties of the dibenzoxepine 16 are consistent with its structural analogy to the colchicinoids, e.g.14 and 15. The interaction of colchicine 15 with tubulin has long been under intense scrutiny, now informed by the 1SA0 crystal structure, which gives a detailed picture of the interaction of N-deacetyl-N-(2-mercaptoacetyl)colchicine (DAMA-colchicine) 73 with the αβ-tubulin heterodimer.24 A comparison of the tubulin-bound 73 with a model of the dibenzoxepine 16 (Fig. 2) reveals some clear parallels. We speculate that, as has been proposed for CA-4 1,50 the phenolic hydroxyl of 16 is suitably placed to emulate the side-chain N–H of the colchicinoids in H-bonding to the carbonyl oxygen of the residue Thr179 located on the adjacent α-tubulin chain, while the 3-methoxy group of 16 can interact with the side-chain nitrogen of the β-tubulin residue Lys352. In the absence of substituents at C(1), C(5) and C(7), the biaryl unit of 16 has a configurationally unbiased axis of the tropos type,51i.e. whose low inversion barrier renders it free to adopt the (aR) arrangement required for binding. It is therefore proposed that, while chemically distinct from CA-4 1 and the colchicinoids 14 and 15, the dibenzoxepine 16 binds to tubulin in a similar manner.
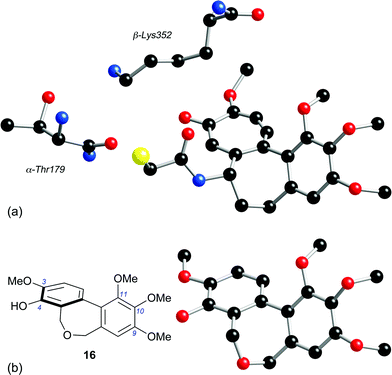 | ||
| Fig. 2 (a) The tubulin-binding conformation of DAMA-colchicine 73, extracted from the crystal structure of the DAMA-colchicine/tubulin:RB3 conjugate (ref. 24), together with the nearby β-Lys352 and α-Thr179 residues (numbering system from ref. 24). (b) The energy-minimised structure of dibenzoxepine 16 (MacroModel 8, MM3 force field). | ||
Conclusion
In the course of this study we have synthesised a series of heterocyclic colchinol analogues and monitored their ability to inhibit microtubule assembly and the in vitrogrowth of K562 human chronic myelogenous leukemia cells, leading us to identify 5,7-dihydro-3,9,10,11-tetramethoxydibenz[c,e]oxepin-4-ol 16 as a potent tubulin-binding and cytotoxic agent. Our results suggest that replacing the central methylene group in the three-atom bridge of colchinol analogues with an oxygen atom does not diminish the ability of suitably substituted derivatives of the system to bind to tubulin and, by inference, interfere with cell division processes and the integrity of tumour neovasculature. We consider the structure 16 to be the leading member of a new series of potential therapeutic agents based on the hitherto unexploited dibenz[c,e]oxepine pharmacophore, and are currently pursuing this principle in the context of a search for clinically useful antitumour agents and VDAs.Experimental
Melting points were determined using Kofler hot-stage, Buchi 512 or Electrothermal 9100 equipment and are uncorrected. Unless otherwise indicated, IR spectra were recorded for neat thin films on NaCl plates, using Perkin–Elmer 1710FT or Nicolet Nexus 670/870 spectrometers. NMR spectra were measured on Bruker AC300 or DPX400 instruments, and assigned with the aid of COSY, HMBC, HSQC and DEPT spectra where appropriate. Coupling constants (J values) are quoted to the nearest 0.1 Hz. Low-resolution mass spectra were measured on a Micromass LCT instrument using a Waters 2790 separations module with electrospray (ES+) ionisation and TOF fragment detection, or a Kratos MS-50 spectrometer with FAB ionisation. High-resolution mass measurements were obtained using ThermoFinnigan MAT95XP or Kratos Concept S1 instruments. Data for most of the peaks of intensity <20% of that of the base peak are omitted. Elemental analyses were carried out by the University of Manchester microanalytical service.Starting materials and solvents were routinely purified by conventional techniques.52 Most reactions were carried out under nitrogen or, when appropriate, argon dried by passage through an anhydrous CaCl2 drying tube and freed from traces of oxygen using an Oxysept cartridge (both Aldrich). Tetrahydrofuran (THF) and N,N,N′,N′-tetramethylethylenediamine (TMEDA) were dried using sodium benzophenone ketyl under argon. Organic solutions were usually dried using anhydrous MgSO4 and concentrated by rotary evaporation under reduced pressure. Analytical thin layer chromatography (TLC) was carried out on Merck silica gel 60 on aluminium plates containing a 254 nm fluorescent indicator. The chromatograms were visualised by the use of UV light or the following developing agents; ethanolic vanillin or potassium permanganate. Unless otherwise indicated, preparative (column) chromatography was carried out using the flash technique53 on 60H silica gel (Merck 9385). Compositions of solvent mixtures are quoted as ratios of volume. ‘Petroleum’ refers to a light petroleum fraction, b.p. 60–80 °C, unless otherwise stated. ‘Ether’ refers to diethyl ether. The preparative routes to the dibenzoxepines 16, 51 and 56 have also been described in a patent application.32 Compounds 18,3420,346331 and 67–7031 were prepared using published procedures.
General procedure for alcohols 19, 25, 28 and 32
To a stirred solution of n-BuLi (2.5 eq.) in dry ether (10 mL mmol−1 of alcohol) at −78 °C was added TMEDA (2.5 eq.) and stirring was continued for 10 min. The neat alcohol was added and the solution was allowed to warm to RT. After stirring for 3 h, the reaction was cooled to −78 °C and the trialkyltin chloride (1.5 eq.) was added. The reaction was warmed to RT and stirred for 2 h. Water (10 mL mmol−1 of alcohol) was added and the mixture was extracted with ether (3 × 10 mL mmol−1 of alcohol). The combined organic extract was washed with brine (20 mL/mmol of alcohol), dried, evaporated, and the residue purified by chromatography.
3,4,5-Trimethoxy-2-tributylstannylbenzyl alcohol19 was prepared from 17 (4.00 g, 20.2 mmol). Chromatography (hexane–ethyl acetate, 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) gave the title compound19 (4.20 g, 43%) as a colourless oil. Elemental analysis, 1H NMR spectroscopy and MS indicated the presence of residual Bu3SnCl. Other data: νmax/cm−1 3464, 2955, 2928, 2866, 2854, 1584, 1561, 1460, 1379, 1313, 1196, 1157, 1099; δH (400 MHz, CDCl3) 6.81 (1 H, s, 6-H), 4.54 (2 H, d, J 5.7 Hz, CH2O), 3.85 (3 H, s, OMe), 3.84 (3 H, s, OMe), 3.80 (3 H, s, OMe), 1.71 (1 H, br t, J 5.7 Hz, OH), 1.52 (6 H, m, 3 × CH2CH2Sn), 1.34 (6 H, m, 3 × CH2CH3), 1.09 (6 H, t, J 8.3 Hz, 3 × CH2Sn), 0.90 (9 H, t, J 7.3 Hz, 3 × CH2CH3); δC (100 MHz, CDCl3, DEPT-135) 12.1 (CH2), 14.1 (CH3), 27.8 (CH2), 29.6 (CH2), 56.3 (CH3), 60.8 (CH3), 61.0 (CH3), 67.4 (CH2), 108.4 (CH); m/z (ES) 489 (MH+, 100%), 332, 291; Rf 0.30 (hexane–ethyl acetate, 6:1).
:1) gave the title compound19 (4.20 g, 43%) as a colourless oil. Elemental analysis, 1H NMR spectroscopy and MS indicated the presence of residual Bu3SnCl. Other data: νmax/cm−1 3464, 2955, 2928, 2866, 2854, 1584, 1561, 1460, 1379, 1313, 1196, 1157, 1099; δH (400 MHz, CDCl3) 6.81 (1 H, s, 6-H), 4.54 (2 H, d, J 5.7 Hz, CH2O), 3.85 (3 H, s, OMe), 3.84 (3 H, s, OMe), 3.80 (3 H, s, OMe), 1.71 (1 H, br t, J 5.7 Hz, OH), 1.52 (6 H, m, 3 × CH2CH2Sn), 1.34 (6 H, m, 3 × CH2CH3), 1.09 (6 H, t, J 8.3 Hz, 3 × CH2Sn), 0.90 (9 H, t, J 7.3 Hz, 3 × CH2CH3); δC (100 MHz, CDCl3, DEPT-135) 12.1 (CH2), 14.1 (CH3), 27.8 (CH2), 29.6 (CH2), 56.3 (CH3), 60.8 (CH3), 61.0 (CH3), 67.4 (CH2), 108.4 (CH); m/z (ES) 489 (MH+, 100%), 332, 291; Rf 0.30 (hexane–ethyl acetate, 6:1).
3,4-Dimethoxy-2-(tributylstannyl)benzyl alcohol25 was prepared from 24 (3.40 g, 20.2 mmol). Chromatography (hexane–ethyl acetate, 6:1) gave the title compound25 (3.60 g, 39%) as a colourless viscous oil (Found: C, 55.0; H, 8.5; Sn, 25.9. C21H38O3Sn requires C, 55.16; H, 8.38; Sn, 25.96%); νmax/cm−1 3367, 2951, 2924, 2870, 2850, 1592, 1464, 1386, 1293, 1269, 1200, 1138, 1037, 870; δH (400 MHz, CDCl3) 7.14 (1 H, d, J 8.1 Hz, 5-H), 6.89 (1 H, d, J 8.1 Hz, 6-H), 4.55 (2 H, d, J 5.7 Hz, CH2O), 3.88 (3 H, s, OMe), 3.85 (3 H, s, OMe), 1.71 (1 H, br t, J 5.7 Hz, OH), 1.52 (6 H, m, 3 × CH2CH2Sn), 1.34 (6 H, m, 3 × CH2CH3), 1.09 (6 H, t, J 8.3 Hz, 3 × CH2Sn), 0.90 (9 H, t, J 7.3 Hz, 3 × CH2CH3); Rf 0.40 (hexane–ethyl acetate, 4:1).
