Nucleotidylation of unsaturated carbasugar in validamycin biosynthesis†
Jongtae
Yang
a,
Hui
Xu
ab,
Yirong
Zhang
b,
Linquan
Bai
b,
Zixin
Deng
b and
Taifo
Mahmud
*a
aDepartment of Pharmaceutical Sciences, Oregon State University, Corvallis, OR 97331, USA. E-mail: Taifo.Mahmud@oregonstate.edu; Fax: (+1) 541-737-3999
bLaboratory of Microbial Metabolism and School of Life Sciences & Biotechnology, Shanghai Jiao Tong University, Shanghai, 200030, China
First published on 27th October 2010
Abstract
Validamycin A is a member of microbial-derived C7N-aminocyclitol family of natural products that is widely used as crop protectant and the precursor of the antidiabetic drug voglibose. Its biosynthetic gene clusters have been identified in several Streptomyces hygroscopicus strains, and a number of genes within the clusters have been functionally analyzed. Of these genes, valB, which encodes a sugar nucleotidyltransferase, was found through inactivation study to be essential for validamycin biosynthesis, but its role was unclear. To characterize the role of ValB in validamycin biosynthesis, four carbasugar phosphate analogues were synthesized and tested as substrate for ValB. The results showed that ValB efficiently catalyzes the conversion of valienol 1-phosphate to its nucleotidyl diphosphate derivatives, whereas other unsaturated carbasugar phosphates were found to be not the preferred substrate. ValB requires Mg2+, Mn2+, or Co2+ for its optimal activity and uses the purine-based nucleotidyltriphosphates (ATP and GTP) more efficiently than the pyrimidine-based NTPs (CTP, dTTP, and UTP) as nucleotidyl donor. ValB represents the first member of unsaturated carbasugar nucleotidyltransferases involved in natural products biosynthesis. Its characterization not only expands our understanding of aminocyclitol-derived natural products biosynthesis, but may also facilitate the development of new tools for chemoenzymatic synthesis of carbohydrate mimetics.
Introduction
Carbohydrates are indispensable elements in living organisms, not only as molecules for energy storage but also as mediators of many complex biological processes.1–3 Their structural diversity enables them to encode information for specific molecular transactions such as cell–cell recognition and molecular targeting.1–3 Due to their ubiquitous involvement in many biological processes, including immune response, cancer, and inflammation, developing new carbohydrate mimetics (e.g., carbasugars, aminosugars, etc) can provide a new roadmap for drug discovery.4–6Among the naturally derived carbasugars are the C7N-aminocyclitol family of natural products, such as the α-glucosidase inhibitor acarbose and the antifungal agent validamycin A. They have attracted attention due to their important biomedical and agricultural uses.7Validamycin A is a bacterial-derived crop protectant that is widely used to treat sheath blight disease in rice plants. The core aminocyclitol unit, valienamine, is used as the semi-synthetic precursor of the antidiabetic drug voglibose. Prompted by the identification of the biosynthetic gene cluster, investigations into validamycin biosynthesis have revealed the involvement of a number of unusual enzymes that exclusively recognize carbasugars (cyclitols) as substrates.8 The pathway leads to a condensation between two cyclitol intermediates by an undetermined coupling mechanism. The lack of clear understanding of this coupling reaction has led to speculation about how the nitrogen bridge in validamycin A is formed.8–13
It has been speculated that the core validoxylamine unit of validamycin is formed by a condensation between a nucleotidyl unsaturated carbasugar, such as NDP-valienol (3a) or NDP-1-epi-valienol (3b) (Scheme 1), and an amino-bearing carbasugar.93a and 3b may be derived from valienol 1-phosphate (1a) or 1-epi-valienol 1-phosphate (1b), respectively, catalyzed by a carbasugar nucleotidyltransferase. A similar reaction has been proposed in the acarbose pathway involving 1-epi-valienol 1,7-diphosphate (2b) and NDP-1-epi-valienol 7-phosphate (4b) as the substrate and the product of the putative nucleotidyltransferase AcbR.11,14 However, no biochemical evidence is available to support this notion.
 | ||
| Scheme 1 Proposed biosynthetic pathways to validamycin involving either valienol 1-phosphate or 1-epi-valienol 1-phosphate (A) and to acarbose involving either valienol 1,7-diphosphate or 1-epi-valienol 1,7-diphosphate (B). | ||
Although sugar 1-phosphate nucleotidyltransferases have been widely studied, little is known about their involvement in carbasugar or aminocyclitol biosynthesis. The only report that directly implicated a sugar 1-phosphate nucleotidyltransferase in NDP-carbasugar formation was that of the Escherichia coliglucose 1-phosphate uridylyltransferase, which could catalyze the conversion of carbaglucose 1-phosphate to its UDP-product, albeit at a lower turnover rate than observed with the natural substrate, glucose 1-phosphate.15 While it is unclear if other nucleotidyltransferases are tolerant to ring oxygen substitutions, the corresponding enzyme from yeast did not recognize carbaglucose 1-phosphate as substrate.15
A gene (valB) encoding a sugar 1-phosphate nucleotidyltransferase homologous to AcbR was identified in the validamycin biosynthetic gene cluster of Streptomyces hygroscopicus subsp. jinggangensis 5008.16 While the catalytic function of ValB was unclear, its homologue (VldB, 99% identity) from another validamycin producing strain was reported to convert glucose 1-phosphate to UDP-glucose.13 It was then postulated that VldB is responsible for the formation of pathway specific UDP-glucose, which is important for the glucosylation of validoxylamine A to validamycin A. As this result is inconsistent with the notion that ValB, and also AcbR, is involved in carbasugar nucleotidylation, we decided to investigate the actual role of ValB in validamycin biosynthesis. The present paper reports our results on the inactivation of valB in S. hygroscopicus 5008, complementation experiments, synthesis of all four possible substrates (1a, 1b, valienol 1,7-diphosphate (2a), and 2b) for ValB, as well as functional characterization of recombinant ValB. The results not only provide insight into the role of ValB in validamycin biosynthesis but may also facilitate the development of new tools for the production of carbohydrate mimetics.
Results
Inactivation of valB in S. hygroscopicus 5008 abolishes production of validamycin A and validoxylamine A
Whereas Kim and co-workers have demonstrated that VldB could convert glucose 1-phosphate to UDP-glucose,13 the question remains as to what is the exact role of ValB in validamycin biosynthesis. Is ValB really responsible for the formation of UDP-glucose, the substrate for the glycosyltransferase ValG,17 or is it involved in the formation of NDP-cyclitol, setting the stage for a subsequent coupling reaction.9 To address this question, the valBgene was inactivated by replacing a 936 bp internal DNA fragment of valB with oriT-aac(3)IV in S. hygroscopicus 5008. This was achieved by using a pHZ1358-derived plasmid pJTU702 in which oriT-aac(3)IV had been inserted between a 3.2 kb left flanking and a 1.5 kb right flanking sequence of the 936 bp DNA fragment to be replaced (Fig. 1A). The plasmid pJTU702 was introduced into the wild-type strain of S. hygroscopicus 5008 through conjugation18 and two thiostrepton-sensitive apramycin-resistant transformants (ZYR-2-1 and ZYR-2-2) were obtained. Total DNA from these