2-Trimethylstannyl-4-(O-silyldimethyl-t-butyl)-3-methoxybenzyl alcohol28 was prepared from 2755 (2.00 g, 7.45 mmol). Chromatography (hexane–ethyl acetate, 10:1) gave the crude stannane 28 (0.90 g, 28%) as a yellow viscous oil which was used without further purification; νmax/cm−1 3309, 2955, 2928, 2854, 1581, 1460, 1383, 1282, 1196, 1134, 1002, 948, 893, 819; δH (400 MHz, CDCl3) 7.01 (1 H, d, J 8.0 Hz, 5-H), 6.79 (1 H, d, J 8.0 Hz, 6-H), 4.53 (2 H, d, J 5.4 Hz, CH2), 3.76 (3 H, s, OMe), 1.48 (1 H, br t, J 5.4 Hz, OH), 1.00 (9 H, s, CMe3), 0.38 (9 H, SnMe3), 0.18 (6 H, s, SiMe2); δC (100 MHz, CDCl3, DEPT-135) −5.7 (CH3), −4.1 (CH3), 26.2 (CH3), 60.8 (CH3), 66.8 (CH2), 122.1 (CH), 124.8 (CH); m/z (ES) 415 (MH+–OH, 100%) (HRMS could not be performed due to low ionisation levels); Rf 0.30 (hexane–ethyl acetate, 8:1). Other fractions provided a sample of the ether 29 (0.50 g, 11%) as a colourless solid, m.p. 54–56 °C, which was identified from the following data: νmax/cm−1 3320, 2963, 2928, 2854, 1581, 1464, 1386, 1285, 1196, 1134, 1006, 948, 893, 831, 769; δH (400 MHz, CDCl3) 6.97 (1 H, d, J 8.0 Hz, 5-H), 6.79 (1 H, d, J 8.0 Hz, 6-H), 4.53 (2 H, d, J 4.6 Hz, OCH2), 3.77 (3 H, s, OMe), 1.51 (1 H, br m, OH), 0.97 (9 H, s, CMe3), 0.33 (9 H, s, ArSnMe3), 0.22 (3 H, s, SiMe), 0.05 (9 H, s, CH2SnMe3), 0.00 (1 H, d, J 19.4 Hz, CHSnMe3), −0.07 (1 H, d, J 19.4 Hz, CHSnMe3); δC (100 MHz, CDCl3, DEPT-135) −8.4 (CH2), −7.3 (CH3), −5.7 (CH3), −2.5 (CH3), 26.6 (CH3), 60.8 (CH3), 66.8 (OCH2), 121.9 (CH), 124.7 (CH); m/z (ES) 577 (MH+–OH, 100%) (HRMS could not be performed due to low ionisation levels); Rf 0.35 (hexane–ethyl acetate, 8:1).
2-Trimethylstannyl-3-methoxy-4-methoxymethylbenzyl alcohol32 was prepared from 3156,57 (3.80 g, 19.2 mmol). Chromatography (hexane–ethyl acetate, 5:1 to 3:1) gave the title compound32 (1.60 g, 23%) as a colourless viscous oil (Found: C, 43.5; H, 6.4; Sn, 32.7. C13H22O4Sn requires C, 43.25; H, 6.14; Sn, 32.88%); νmax/cm−1 3421, 2971, 2936, 2909, 1464, 1394, 1262, 1192, 1157, 1134, 1080, 1002, 773; δH (400 MHz, CDCl3) 7.12 (1 H, d, J 8.2 Hz, 5-H), 7.07 (1 H, d, J 8.2 Hz, 6-H), 5.20 (2 H, s, OCH2O), 4.55 (2 H, s, ArCH2O), 3.88 (3 H, s, ArOMe), 3.52 (3 H, s, CH2OMe), 1.70 (1 H, br s, OH), 0.38 (9 H, SnMe3); δC (100 MHz, CDCl3, DEPT-135) −5.6 (CH3), 56.6 (CH3), 61.3 (CH3), 66.7 (CH2), 95.3 (CH2), 117.3 (CH), 124.8 (CH); m/z (ES) 345 (MH+–OH, 70%), 315 (55), 206 (100); Rf 0.35 (hexane–ethyl acetate, 4:1). Other fractions provided a sample of the isomeric stannane 33 (1.30 g, 19%) as a colourless oil, δH (400 MHz, CDCl3) 6.96 (1 H, d, J 1.9 Hz, ArH), 6.94 (1 H, d, J 1.9 Hz, ArH), 5.20 (2 H, s, OCH2O), 4.55 (2 H, s, ArCH2O), 3.85 (3 H, s, ArOMe), 3.50 (3 H, s, CH2OMe), 1.65 (1 H, br s, OH), 0.35 (9 H, SnMe3); δC (75 MHz, CDCl3, DEPT-135) −8.1 (CH3), 55.95 (CH3), 58.0 (CH3), 65.6 (CH2), 99.1 (CH2), 112.6 (CH), 126.8 (CH); m/z (ES) 345 (MH+–OH, 70%), 315 (55), 206 (100); Rf 0.18 (hexane–ethyl acetate, 4:1).
3,4,5-Trimethoxy-2-tributylstannylbenzyl bromide 21
A solution of triphenylphosphine (1.62 g, 6.2 mmol) in dry DCM (50 mL) at 0 °C was treated dropwise with bromine (0.32 mL, 1.0 g, 6.2 mmol). The yellow colour that persisted was discharged by the addition of a few more crystals of triphenylphosphine, and the solution was stirred for a further 20 min. Imidazole (450 mg, 6.6 mmol) was then added in one portion and the stirring continued for a further 20 min. A solution of the alcohol 19 (2.31 g, 4.74 mmol) in DCM (10 mL) was added and the mixture was stirred at 0 °C for 20 min and RT for 1 h. Pentane (7 mL) was added and the white suspension filtered through a cake of silica gel (ca. 10 g), rinsing with ether (100 mL). Evaporation of the filtrate and chromatography of the residue (110 g silica gel, hexane–ethyl acetate, 6:1) gave the title compound21 (1.80 g, 69%) as a colourless oil which was used without further purification; δH (400 MHz, CDCl3) 6.68 (1 H, s, 6-H), 4.37 (2 H, s, CH2Br), 3.81 (3 H, s, OMe), 3.79 (3 H, s, OMe), 3.75 (3 H, s, OMe), 1.45 (6 H, m, 3 × CH2CH2Sn), 1.27 (6 H, m, 3 × CH2CH3), 1.09 (6 H, t, J 8.3 Hz, 3 × CH2Sn), 0.90 (9 H, t, J 7.3 Hz, 3 × CH2CH3); δC (100 MHz, CDCl3, DEPT-135) 12.2 (CH2), 14.0 (CH3), 27.8 (CH2), 29.5 (CH2), 37.9 (CH2), 56.4 (CH3), 60.8 (CH3), 61.0 (CH3), 110.3 (CH); Rf 0.50 (hexane–ethyl acetate, 8:1).
4,5-Dimethoxy-2-trimethylstannylbenzyl alcohol 35
A stirred solution of 2-bromo-4,5-dimethoxybenzyl alcohol 34 (2.90 g, 11.7 mmol) in THF (10 mL) at −78 °C was treated dropwise with n-BuLi (2.5 M in hexanes, 11.3 mL, 28.3 mmol). The solution was stirred for 1 h at −78 °C, treated dropwise with a solution of trimethyltin chloride (3.50 g, 17.6 mmol) in THF (4 mL), stirred at −78 °C for a further 1 h and then allowed to warm to RT and stirred overnight. The mixture was poured into 1 M sulfuric acid (50 mL), extracted with ether (3 × 50 mL), and the combined extracts dried and evaporated. Chromatography of the residue (ethyl acetate–hexane, 8:1 to 1:1) gave the title compound35 (2.30 g, 59%) as a colourless oil (Found: C, 43.6; H, 6.3; Sn, 35.8. C12H20O3Sn requires C, 43.54; H, 6.09; Sn, 35.86%); νmax/cm−1 3499, 2959, 1577, 1495, 1452, 1324, 1289, 1251, 1045, 866; δH (300 MHz, CDCl3) 6.97 (1 H, s, ArH), 6.89 (1 H, s, ArH), 4.97 (2 H, d, J 5.7 Hz, CH2O), 3.87 (3 H, s, OMe), 3.84 (3 H, s, OMe), 1.91 (1 H, t, J 5.7 Hz, OH), 0.28 (9 H, s, SnMe3); m/z (FAB) 332 (M+, 5%), 315 (M+–OH, 100); Rf 0.40 (hexane–ethyl acetate, 1:1).
General procedure for bis(trialkylstannylbenzyl) ethers 36–39
To a suspension of NaH [1.3 eq., pre-washed with pentane (6 mL mmol−1) and THF (6 mL mmol−1)] in anhydrous DMF (1 mL mmol−1 of alcohol) at 0 °C was added the alcohol (1.1 eq.) as a solution in anhydrous DMF (1 mL mmol−1) dropwise and the mixture was stirred for 10 min. The bromide was added as a solution in DMF (1 mL mmol−1) and the solution was stirred at 0 °C for 20 min, then at RT for 16 h. DCM (10 mL mmol−1) and water (10 mL mmol−1) was added and the mixture was extracted with DCM (2 × 5 mL mmol−1 of alcohol). The combined extract was washed with brine (10 mL/mmol of alcohol), dried, evaporated, and the residue purified by chromatography.