two mutant strains and from the wild-type strain was used as templates for PCR analysis using two primers, ValB-Det-F and ValB-Det-R. The two mutants gave a 1.8 kb PCR product, while the wild-type strain gave a 1.4 kb PCR product, which confirmed that a 936 bp DNA fragment internal to valB had been replaced by the 1384 bp oriT-aac(3)IV cassette in these mutants (Fig. 1B). Fermentation broths of the mutants were analyzed by bioassay and HPLC. No inhibition of the fungus Pellicularia sasakii could be detected in the bioassay (Fig. 1C) and no peak corresponding to validamycin A and validoxylamine A was detected by HPLC analysis, indicating a complete loss of production of both compounds in the valB mutants (Fig. 1D). The lack of validoxylamine A formation in the mutant strains indicated that ValB is involved in the formation of the core aminocyclitol unit, rather than in UDP-glucose formation.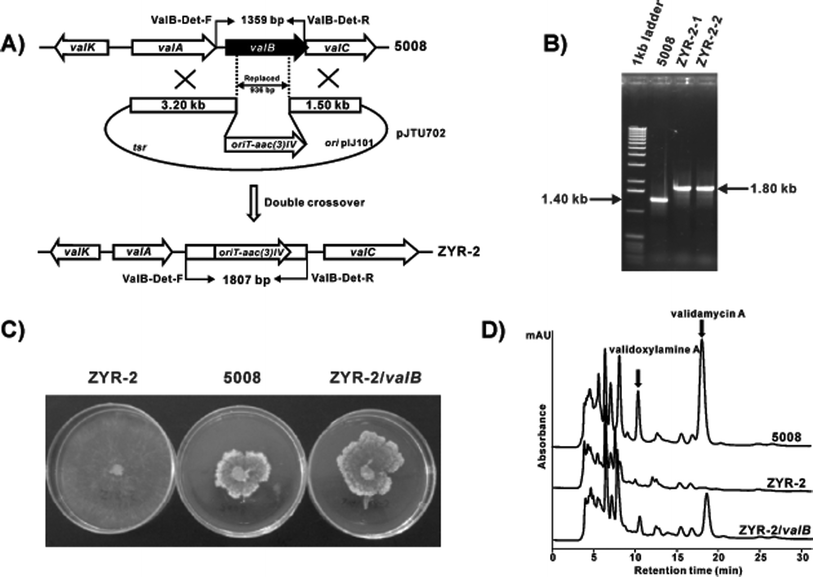 | ||
| Fig. 1 Construction and analysis of valB mutant strains of S. hygroscopicus 5008. A, Schematic representation for the replacement of a 936 bp fragment of valB with oriT-aac(3)IV. B, Gel electrophoresis of PCR amplified products of the wild-type and the mutant strains. C, Bioassay comparison between the wild-type 5008, the valB mutant ZYR-2, and the valB mutant complemented with cloned valB (ZYR-2/valB) using Pellicularia sasakii as the indicator strain. Growth inhibition was determined by the appearance of abnormal branching of the mycelia and the formation of a thick border. D, HPLC analysis of the wild-type 5008, the valB mutant ZYR-2, and the valB mutant complemented with cloned valB (ZYR-2/valB). Validamycin A and validoxylamine A are indicated with arrows. | ||
Restoration of validamycin A and validoxylamine A production in ZYR-2 through complementation with cloned valB
To confirm the critical role of valB in validoxylamine A biosynthesis, the PCR-amplified valBgene was cloned onto the integrative vector pPM927 downstream of the PermE* promoter, and the product subsequently introduced into the valB mutant ZYR-2-1 by conjugation. Fermentation broths of the ZYR-2 strain complemented with valB regained inhibitory activity in the bioassay (Fig. 1C) and HPLC analysis of the fermentation extracts clearly demonstrated that validoxylamine A and validamycin A productions were restored to about 50% of those of the wild-type strain (Fig. 1D).Chemical synthesis of valienol phosphates
As the inactivation experiments suggested the involvement of ValB in the formation of validoxylamine A, we attempted to identify the substrate for this enzyme by preparing and testing a number of carbasugar compounds. Despite that numerous strategies have been applied to the synthesis of carbocyclic framework of carbasugars, both in racemic and enantiomerically pure forms,7,19 the synthesis of unsaturated carbasugar phosphates, such as valienol phosphates and its analogues, has never been reported. In order to gain access to these unsaturated carbasugar phosphates chemical syntheses were carried out using a ring-closing metathesis strategy as shown in Scheme 2. The key intermediate tetrabenzylated diene 7 was synthesized from commercially available 2,3,4,6-tetra-O-benzyl-D-glucopyranose (5) in five steps according to a literature procedure.19Ring-closing metathesis19 of 7 mediated by Grubbs' 2nd generation catalyst20 furnished, after column chromatography, the tetrabenzylated valienols 8a and 8b in 21% and 57% yields, respectively (Scheme 2). Alcohols 8a and 8b were then individually treated with dibenzyl diisopropyl phosphoramidite in the presence of 1H-tetrazole to form the phosphite intermediates, which were readily oxidized by m-chloroperoxybenzoic acid (m-CPBA) to afford phosphate compounds 9a and 9b, respectively.21 Finally, debenzylation was achieved by treating the compounds with BBr322 at −40 °C to give the desired products 1a and 1b. | ||
| Scheme 2 Synthesis of valienol 1-phosphate 1a, 1-epi-valienol 1-phosphate 1b, valienol 1,7-diphosphate 2a, and 1-epi-valienol 1,7-diphosphate 2b. a) EtSH, TFA; b) DMSO, Ac2O; c) Ph3PCH3Br, t-BuOK, benzene; d) HgCl2, HgO, aq. CH3CN; e) vinylmagnesium bromide, THF, 0 °C; f) Grubbs' 2nd gen. catalyst, CH2Cl2, reflux; g) iPr2NP(OBn)2, 1H-tetrazole, CH2Cl2, m-CPBA; h) BBr3, CH2Cl2; i) ZnCl2, Ac2O, AcOH; j) NaOMe, MeOH. | ||
The synthesis of valienol 1,7-diphosphate analogues also proceeded from compounds 8a and 8b. Regioselective debenzylation of 8a and 8b using ZnCl2, Ac2O and AcOH, followed by a deacetylation step furnished diols 10a and 10b, respectively.23 The two free alcohol functionalities were then phosphorylated and the product debenzylated as described for the synthesis of 1a and 1b to afford valienol 1,7-diphosphate (2a) and 1-epi-valienol 1,7-diphosphate (2b).
Preparation of recombinant ValB and initial characterization of ValB with different valienol phosphates and NTPs
With the potential substrates for the enzyme in hand, the recombinant ValB protein was then prepared by cloning the 1.1 kb valBgene into expression vector pRSET B (Invitrogen) and the resulting plasmid was introduced into E. coli BL21 Gold(DE3)/pLysS by heat-pulse transformation. Gene expression was induced by isopropyl β-D-1-thiogalactopyranoside (IPTG) and the recombinant protein was purified through a Ni-NTA column and dialyzed against 60 mM Tris-HCl pH 8 to give >80% pure 45 kDa protein (Fig. 2A). | ||
| Fig. 2 Preparation and characterization of recombinant ValB. A, SDS PAGE Analysis of ValB (M = protein marker, W = whole cell extract, S = soluble protein, P = purified protein). B–C, Relative activity of ValB based on coupled colorimetric assays using four different valienol phosphate substrates (B) and five different NTPs (C). D–E, MS analysis of ValB using four different valienol phosphate substrates (D) and five different NTPs (E). | ||
Preliminary enzymatic reactions of ValB were carried out using the four different substrates (1a, 1b, 2a, and 2b) and all five naturally occurring NTPs (ATP, GTP, CTP, dTTP, and UTP). Analysis of the products by TLC and MS revealed that only 1a was converted to the corresponding NDP-valienols. Similar experiments with glucose 1-phosphate also gave NDP-glucose products, suggesting that ValB has somewhat relaxed substrate specificity. However, due to their instability, attempts to analyze the NDP-valienol products by HPLC or to isolate the products by chromatography were unsuccessful. We observed that during prolonged incubation and/or chromatography, the products degraded to valienol. Therefore, an alternative assay for the enzyme reaction was developed.