Tributyl(2-((3,4,5-trimethoxy-2-(trimethylstannyl)benzyloxy)methyl)phenyl)stannane36 was prepared from alcohol 2354 (960 mg, 2.42 mmol) and bromide 2034 (930 mg, 2.19 mmol). Chromatography (hexane–ethyl acetate, 10:1) gave the title compound36 (1.08 g, 67%) as a clear oil (Found: C, 51.9; H, 7.4; Sn, 31.8. C32H54O4Sn2 requires C, 51.92; H, 7.35; Sn, 32.08%); νmax/cm−1 3048, 2955, 2924, 2847, 1584, 1561, 1480, 1464, 1375, 1351, 1313, 1192, 1161, 1049, 1017; δH (300 MHz, CDCl3) 7.48 (1 H, d, J 6.8 Hz, ArH), 7.30 (1 H, t, J 6.7 Hz, ArH), 7.31–7.25 (2 H, m, ArH), 6.76 (1 H, s, 5-H), 4.46 (2 H, s, CH2O), 4.39 (2 H, s, CH2O), 3.67 (3 H, s, OMe), 3.65 (6 H, s, 2 × OMe), 1.52 (6 H, m, 3 × CH2CH2Sn), 1.31 (6 H, m, 3 × CH2CH3), 1.07 (6 H, t, J 8.2 Hz, 3 × CH2Sn), 0.90 (9 H, t, J 7.3 Hz, 3 × CH2CH3), 0.26 (9 H, s, SnMe3); δC (100 MHz, CDCl3, DEPT-135) −6.3 (CH3), 10.7 (CH2), 14.1 (CH3), 27.8 (CH2), 29.6 (CH2), 56.4 (CH3), 60.9 (CH3), 61.2 (CH3), 73.2 (CH2), 74.0 (CH2), 109.0 (CH), 127.4 (2 × CH), 128.4 (CH), 137.3 (CH); m/z (ES) 741 (MH+, 100%); Rf 0.55 (hexane–ethyl acetate, 6:1).
Tributyl(6-((3,4-dimethoxy-2-(tributylstannyl)benzyloxy)methyl)-2,3,4-trimethoxyphenyl)stannane37 was prepared from alcohol 25 (1.60 g, 3.50 mmol) and bromide 21 (1.70 g, 3.09 mmol). Chromatography (hexane–ethyl acetate, 10:1) gave the title compound37 (1.00 g, 35%) as a clear oil (Found: C, 55.9; H, 8.4; Sn, 25.9. C43H76O6Sn2 requires C, 55.74; H, 8.27; Sn, 25.63%); νmax/cm−1 2951, 2928, 2847, 1584, 1565, 1464, 1375, 1317, 1262, 1103, 1041, 1013; δH (400 MHz, CDCl3) 7.10 (1 H, d, J 8.1 Hz, 6′-H), 6.86 (1 H, t, J 8.1 Hz, 5′-H), 6.85 (1 H, s, 5-H), 4.40 (2 H, s, OCH2), 4.33 (2 H, s, OCH2) 3.88 (9 H, br s, 3 × OMe), 3.85 (6 H, br s, 2 × OMe), 1.52 (12 H, m, 6 × CH2CH2Sn), 1.31 (12 H, m, 6 × CH2CH3), 1.10 (6 H, t, J 8.5 Hz, 3 × CH2Sn), 1.04 (6 H, t, J 8.2 Hz, 3 × CH2Sn), 0.89 (18 H, m, 6 × CH2CH3); δC (100 MHz, CDCl3, DEPT-135) 12.1 (CH2), 12.15 (CH2), 14.1 (CH3), 14.12 (CH3), 27.8 (CH2), 27.9 (CH2), 29.6 (CH2), 29.7 (CH2), 55.8 (CH3), 56.3 (CH3), 60.8 (CH3), 61.0 (CH3), 61.0 (CH3), 73.1 (CH2), 73.4 (CH2), 108.2 (CH), 112.6 (CH), 125.4 (CH); Rf 0.45 (hexane–ethyl acetate, 6:1).
(2-Methoxy-3-(methoxymethoxy)-6-((3,4,5-trimethoxy-2-(trimethylstannyl)-benzyloxy)methyl)phenyl)trimethylstannane38 was prepared from alcohol 32 (1.52 g, 4.21 mmol) and bromide 2034 (2.40 g, 5.7 mmol). Chromatography (hexane–ethyl acetate, 6:1) gave the title compound38 (2.40 g, 81%) as a clear oil (Found: C, 44.3; H, 6.2; Sn, 34.0. C26H42O7Sn2 requires C, 44.36; H, 6.01; Sn, 33.72%); νmax/cm−1 2932, 2850, 1584, 1561, 1468, 1375, 1317, 1262, 1192, 1161, 1103, 1014, 767; δH (300 MHz, CDCl3) 7.11 (1 H, d, J 8.7 Hz, 6-H), 7.03 (1 H, d, J 8.7 Hz, 5-H), 6.75 (1 H, s, 6′-H), 5.23 (2 H, s, OCH2O), 4.40 (2 H, s, ArCH2O), 4.35 (2 H, s, ArCH2O), 3.85–3.75 (12 H, 4 × s, 4 × OMe), 3.54 (3 H, s, OCH2OMe), 0.32 (9 H, s, SnMe3), 0.26 (9 H, s, SnMe3); δC (100 MHz, CDCl3, DEPT-135) −7.7 (CH3), −5.7 (CH3), 56.2 (CH3), 56.4 (CH3), 56.6 (CH3), 60.9 (CH3), 61.3 (CH3), 72.7 (CH2), 73.2 (CH2), 95.4 (CH2), 109.0 (CH), 117.0 (CH), 125.7 (CH); m/z (ES) 727 (MNa+, 100%); Rf 0.38 (hexane–ethyl acetate, 5:1).
(4,5-Dimethoxy-2-((3,4,5-trimethoxy-2-(trimethylstannyl)benzyloxy)methyl)phenyl)-trimethylstannane39 was prepared from alcohol 35 (1.00 g, 3.02 mmol) and bromide 2034 (1.20 g, 2.83 mmol). Chromatography (hexane–ethyl acetate, 6:1) gave the title compound39 (1.20 g, 63%) as a clear oil (Found: C, 44.7; H, 6.1; Sn, 35.2. C25H40O6Sn2 requires C, 44.55; H, 5.98; Sn, 35.23%); νmax/cm−1 2932, 2909, 2839, 1584, 1557, 1499, 1460, 1379, 1313, 1250, 1161, 1045, 1014, 909, 773, 730; δH (300 MHz, CDCl3) 6.97 (1 H, s, ArH), 6.87 (1 H, s, Ar), 6.71 (1 H, s, ArH), 4.40 (2 H, s, CH2), 4.37 (2 H, s, CH2), 3.90 (3 H, s, OMe), 3.85 (6 H, s, 2 × OMe), 3.83 (3 H, s, OMe), 3.80 (3 H, s, OMe), 0.26 (9 H, s, SnMe3), 0.25 (9 H, s, SnMe3); δC (100 MHz, CDCl3, DEPT-135) −7.7 (CH3), −5.7 (CH3), 56.2 (CH3), 56.4 (CH3), 56.4 (CH3), 60.9 (CH3), 61.3 (CH3), 73.1 (CH2), 73.4 (CH2), 109.3 (CH), 112.4 (CH), 119.1 (CH); m/z (ES) 697 (MNa+, 100%); Rf 0.38 (hexane–ethyl acetate, 6:1).
General procedure for dibenz[c,e]oxepines 40–43
To a suspension of CuCl (5 eq.) in anhydrous DMF (18 mL mmol−1 of ether) under argon was added dropwise the ether (1 eq.) as a solution in anhydrous DMF (30 mL mmol−1) and the mixture was stirred for 2 h. Saturated NH4Cl solution (pH 8, ca. 20 mL mmol−1) was added and the mixture was stirred in an open vessel until deep blue. The mixture was extracted with ether (2 × 40 mL mmol−1 of ether). The combined extract was washed with brine (10 mL/mmol of ether), dried, evaporated, and the residue purified by chromatography.
5,7-Dihydro-1,2,3-trimethoxydibenz[c,e]oxepine40 was prepared from the ether 36 (1.00 g, 1.35 mmol). Chromatography (hexane–ethyl acetate, 6:1) gave a sample of 40 (320 mg, 83%) as a clear oil which crystallised on standing and was shown by 1H NMR spectroscopy to contain a co-eluting by-product. Crystallisation (ether–hexane) gave the pure title compound40 (100 mg, 26%), m.p. 79–80 °C (Found: C, 71.4; H, 6.3. C17H18O4 requires C, 71.31; H, 6.34%); νmax/cm−1 2955, 2932, 2854, 1596, 1487, 1460, 1452, 1402, 1332, 1254, 1231, 1192, 1146, 1118, 1091, 1052, 1005; δH (300 MHz, CDCl3) 7.64 (1 H, d, J 7.8 Hz, 11-H), 7.40–7.27 (3 H, m, 8,9,10-H), 6.70 (1 H, s, 4-H), 4.42 (1 H, br m, 5-H), 4.31 (1 H, br m, 5-H), 4.10 (1 H, br m, 7-H), 3.96 (1 H, br m, 7-H), 3.88 (3 H, s, OMe), 3.86 (3 H, s, OMe), 3.60 (3 H, s, OMe); δC (100 MHz, CDCl3, DEPT-135) 56.5 (CH3), 61.3 (CH3), 61.5 (CH3), 67.91 (CH2), 67.93 (CH2), 109.1 (CH), 128.1 (CH), 128.4 (CH), 129.9 (CH), 130.0 (CH); m/z (ES) 287 (MH+, 23%), 257 (100), 224 (30); Rf 0.18 (hexane–ethyl acetate, 6:1).
5,7-Dihydro-1,2,3,10,11-pentamethoxydibenz[c,e]oxepine41 was prepared from the ether 37 (1.00 g, 1.08 mmol). Chromatography (hexane–ethyl acetate, 6:1) followed by crystallisation (ethyl acetate) gave the title compound41 (130 mg, 35%) as colourless prisms, m.p. 159–160 °C (Found: C, 66.0; H, 6.6. C19H22O6 requires C, 65.88; H, 6.40%); νmax/cm−1 2994, 2963, 2940, 2858, 1600, 1577, 1487, 1460, 1410, 1324, 1274, 1196, 1157, 1111, 1060, 1010; δH (400 MHz, CDCl3) 7.11 (1 H, d, J 8.2 Hz, 9-H), 6.97 (1 H, d, J 8.2 Hz, 8-H), 6.75 (1 H, s, 4-H), 4.43 (1 H, d, J 11.3 Hz, 5-H), 4.38 (1 H, d, J 11.3 Hz, 5-H), 4.07 (1 H, d, J 11.2 Hz, 7-H), 4.06 (1 H, d, J 11.2 Hz, 7-H), 3.94 (9 H, s, 3 × OMe), 3.78 (3 H, s, OMe), 3.65 (3 H, s, OMe); δC (100 MHz, CDCl3, DEPT-135) 56.3 (CH3), 56.4 (CH3), 60.8 (CH3), 61.2 (CH3), 61.3 (CH3), 67.4 (CH2), 67.6 (CH2), 108.1 (CH), 112.4 (CH), 124.8 (CH); m/z (ES) 410 [M(MeCN)Na+, 100%], 369 (MNa+, 12), 347 (MH+, 25), 317 (32); Rf 0.24 (hexane–ethyl acetate, 4:1).
5,7-Dihydro-1,2,3,11-tetramethoxy-10-methoxymethoxydibenz[c,e]oxepine42 was prepared from the ether 38 (2.40 g, 3.41 mmol). Chromatography (ether–petroleum, 1:1) followed by crystallisation (ethyl acetate–hexane) gave the title compound42 (650 mg, 51%) as colourless crystals, m.p. 76–77 °C (Found: C, 63.6; H, 6.6. C20H24O7 requires C, 63.82; H, 6.43%); νmax/cm−1 2944, 2854, 1596, 1487, 1464, 1406, 1332, 1266, 1161, 1115, 1068; δH (300 MHz, CDCl3) 7.17 (1 H, d, J 8.2 Hz, 9-H), 7.04 (1 H, d, J 8.2 Hz, 8-H), 6.71 (1 H, s, 4-H), 5.28 (1 H, d, J 6.7 Hz, OCHOMe), 5.25 (1 H, d, J 6.7 Hz, OCHOMe), 4.40 (1 H, d, J 11.3 Hz, 5-H), 4.35 (1 H, d, J 11.3 Hz, 5-H), 4.05 (1 H, d, J 11.2 Hz, 7-H), 4.01 (1 H, d, J 11.2 Hz, 7-H), 3.91 (3 H, s, ArOMe), 3.91 (3 H, s, ArOMe), 3.73 (3 H, s, ArOMe), 3.63 (3 H, s, ArOMe), 3.54 (3 H, s, OCH2OMe); δC (100 MHz, CDCl3, DEPT-135) 56.4 (CH3), 56.7 (CH3), 61.0 (CH3), 61.2 (CH3), 61.3 (CH3), 67.4 (CH2), 67.6 (CH2), 95.8 (CH2), 108.2 (CH), 116.9 (CH), 124.9 (CH); m/z (ES) 440 [M(MeCN)Na+, 100%], 399 (MNa+, 54); Rf 0.25 (hexane–ethyl acetate, 4:1).
5,7-Dihydro-1,2,3,9,10-pentamethoxydibenz[c,e]oxepine43 was prepared from the ether 39 (1.10 g, 1.63 mmol). Chromatography (hexane–ethyl acetate, 6:1) followed by crystallisation (ethyl acetate–hexane) gave the title compound43 (186 mg, 33%) as colourless crystals, m.p. 135–136 °C (lit.58 124–125 °C) (Found: C, 66.1; H, 6.6. C19H22O6 requires C, 65.88; H, 6.40%); νmax/cm−1 2936, 2854, 1608, 1515, 1491, 1460, 1410, 1375, 1328, 1247, 1122, 1091, 1049, 1014, 990, 854, 734; δH (300 MHz, CDCl3) 7.30 (1 H, s, 11-H), 6.93 (1 H, s, 8-H), 6.78 (1 H, s, 4-H), 4.42 (2 H, br m, 5-H2), 4.17–4.07 (1 H, br m, 7-H), 4.05–3.95 (1 H, br m, 7-H), 3.99–3.93 (12 H, 4 × s, 4 × OMe), 3.68 (3 H, s, ArOMe); δC (100 MHz, CDCl3, DEPT-135) 56.3 (CH3), 56.4 (CH3), 61.2 (CH3), 61.6 (CH3), 67.7 (CH2), 68.0 (CH2), 109.2 (CH), 112.5 (CH), 112.9 (CH) (in accord with published data58); m/z (ES) 410 [M(MeCN)Na+, 100%], 371 (45), 347 (MH+, 5), 317 (94), 302 (20); Rf 0.20 (hexane–ethyl acetate, 4:1).
5,7-Dihydro-10-hydroxy-1,2,3,11-tetramethoxydibenz[c,e]oxepine 44
To a solution of the oxepine 42 (100 mg, 0.27 mmol) in dry DCM (2 mL) was added Montmorillonite clay K10 (100 mg, washed with dry DCM, dried in vacuo).36 The mixture was stirred at RT for 3 h and then concentrated in vacuo. Chromatography of the residue (10 g silica gel, hexane–ethyl acetate, 3:2) gave the title compound44 (55 mg, 62%) as a colourless solid, m.p. 166–167 °C (Found: C, 65.2; H, 6.0. C18H20O6 requires C, 65.05; H, 6.07%); νmax/cm−1 3379, 2941, 2862, 1598, 1489, 1465, 1406, 1328, 1259, 1194, 1150, 1112, 1064; δH (300 MHz, CDCl3) 7.09 (1 H, d, J 8.1 Hz, 9-H), 7.00 (1 H, d, J 8.1 Hz, 8-H), 6.78 (1 H, s, 4-H), 6.00 (1 H, s, OH), 4.43 (1 H, d, J 11.4 Hz, 5-H), 4.39 (1 H, d, J 11.3 Hz, 7-H), 4.09 (1 H, d, J 11.3 Hz, 7-H), 4.02 (1 H, d, J 11.4 Hz, 5-H), 3.95 (3 H, s, OMe), 3.95 (3 H, s, OMe), 3.70 (3 H, s, OMe), 3.43 (3 H, s, OMe); δC (75 MHz, CDCl3) 56.4 (CH3), 60.8 (CH3), 61.4 (CH3), 61.5 (CH3), 67.4 (CH2), 67.6 (CH2), 108.5 (CH), 115.1 (CH), 123.1 (CH), 125.6 (C), 128.5 (C), 129.1 (C), 131.3 (C), 142.8 (C), 144.7 (C), 149.4 (C), 151.5 (C), 154.1 (C); m/z (ES) 396 [M(MeCN)Na+, 100%], 303 (25), 286 (23); Rf 0.23 (hexane–ethyl acetate, 7:1).
Dibenz[c,e]oxepines 16, 51 and 56via Ullmann cross-coupling reactions
The sequences leading to 16, 51 and 56 proceeded along conventional lines, as illustrated by the route to 16 (Scheme 5) below. Full details of Schemes 3 and 4 are provided in the ESI.†5,7-Dihydro-1,2,3,9-tetramethoxydibenz[c,e]oxepine 51
A solution of 50 (170 mg, 0.51 mmol) in THF (2 mL), 2 M hydrochloric acid (2 mL) and conc. hydrochloric acid (1 mL) was stirred under reflux for 3 h. Water (15 mL) and ethyl acetate (15 mL) were added to the reaction, the layers were separated and the aqueous layer was extracted with ethyl acetate (2 × 10 mL). The combined organic extract was dried over Na2SO4 and concentrated in vacuo. Chromatography of the residue (20 g silica gel, hexane–ethyl acetate, 4:1) gave the title compound51 (150 mg, 93%) as a colourless solid, m.p. 151–153 °C (Found: C, 68.5; H, 6.5. C18H20O5 requires C, 68.34; H, 6.37%); νmax/cm−1 2963, 2936, 2858, 2839, 1612, 1491, 1456, 1332, 1243, 1150, 1104, 1052, 1006; δH (300 MHz, CDCl3) 7.63 (1 H, d, J 8.4 Hz, 11-H), 6.98 (1 H, dd, J 2.6, 8.4 Hz, 10-H), 6.96 (1 H, d, J 2.6 Hz, 8-H), 6.75 (1 H, s, 4-H), 4.42 (2 H, m), 4.08 (2 H, m), 3.94 (3 H, s, ArOMe), 3.91 (3 H, s, ArOMe), 3.86 (3 H, s, ArOMe), 3.65 (3 H, s, ArOMe); δC (75 MHz, CDCl3) 55.7 (CH3), 56.4 (CH3), 61.2 (CH3), 61.5 (CH3) [one CH2 signal obscured], 68.1 (CH2), 109.1 (CH), 114.2 (CH), 114.8 (CH), 126.7 (C), 129.7 (C), 131.1 (CH), 131.4 (C), 136.8 (C), 143.1 (C), 150.9 (C), 153.1 (C), 159.4 (C); m/z (ES) 380 [M(MeCN)Na+, 100%], 287 (MH+–CH2O, 100); Rf 0.39 (hexane–ethyl acetate, 3:1).
5,7-Dihydro-1,2,3-trimethoxybenzo[d][1,3]dioxolo[4,5-h][2]benzoxepine 56
A solution of 55 (170 mg, 0.48 mmol) in THF (2 mL), 2 M hydrochloric acid (2 mL) and conc. hydrochloric acid (1 mL) was stirred under reflux for 3 h. Water (15 mL) and ethyl acetate (15 mL) were added, the layers were separated and the aqueous layer was extracted with ethyl acetate (2 × 10 mL). The combined organic extract was dried over Na2SO4 and concentrated in vacuo. Chromatography of the residue (20 g silica gel, hexane–ethyl acetate, 4:1) followed by crystallisation (ethyl acetate) gave the title compound56 (103 mg, 64%) as large clear crystals, m.p. 154–156 °C (Found: C, 65.3; H, 5.5. C18H18O6 requires C, 65.45; H, 5.49%); νmax/cm−1 2967, 2932, 2866, 1600, 1484, 1460, 1414, 1324, 1239, 1146, 1107, 1045; δH (300 MHz, CDCl3) 7.21 (1 H, s, 12-H), 6.98 (1 H, s, 8-H), 6.75 (1 H, s, 4-H), 6.04 (2 H, d, J 4.8 Hz, 10-H2), 4.40 (2 H, d, J 11.2 Hz, 5-H2), 4.04 (1 H, d, J 10.8 Hz, 7-HA), 4.01 (1 H, d, J 10.8 Hz, 7-HB), 3.96 (3 H, s, OMe), 3.91 (3 H, s, OMe), 3.71 (3 H, s, OMe); δC (75 MHz, CDCl3) 56.3 (CH3), 61.2 (CH3), 61.5 (CH3), 67.6 (CH2), 67.8 (CH2), 101.6 (CH2), 109.0 (CH), 109.9 (CH), 110.2 (CH), 126.8 (C), 129.5 (C), 131.3 (C), 131.7 (C), 143.0 (C), 147.3 (C), 147.7 (C), 150.8 (C), 153.3 (C); m/z (ES) 301 (MH+–CH2O, 100%); Rf 0.28 (hexane–ethyl acetate, 4:1).
3-Bromo-2-formyl-6-methoxyphenyl methanesulfonate 5832
To a stirred solution of 6-bromo-2-hydroxy-3-methoxybenzaldehyde 57 (0.33 g, 1.43 mmol) and triethylamine (0.17 g, 1.71 mmol) in DCM (5 mL) was added methanesulfonyl chloride (0.19 g, 2.3 mmol) at 0 °C. The reaction was stirred at 0 °C for 10 min and then at RT for 30 min, by which time a brown colour had developed. Water (20 mL) was added, the mixture was extracted with ethyl acetate (2 × 20 mL). The combined extract was washed with brine, dried and evaporated to give a crude solid (0.42 g). Chromatography (60 g silica gel, hexane–ethyl acetate, 3:1) gave the mesylate 58 (320 mg, 72%) and 5-bromo-8-methoxybenzo[e][1,2]oxathiine 2,2-dioxide 59 (90 mg, 22%) as white solids. The title compound58 had m.p. 95–97 °C; νmax/cm−1 1705, 1565, 1468, 1399, 1363, 1293, 1219, 1165, 1130, 971, 878, 800; δH (400 MHz, CDCl3) 10.28 (1 H, s, CHO), 7.55 (1 H, d, J 8.9 Hz, 4-H), 7.09 (1 H, d, J 8.9 Hz, 5-H), 3.93 (3 H, s, OMe), 3.38 (3 H, s, SMe); δC (100 MHz, CDCl3) 40.1 (CH3), 56.9 (CH3), 115.2 (C), 118.1 (CH), 128.9 (C), 133.9 (CH), 138.6 (C), 152.6 (C), 189.8 (CH); m/z (ES) 374/372 [M(MeCN)Na+, 100%], 333/331 (MNa+, 80); Rf 0.35 (hexane–ethyl acetate, 1:1). 5-Bromo-8-methoxybenzo[e][1,2]oxathiine 2,2-dioxide 59 had m.p. 186–188 °C (Found: C, 37.3; H, 2.2; S, 10.9; Br, 27.2. C9H7BrO4S requires C, 37.13; H, 2.42; S, 11.01; Br, 27.45%); νmax/cm−1 (nujol mull) 3091, 2959, 2924, 2854, 1608, 1569, 1468, 1367, 1309, 1270, 1169, 1080, 909; δH (300 MHz, CDCl3) 7.62 (1 H, d, J 10.5 Hz, 4-H), 7.47 (1 H, d, J 10.5 Hz, 6-H), 6.96 (1 H, d, J 10.5 Hz, 7-H), 6.88 (1 H, d, J 10.5 Hz, 3-H), 3.88 (3 H, s, OMe); δC (75 MHz, CDCl3) 57.0 (CH3), 113.7 (C), 116.2 (CH), 120.3 (C), 123.9 (CH), 129.9 (CH), 135.8 (CH), 142.0 (C), 149.1 (C); Rf 0.60 (hexane–ethyl acetate, 1:1).
2,6′-Diformyl-4,2′,3′,4′-tetramethoxybiphenyl-3-yl methanesulfonate 60
To a suspension of copper bronze (0.646 g, 10 mmol) in anhydrous DMF (2 mL) was added a solution of 2-bromo-3,4,5-trimethoxybenzaldehyde 4538 (0.80 g, 2.91 mmol) and 58 (0.30 g, 0.97 mmol) in anhydrous DMF (1 mL) and the suspension was stirred at 165 °C for 3 h. TLC showed the presence of unreacted 58 and more 45 (100 mg) was added. After a further 1 h at 165 °C the reaction, now complete, was cooled and diluted with ethyl acetate (20 mL). The resulting suspension was filtered through Celite® (3 g) and the filtrate concentrated in vacuo. Chromatography (70 g silica gel, hexane–ethyl acetate, 2:1 to 1:1) and crystallisation from ethyl acetate yielded the title compound60 (180 mg, 44%), m.p. 131–132 °C (Found: C, 53.7; H, 4.8; S, 7.8. C19H20O9S requires C, 53.77; H, 4.75; S, 7.56%); νmax/cm−1 2934, 2843, 1701, 1682, 1588, 1561, 1480, 1371, 1336, 1285, 1169, 1111, 1072; δH (400 MHz, CDCl3) 10.15 (1 H, s, 2-CHO), 9.61 (1 H, s, 6′-CHO), 7.34 (1 H, s, 5′-H), 7.26 (1 H, d, J 8.5 Hz, 6-H), 7.15 (1 H, d, J 8.5 Hz, 5-H), 4.00 (3 H, s, OMe), 3.958 (3 H, s, OMe), 3.955 (3 H, s, OMe), 3.55 (3 H, s, OMe), 3.41 (3 H, s, SMe); δC (100 MHz, CDCl3) 40.0 (CH3), 56.2 (CH3), 56.6 (CH3), 60.9 (CH3), 61.2 (CH3), 105.9 (CH), 116.4 (CH), 127.3 (C), 129.7 (C), 130.3 (C), 130.7 (C), 132.1 (CH), 139.5 (C), 147.3 (C), 150.8 (C), 152.3 (C), 153.8 (C), 189.0 (CH), 190.2 (CH); m/z (ES+) 447 (MNa+, 100%); Rf 0.20 (ethyl acetate–hexane, 2:1), 0.30 (ether).
2,6′-Bis(hydroxymethyl)-4,2′,3′,4′-tetramethoxybiphenyl-3-yl methanesulfonate 61
To a solution of 60 (170 mg, 0.40 mmol) in methanol (4 mL) was added sodium borohydride (45 mg, 1.20 mmol) and the solution was stirred at RT for 1 h. Water (20 mL) and ethyl acetate (20 mL) were added to the reaction, the layers were separated and the aqueous layer was extracted with ethyl acetate (2 × 20 mL). The combined organic extract was dried over Na2SO4 and filtered. The solvent was removed in vacuo and the residue purified by chromatography (25 g silica gel, hexane–ethyl acetate, 1:2), followed by crystallisation (ethyl acetate), which yielded the title compound61 (164 mg, 96%) as a white crystals, m.p. 124–126 °C (Found: C, 53.30; H, 5.67; S, 7.26. C19H24O9S requires C, 53.26; H, 5.65; S, 7.48%); νmax/cm−1 3219, 2947, 1607, 1484, 1410, 1363, 1328, 1278, 1161, 1111, 1006, 889; δH (400 MHz, CDCl3) 7.07 (1 H, d, J 8.5 Hz, 6-H), 7.02 (1 H, d, J 8.5 Hz, 5-H), 6.89 (1 H, s, 5′-H), 4.62 (1 H, dd, J 4.2, 12.0 Hz, CHOH), 4.26–4.15 (3 H, m, CHOH and CH2OH), 3.94 (3 H, s, OMe), 3.91 (3 H, s, OMe), 3.88 (3 H, s, OMe), 3.55 (3 H, s, OMe), 3.51–3.46 (2 H, m, 2 × OH), 3.40 (3 H, s, SMe); δC (100 MHz, CDCl3) 39.3 (CH3), 56.1 (CH3), 56.2 (CH3), 57.5 (CH2), 61.0 (CH3), 61.1 (CH3), 62.5 (CH2), 108.7 (CH), 112.2 (CH), 125.1 (C), 130.1 (CH), 130.2 (C), 135.0 (C), 136.0 (C), 137.8 (C), 141.5 (C), 150.9 (C), 151.3 (C), 153.6 (C); m/z (ES) 451 (MNa+, 100%); Rf 0.18 (hexane–ethyl acetate, 1:2).
5,7-Dihydro-3,9,10,11-tetramethoxydibenz[c,e]oxepin-4-yl methanesulfonate 62
A solution of 61 (155 mg, 0.362 mmol) in THF (2 mL), 2 M hydrochloric acid (2 mL) and conc. hydrochloric acid (1 mL) was stirred under reflux for 3 h. Water (15 mL) and ethyl acetate (15 mL) were added to the reaction, the layers were separated and the aqueous layer was extracted with ethyl acetate (2 × 10 mL). The extracts were combined, dried over Na2SO4, filtered and concentrated. Chromatography of the residue (20 g silica gel, hexane–ethyl acetate, gradient 3:1 to 1:1) gave the title compound62 (110 mg, 74%) as a colourless solid, m.p. 158–161 °C (MeOH) (Found: C, 55.61; H, 5.34; S, 7.74. C19H22O8S requires C, 55.60; H, 5.40; S, 7.81%); νmax/cm−1 2940, 1604, 1573, 1484, 1460, 1367, 1282, 1161, 1118, 1072, 1060, 909, 831; δH (400 MHz, CDCl3) 7.61 (1 H, d, J 8.7 Hz, 1-H), 7.09 (1 H, d, J 8.7 Hz, 2-H), 6.75 (1 H, s, 8-H), 5.01 (1 H, br d, J 11.3 Hz, 5-H), 4.42 (1 H, br d, J 11.3 Hz, 7-H), 4.01 (1 H, br d, J 11.3 Hz, 7-H), 3.96 (3 H, s, OMe), 3.94 (3 H, s, OMe), 3.92 (3 H, s, OMe), 3.90 (1 H, br d, J 11.3 Hz, 5-H), 3.68 (3 H, s, OMe), 3.39 (3 H, s, SMe); δC (100 MHz, CDCl3) 39.5 (CH3), 56.2 (CH3), 56.3 (CH3), 60.4 (5-CH2), 61.1 (CH3), 61.2 (CH3), 67.9 (7-CH2), 108.9 (CH), 112.3 (CH), 125.4 (C), 129.0 (CH), 130.2 (C), 130.8 (C), 131.0 (C), 137.1 (C), 142.8 (C), 150.6 (C), 151.2 (C), 153.3 (C); m/z (ES) 433 (MNa+, 100%), 411 (MH+, 58); Rf 0.31 (hexane–ethyl acetate, 1:1).
5,7-Dihydro-3,9,10,11-tetramethoxydibenz[c,e]oxepin-4-ol 16
To a solution of 62 (85 mg, 0.231 mmol) in 1,4-dioxane (0.25 mL) and methanol (0.25 mL) was added aqueous 3 M potassium hydroxide (0.5 mL) and the mixture was stirred overnight at 50 °C. The solution was diluted with 2 M aqueous hydrochloric acid (2 mL) at 0 °C, extracted with DCM (3 × 5 mL), washed with saturated aqueous sodium hydrogen carbonate (5 mL), dried and evaporated. The residual white solid was chromatographed (30 g silica gel, ethyl acetate–hexane, 1:2) to obtain the title compound16 (62 mg, 90%), m.p. 145–147 °C (Found: C, 64.8; H, 6.1. C18H20O6 requires C, 65.05; H, 6.07%); νmax/cm−1 3394, 2932, 2858, 1600, 1480, 1344, 1270, 1250, 1150, 1115, 1087, 1056; δH (400 MHz, CDCl3) 7.21 (1 H, d, J 8.4 Hz, 1-H), 6.94 (1 H, d, J 8.4 Hz, 2-H), 6.75 (1 H, s, 8-H), 5.91 (1 H, s, 4-OH), 5.15 (1 H, d, J 11.1 Hz, 5-CH), 4.40 (1 H, d, J 11.0 Hz, 7-CH), 4.05 (1 H, d, J 11.0 Hz, 7-CH), 3.95 (3 H, s, OMe), 3.94 (3 H, s, OMe), 3.91 (3 H, s, OMe), 3.85 (1 H, d, J 11.1 Hz, 5-CH), 3.66 (3 H, s, OMe); δC (100 MHz, CDCl3) 56.1 (CH3), 56.2 (CH3), 59.4 (5-CH2), 61.0 (CH3), 61.2 (CH3), 67.9 (7-CH2), 108.9 (CH), 110.1 (CH), 120.9 (CH), 121.2 (C), 126.5 (C), 130.7 (C), 131.3 (C), 142.7 (C), 143.5 (C), 145.7 (C), 150.8 (C), 152.8 (C); m/z (ES) 396 [M(MeCN)Na+, 55%], 355 (MNa+, 4), 315 (MH+–H2O, 100), 303 (MH+–CH2O, 17); Rf 0.16 (acetone–hexane, 1:4); Rf 0.41 (ethyl acetate–hexane, 1:1).
6-Benzyl-6,7-dihydro-1,2,3,9,10,11-hexamethoxy-5H-dibenz[c,e]azepine (±)-66
To a solution of the 6,6′-bis(bromomethyl)-2,2′,3,3′,4,4′-hexamethoxybiphenyl 65, prepared from the diol 64 (134 mg, 0.34 mmol) as described,31 in dry DMF (1 mL) at 0 °C under argon was added benzylamine (146 mg, 1.36 mmol) and triethylamine (103 mg, 1.02 mmol) and the mixture was stirred overnight at RT. Water (10 mL) was added and the mixture extracted with ethyl acetate (3 × 10 mL). The combined organic extract was washed with brine (10 mL), dried and evaporated in vacuo. Chromatography of the residue (25 g silica gel, hexane–ethyl acetate, 3:2) gave the title compound66 (155 mg, 98%) as a colourless crystalline solid, m.p. 117–118 °C, which darkened in air (Found: M + H+, 466.2216; C27H32O6N requires 466.2225); νmax/cm−1 2940, 2835, 1600, 1577, 1495, 1456, 1406, 1317, 1231, 1130, 1099, 734, 699; δH (400 MHz, CDCl3) 7.43 (2 H, d, J 7.1 Hz, 2′,6′-H), 7.38–7.27 (3 H, m, 3′,4′,5′-H), 6.62 (2 H, s, 4,8-H), 3.91 (6 H, s, 2 × OMe), 3.90 (6 H, s, 2 × OMe), 3.73 (6 H, s, 2 × OMe), 3.75–3.65 (2 H, br m, 12-H2), 3.45 (2 H, d, J 12.0 Hz, 5,7-HA), 3.08 (2 H, d, J 12.0 Hz, 5,7-HB); δC (100 MHz, CDCl3) 55.4 (CH2), 56.4 (CH3), 60.0 (CH2), 61.1 (CH3), 61.4 (CH3), 108.5 (CH), 123.2 (C), 127.7 (CH), 128.9 (CH), 129.7 (CH), 130.6 (C), 142.0 (C), 151.8 (C), 153.1 (C) (1 C coincident); m/z (ES) 466 (MH+, 100%); Rf 0.22 (hexane–ethyl acetate, 3:2).
Biological evaluation
Molecular mechanics calculation (Fig. 2b)
The model of 16 (Fig. 2b) was generated on a Mac Mini 2.6 GHz Intel Core 2 Duo running Linux (Fedora Core 12, x86 64-bit), using MacroModel v. 8.0 (Maestro v. 9.0.211 interface) with the MM3 force field and Monte Carlo conformational search (csearch) method (1000 iterations). For the calculation, the dihedral angles (C–O–Cn–Cn−1) associated with the methoxy groups at C(n) [where n = 9, 10 or 11] were constrained (force constant 5000) so as to match those of the DAMA-colchicine 73. The set dihedral angles were: n = 9, 100.0° (99.7° after minimisation); n = 10, 79.1° (79.1° after minimisation); n = 11, 78.8° (78.6° after minimisation). The csearch parameters were: no solvent; PRCG method; convergence on gradient; max. number of iterations 3000; convergence threshold 0.0200.Acknowledgements
We thank the Association for International Cancer Research (AICR) for financial support of this work (grant ref. 00-090). We are also grateful to Alan McGown (University of Salford) for valuable discussions, Edward Piers (University of British Columbia) for his kind provision of experimental details, Darren Cook (University of Salford) for help with compound testing, and Val Boote and Steve Kelly (University of Manchester) for assistance with MS and NMR measurements. We also wish to acknowledge the use of the EPSRC's Chemical Database Service at Daresbury.59References
- (a) N. Ferrara, H.-P. Gerber and J. LeCouter, Nat. Med., 2003, 9, 669–676 CrossRef CAS; (b) D. Ribattia, A. Vaccab and M. Presta, Gen. Pharmacol., 2002, 35, 227–231.
- (a) K. J. Kim, B. Li, J. Winer, M. Armanini, N. Gillett, H. S. Phillips and N. Ferrara, Nature, 1993, 362, 841–844 CrossRef CAS; (b) L. G. Presta, H. Chen, H., S. J. O'Connor, V. Chisholm, Y. G. Meng, L. Krummen, M. Winkler and N. Ferrara, Cancer Res., 1997, 57, 4593–4599 CAS; (c) N. Ferrara, K. J. Hillan, H.-P. Gerber and W. Novotny, Nat. Rev. Drug Discovery, 2004, 3, 391–400 CrossRef CAS.
- For pertinent discussions, see (a) P. Carmeliet and R. K. Jain, Nature, 2000, 407, 249–257 CrossRef CAS; (b) G. M. Tozer, Br. J. Radiol., 2003, 76, S23–S35 Search PubMed; (c) R. K. Jain, Science, 2005, 307, 58–62 CrossRef CAS; (d) S. Yano, Y. Matsumori, K. Ikuta, H. Ogino, T. Doljinsuren and S. Sone, Int. J. Clin. Oncol., 2006, 11, 73–81 CrossRef CAS; (e) S. Cébe-Suarez, A. Zehnder-Fjällman and K. Ballmer-Hofer, Cell. Mol. Life Sci., 2006, 63, 601–615 CrossRef CAS; (f) A. Bozec, S. Lassalle, J. Gugenheim, J.-L. Fischel, P. Formento, P. Hofman and G. Milano, Br. J. Cancer, 2006, 95, 722–728 CrossRef CAS; (g) L. M. Ellis and D. J. Hicklin, Nat. Rev. Cancer, 2008, 8, 579–591 CrossRef CAS; (h) J. M. L. Roodhart, M. H. G. Langenberg, L. G. M. Daenen and E. E. Voest, Biochim. Biophys. Acta Rev. Cancer, 2009, 1796, 41–49 Search PubMed.
- For reviews, see (a) P. E. Thorpe, Clin. Cancer Res., 2004, 10, 415–427; (b) G. M. Tozer, C. Kanthou and B. C. Baguley, Nat. Rev. Cancer, 2005, 5, 423–435 CrossRef CAS; (c) C. Kanthou and G. M. Tozer, International Journal of Experimental Pathology, 2009, 90, 284–294 Search PubMed.
- J. W. Lippert, Bioorg. Med. Chem., 2007, 15, 605–615 CrossRef.
- G. R. Pettit, S. B. Singh, M. R. Boyd, E. Hamel, R. K. Pettit, J. M. Schmidt and F. Hogan, J. Med. Chem., 1995, 38, 1666–1672 CrossRef CAS and references cited therein.
- (a) G. G. Dark, S. A. Hill, V. E. Prise, G. M. Tozer, G. R. Pettit and D. J. Chaplin, Cancer Res., 1997, 57, 1829–1834 CAS; (b) S. M. Galbraith, R. J. Maxwell, M. A. Lodge, G. M. Tozer, J. Wilson, N. J. Taylor, J. J. Stirling, L. Sena, A. R. Padhani and G. S. Rustin, J. Clin. Oncol., 2003, 21, 2831–2842 CrossRef CAS.
- (a) I. G. Kirwan, P. M. Loadman, D. J. Swaine, D. A. Anthoney, G. R. Pettit, J. W. Lippert, S. D. Shnyder, P. A. Cooper and M. C. Bibby, Clin. Cancer Res., 2004, 10, 1446–1453 CrossRef CAS; (b) Y. Sheng, J. Hua, K. G. Pinney, C. M. Garner, R. R. Kane, J. A. Prezioso, D. J. Chaplin and K. Edvardsen, Int. J. Cancer, 2004, 111, 604–610 CrossRef CAS; (c) H. W. Salmon and D. W. Siemann, Clin. Cancer Res., 2006, 12, 4090–4094 CrossRef CAS; (d) G. R. Pettit, A. J. Thornhill, B. R. Moser and F. Hogan, J. Nat. Prod., 2008, 71, 1561–1563 CrossRef CAS.
- (a) K. Hori, S. Saito and K. Kubota, Br. J. Cancer, 2002, 86, 1604–1614 CrossRef CAS; (b) K. Hori and S. Saito, Br. J. Cancer, 2003, 89, 1334–1344 CrossRef CAS; (c) A. Delmonte and C. Sessa, Expert Opin. Invest. Drugs, 2009, 18, 1541–1548 CrossRef CAS.
- (a) K. Miyazaki, M. Kobayashi, T. Natsume, M. Gondo, T. Mikami, K. Sakakibara and S. Tsukagoshi, Chem. Pharm. Bull., 1995, 43, 1706–1718 CAS; (b) N. Yamamoto, M. Andoh, M. Kawahara, M. Fukuoka and H. Niitani, Cancer Sci., 2009, 100, 316–321 CrossRef CAS and references cited therein.
- (a) G. J. S. Rustin, C. Bradley, S. Galbraith, M. Stratford, P. Loadman, S. Waller, K. Bellenger, L. Gumbrell, L. Folkes and G. Halbert, Br. J. Cancer, 2003, 88, 1160–1167 CrossRef CAS; (b) M. B. Jameson, P. I. Thompson, B. C. Baguley, B. D. Evans, V. J. Harvey, D. J. Porter, M. R. McCrystal, M. Small, K. Bellenger, L. Gumbrell, G. W. Halbert and P. Kestell, Br. J. Cancer, 2003, 88, 1844–1850 CrossRef CAS; (c) B. G. Siim, A. E. Lee, S. Shalal-Zwain, F. B. Pruijn, M. J. McKeage and W. R. Wilson, Cancer Chemother. Pharmacol., 2003, 51, 43–52 CrossRef CAS.
- (a) S. Kasibhatla, V. Baichwal, S. X. Cai, B. Roth, I. Skvortsova, S. Skvortsov, P. Lukas, N. M. English, N. Sirisoma, J. Drewe, A. Pervin, B. Tseng, R. O. Carlson and C. M. Pleiman, Cancer Res., 2007, 67, 5865–5871 CrossRef CAS; (b) N. Sirisoma, A. Pervin, H. Zhang, S. Jiang, J. A. Willardsen, M. B. Anderson, G. Mather, C. M. Pleiman, S. Kasibhatla, B. Tseng, J. Drewe and S. X. Cai, J. Med. Chem., 2009, 52, 2341–2351 CrossRef CAS; (c) N. Sirisoma, A. Pervin, H. Zhang, S. Jiang, J. A. Willardsen, M. B. Anderson, G. Mather, C. M. Pleiman, S. Kasibhatla, B. Tseng, J. Drewe and S. X. Cai, Bioorg. Med. Chem. Lett., 2010, 20, 2330–2334 CrossRef CAS.
- (a) J. A. Segreti, J. S. Polakowski, K. A. Koch, K. C. Marsh, J. L. Bauch, S. H. Rosenberg, H. L. Sham, B. F. Cox and G. A. Reinhart, Cancer Chemother. Pharmacol., 2004, 54, 273–281 CAS; (b) Y. Luo, V. P. Hradil, D. J. Frost, S. H. Rosenberg, G. B. Gordon, S. J. Morgan, G. D. Gagne, B. F. Cox, S. K. Tahir and G. B. Fox, Anti-Cancer Drugs, 2009, 20, 483–492 CrossRef CAS.
- B. Nicholson, G. K. Lloyd, B. R. Miller, M. A. Palladino, Y. Kiso, Y. Hayashi and S. T. C. Neuteboom, Anti-Cancer Drugs, 2006, 17, 25–31 CrossRef CAS.
- (a) W. Shi and D. W. Siemann, Anticancer Res., 2005, 25, 3899–3904 CAS; (b) A. M. Traynor, M. S. Gordon, D. Alberti, D. S. Mendelson, M. S. Munsey, G. Wilding, R. E. Gammans and W. L. Read, Invest. New Drugs, 2010, 28, 509–515 CrossRef CAS.
- (a) S. Kasibhatla, H. Gourdeau, K. Meerovitch, J. Drewe, S. Reddy, L. Qiu, H. Zhang, F. Bergeron, D. Bouffard, Q. Yang, J. Herich, S. Lamothe, S. X. Cai and B. Tseng, Mol. Cancer Ther., 2004, 3, 1365–1373 CAS; (b) H. Gourdeau, L. Leblond, B. Hamelin, C. Desputeau, K. Dong, I. Kianicka, D. Custeau, C. Boudreau, L. Geerts, S.-X. Cai, J. Drewe, D. Labrecque, S. Kasibhatla and B. Tseng, Mol. Cancer Ther., 2004, 3, 1375–1383 CAS; (c) W. Kemnitzer, J. Drewe, S. Jiang, H. Zhang, Y. Wang, J. Zhao, S. Jia, J. Herich, D. Labreque, R. Storer, K. Meerovitch, D. Bouffard, R. Rej, R. Denis, C. Blais, S. Lamothe, G. Attardo, H. Gourdeau, B. Tseng, S. Kasibhatla and S. X. Cai, J. Med. Chem., 2004, 47, 6299–6310 CrossRef CAS.
- G. Kremmidiotis, A. F. Leske, T. C. Lavranos, D. Beaumont, J. Gasic, A. Hall, M. O'Callaghan, C. A. Matthews and B. Flynn, Mol. Cancer Ther., 2010, 9, 1562–1573 CrossRef CAS.
- (a) P. D. Davis, G. J. Dougherty, D. C. Blakey, S. M. Galbraith, G. M. Tozer, A. L. Holder, M. A. Naylor, J. Nolan, M. R. Stratford, D. J. Chaplin and S. A. Hill, Cancer Res., 2002, 62, 7247–7253 CAS; (b) D. C. Blakey, F. R. Westwood, M. Walker, G. D. Hughes, P. D. Davis, S. E. Ashton and A. J. Ryan, Clin. Cancer Res., 2002, 8, 1974–1983 CAS; (c) L. V. Beerepoot, S. A. Radema, E. O. Witteveen, T. Thomas, C. Wheeler, S. Kempin and E. E. Voest, J. Clin. Oncol., 2006, 24, 1491–1498 CrossRef CAS; (d) S. Gould, F. R. Westwood, J. O. Curwen, S. E. Ashton, D. W. Roberts, S. C. Lovick and A. J. Ryan, J. Natl. Cancer Inst., 2007, 99, 1724–1728 CrossRef CAS.
- (a) Z. J. Roberts, N. Goutagny, P.-Y. Perera, H. Kato, H. Kumar, T. Kawai, S. Akira, R. Savan, D. van Echo, K. A. Fitzgerald, H. A. Young, L.-M. Ching and S. N. Vogel, J. Exp. Med., 2007, 204, 1559–1569 CrossRef CAS; (b) A. Wallace, D. F. LaRosa, V. Kapoor, J. Sun, G. Cheng, A. Jassar, A. Blouin, L.-M. Ching and S. M. Albelda, Cancer Res., 2007, 67, 7011–7019 CrossRef CAS; (c) S. Gobbi, F. Belluti, A. Bisi, L. Piazzi, A. Rampa, A. Zampiron, M. Barbera, A. Caputo and M. Carrara, Bioorg. Med. Chem., 2006, 14, 4101–4109 CrossRef CAS.
- L.-M. Ching, Z. Cao, C. Kieda, S. Zwain, M. B. Jameson and B. C. Baguley, Br. J. Cancer, 2002, 86, 1937–1942 CrossRef CAS.
- For reviews, see (a) K. H. Downing, Annu. Rev. Cell Dev. Biol., 2000, 16, 89–111 CrossRef CAS; (b) L. A. Amos, Org. Biomol. Chem., 2004, 2, 2153–2160 RSC.
- (a) M. A. Jordan, R. J. Toso, D. Thrower and L. Wilson, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 9552–9556 CrossRef CAS and references cited therein; (b) C. Elie-Caille, F. Severin, J. Helenius, J. Howard, D. J. Muller and A. A. Hyman, Curr. Biol., 2007, 17, 1765–1770 CrossRef CAS; (c) S. Goodin, M. P. Kane and E. H. Rubin, J. Clin. Oncol., 2004, 22, 2015–2025 CrossRef CAS.
- B. Bhattacharyya, D. Panda, S. Gupta and M. Banerjee, Med. Res. Rev., 2008, 28, 155–183 CrossRef CAS.
- R. B. G. Ravelli, B. Gigant, P. A. Curmi, I. Jourdain, S. Lachkar, A. Sobel and M. Knossow, Nature, 2004, 428, 198–202 CrossRef CAS.
- (a) B. Gigant, C. Wang, R. B. G. Ravelli, F. Roussi, M. O. Steinmetz, P. A. Curmi, A. Sobel and M. Knossow, Nature, 2005, 435, 519–522 CrossRef CAS; (b) A. Dorléans, B. Gigant, R. B. G. Ravelli, P. Mailliet, V. Mikol and M. Knossow, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 13775–13779 CrossRef CAS.
- S. M. Galbraith, D. J. Chaplin, F. Lee, M. R. L. Stratford, R. J. Locke, B. Vojnovic and G. M. Tozer, Anticancer Res., 2001, 21, 93–102 CAS.
- (a) C. M. Lin, S. B. Singh, P. S. Chu, R. O. Dempcy, J. M. Schmidt, G. R. Pettit and E. Hamel, Mol. Pharmacol., 1988, 34, 200–208 CAS; (b) C. M. Lin, H. H. Ho, G. R. Pettit and E. Hamel, Biochemistry, 1989, 28, 6984–6991 CrossRef CAS.
- For a review, see B. Bhattacharyya, D. Panda, S. Gupta and M. Banerjee, Med. Res. Rev., 2008, 28, 155–183 Search PubMed.
- T. J. Lampidis, D. Kolonias, N. Savaraj and R. W. Rubin, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 1256–1260 CrossRef CAS.
- (a) W. J. van Heeckeren, S. Bhakta, J. Ortiz, J. Duerk, M. M. Cooney, A. Dowlati, K. McCrae and S. C. Remick, J. Clin. Oncol., 2006, 24, 1485–1488 CrossRef CAS; (b) For a study of the cardiovascular profile of CA-4P 2, see M. M. Cooney, T. Radivoyevitch, A. Dowlati, B. Overmoyer, N. Levitan, K. Robertson, S. L. Levine, K. DeCaro, C. Buchter, A. Taylor, B. S. Stambler and S. C. Remick, Clin. Cancer Res., 2004, 10, 96–100 Search PubMed.
- D. J. Edwards, R. G. Pritchard and T. W. Wallace, Acta Crystallogr., Sect. B: Struct. Sci., 2005, 61, 335–345 CrossRef.
- T. W. Wallace, D. J. Edwards, and J. A. Hadfield, PCT Int. Appl. WO 2008075048, 2008.
- For independent studies along related lines, see (a) V. Colombel, A. Joncour, S. Thoret, J. Dubois, J. Bignon, J. Wdzieczak-Bakala and O. Baudoin, Tetrahedron Lett., 2010, 51, 3127–3129 CrossRef CAS; (b) A. Joncour, J.-M. Liu, A. Décor, S. Thoret, J. Wdzieczak-Bakala, J. Bignon and O. Baudoin, ChemMedChem, 2008, 3, 1731–1739 CrossRef CAS; (c) A. Joncour, A. Décor, S. Thoret, A. Chiaroni and O. Baudoin, Angew. Chem., Int. Ed., 2006, 45, 4149–4152 CrossRef CAS; (d) N. Nicolaus, S. Strauss, J.-M. Neudörfl, A. Prokop and H.-G. Schmalz, Org. Lett., 2009, 11, 341–344 CrossRef CAS.
- E. Piers, J. G. K. Yee and P. L. Gladstone, Org. Lett., 2000, 2, 481–484 CrossRef CAS.
- H. Imanieh, P. Quayle, M. Voaden, J. Conway and S. D. A. Street, Tetrahedron Lett., 1992, 33, 543–546 CrossRef CAS.
- J. P. Deville and V. Behar, J. Org. Chem., 2001, 66, 4097–4098 CrossRef CAS.
- For reviews, see (a) T. D. Nelson and R. D. Crouch, Org. React., 2004, 63, 265–555 CAS; (b) J. Hassan, M. Sévignon, C. Gozzi, E. Schulz and M. Lemaire, Chem. Rev., 2002, 102, 1359–1469 CrossRef CAS; (c) P. E. Fanta, Synthesis, 1974, 9–21 CrossRef CAS.
- E. Brown, J.-P. Robin and R. Dhal, Tetrahedron, 1982, 38, 2569–2579 CrossRef CAS.
- H. Suzuki, T. Enya and Y. Hisamatsu, Synthesis, 1997, 1273–1276 CrossRef.
- (a) F. E. Ziegler, I. Chliwner, K. W. Fowler, S. J. Kanfer, S. J. Kuo and N. D. Sinha, J. Am. Chem. Soc., 1980, 102, 790–798 CrossRef CAS; (b) F. E. Ziegler, K. W. Fowler, W. B. Rodgers and R. T. Wester, Org. Synth., 1987, 65, 108–118 CAS.
- J. M. Insole, J. Chem. Res. (S), 1990, 378–379 Search PubMed; J. M. Insole, J. Chem. Res. (M), 1990, 2831–2867 Search PubMed.
- (a) G. Wu, H.-F. Guo, K. Gao, Y.-N. Liu, K. F. Bastow, S. L. Morris-Natschke, K.-H. Lee and L. Xie, Bioorg. Med. Chem. Lett., 2008, 18, 5272–5276 CrossRef CAS; (b) L. G. Monovich, Y. Le Huérou, M. Rönn and G. A. Molander, J. Am. Chem. Soc., 2000, 122, 52–57 CrossRef CAS.
- I. Lantos and B. Loev, Tetrahedron Lett., 1975, 16, 2011–2014 CrossRef.
- M. Oki, H. Iwamura and T. Nishida, Bull. Chem. Soc. Jpn., 1968, 41, 656–660 CrossRef CAS.
- (a) N. J. Lawrence, A. T. McGown, S. Ducki and J. A. Hadfield, Anti-Cancer Drug Des., 2000, 15, 135–141 CAS; (b) J. A. Woods, J. A. Hadfield, G. R. Pettit, B. W. Fox and A. T. McGown, Br. J. Cancer, 1995, 71, 705–711 CAS.
- (a) J. M. Edmondson, L. S. Armstrong and A. O. Martinez, J. Tissue Cult. Methods, 1988, 11, 15–17 CrossRef CAS; (b) T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS.
- Y. Kataoka, M. Ishikawa, M. Miura, M. Takeshita, R. Fujita, S. Furusawa, M. Takayanagi, Y. Takayanagi and K. Sasaki, Biol. Pharm. Bull., 2001, 24, 612–617 CrossRef CAS.
- O. Boye, A. Brossi, H. J. C. Yeh, E. Hamel, B. Wegrzynski and V. Toome, Can. J. Chem., 1992, 70, 1237–1249 CAS.
- M. G. Banwell, J. M. Cameron, M. Corbett, J. R. Dupuche, E. Hamel, J. N. Lambert, C. M. Lin and M. F. Mackay, Aust. J. Chem., 1992, 45, 1967–1982 CAS.
- S. Ducki, G. Mackenzie, B. Greedy, S. Armitage, J. Fournier, Dit Chabert, E. Bennett, J. Nettles, J. P. Snyder and N. J. Lawrence, Bioorg. Med. Chem., 2009, 17, 7711–7722 CrossRef CAS.
- For a discussion, see K. Mikami, K. Aikawa, Y. Yusa, J. J. Jodry and M. Yamanaka, Synlett, 2002, 1561–1578 Search PubMed.
- D. D. Perrin, W. L. F. Armarego, and D. R. Perrin, Purification of Laboratory Chemicals, 2nd Edition, Pergamon, Oxford, 1980 Search PubMed.
- W. C. Still, M. Kahn and A. Mitra, J. Org. Chem., 1978, 43, 2923–2925 CrossRef CAS.
- N. Meyer and D. Seebach, Chem. Ber., 1980, 113, 1304–1319 CAS.
- S. B. Singh and G. R. Pettit, Synth. Commun., 1987, 17, 877–892 CrossRef CAS.
- S. Mabic, L. Vaysse, C. Benezra and J.-P. Lepoittevin, Synthesis, 1999, 1127–1134 CrossRef CAS.
- Y. Wang, C. A. Mathis, G. Huang, D. P. Holt, M. L. Debnath and W. E. Klunk, J. Labelled Compd. Radiopharm., 2002, 45, 647–664 CrossRef CAS.
- T. Takada, M. Arisawa, M. Gyoten, R. Hamada, H. Tohma and Y. Kita, J. Org. Chem., 1998, 63, 7698–7706 CrossRef CAS.
- D. A. Fletcher, R. F. McMeeking and D. Parkin, J. Chem. Inf. Comput. Sci., 1996, 36, 746–749 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Further experimental details. See DOI: 10.1039/c0ob00500b |
| ‡ Present address: Laboratoire de Chimie des Hétérocycles et des Glucides, ENSCCF, Clermont Université, 63174 Aubière, France. |
| This journal is © The Royal Society of Chemistry 2011 |