Coupled colorimetric assays of ValB
Because the conversion of valienol 1-phosphate and a nucleotidyl triphosphate to the corresponding NDP-valienol produces inorganic pyrophosphate (PPi), measuring the net amount of PPi in the reaction mixture provides an indirect measure of the NDP-valienol being produced. Therefore, a coupled colorimetric assay using EnzChek Pyrophosphate Assay Kit24 (Molecular Probes) was used to measure the amount of PPi in the reactions (Scheme 3). The system includes the enzymes inorganic pyrophosphatase and purine nucleoside phosphorylase (PNP), in which the former enzyme catalyzes the conversion of PPi into two equivalents of inorganic phosphate (Pi), and the latter, in the presence of Pi, converts 2-amino-6-mercapto-7-methylpurine ribonucleoside (MESG) to ribose 1-phosphate and 2-amino-6-mercapto-7-methylpurine. Enzymatic conversion of MESG results in a shift in absorbance maximum from 330 nm for the substrate to 360 nm for the product. | ||
| Scheme 3 Coupled colorimetric assays of ValB. Enzymatic conversion of MESG to ribose 1-phosphate and 2-amino-6-mecapto-7-methylpurine results in a shift in absorbance maximum from 330 nm to 360 nm. | ||
The substrate specificity of ValB was then reevaluated using this assay, in which 1a, 1b, 2a, and 2b were tested in the presence of GTP. After 1 h incubation, EnzChek solution was added and the reaction mixtures were incubated in a 96-well plate for 30 min at 22 °C. The change in absorbance at 360 nm was recorded in triplicate. Consistent with the preliminary results, the colorimetric assay and MS provided compelling evidence that 1a is the preferred substrate for ValB (Fig. 2B and 2D).
Utilization of various nucleotidyltriphosphates by ValB
BLAST analysis of the amino acid sequence of ValB revealed that besides having high sequence identities with other putative carbasugar nucleotidyltransferases, AcbR (from the acarbose pathway in Actinoplanes sp., 64%), GagR (from the acarbose pathway in Streptomyces glaucescens, 67%), and SalF (from the salbostatin pathway in Streptomyces albus, 68%), ValB is also similar to the glucose 1-phosphate adenylyltransferases (GlgC, 27–41%). The result was inconsistent with the report by Kim and co-workers, as it indicated that VldB is a uridylyltransferase enzyme.13 To identify the preferred nucleotidyl donor(s) in ValB reaction, the enzyme was incubated with valienol 1-phosphate in the presence of different NTPs. Colorimetric assay of the products indicated that ValB more efficiently utilized the purine-based nucleotidyltriphosphates, ATP and GTP, than pyrimidine-based NTPs as nucleotidyl donor (Fig. 2C). These results were unambiguously supported by strong signals for both ADP-valienol (m/z 583.93) and GDP-valienol (m/z 599.93) products in the mass spectra (Fig. 2E). Interestingly, ValB also used CTP as nucleotidyl donor, converting valienol 1-phosphate to CDP-valienol (m/z 559.93). On the other hand, only low-level activity of ValB was observed in the colorimetric assay with dTTP or UTP. However, the amounts of the products seemed to be below the MS-detectable levels.Metal ion cofactors for ValB
Most sugar 1-phosphate nucleotidyltransferases require metal ions for their activity.25,26 To assess the metal ion dependence of ValB activity, the reactions were carried out using EDTA-treated protein in the presence of various metal ions. However, light absorption at 360 nm by some metal ions precluded the use of the colorimetric assay for these experiments. Therefore, TLC analysis with detection under UV light at 254 nm (Fig. 3A) and MS (Fig. 3B) were used to analyze the reaction products. The results confirmed that metal ions are indeed essential for ValB activity, as reactions using EDTA-treated ValB alone did not give any product at a detectable level. Of eight metal ions tested, Mg2+, Mn2+, and Co2+ were able to rescue the activity of the EDTA-treated enzyme, whereas Fe2+, Cu2+, Ca2+, Zn2+, and Ni2+ were inactive (Fig. 3). The results suggest that ValB adopts a similar metal ion requirement as sugar 1-phosphate nucleotidyltransferases commonly found in biological systems. | ||
| Fig. 3 Identification of metal ion cofactors for ValB. A, TLC analysis of ValB products with different metal ions under UV light at 254 nm (M-, with no metal ion; BE, with boiled enzyme; AG, ADP-glucose standard; thick arrows show ADP–valienol products). B, MS analysis of ValB products with different metal ions. All reaction mixtures contained EDTA. | ||
Discussion
Sugar nucleotides are ubiquitous in Nature. They play critical roles in primary and secondary metabolism as substrates for many important enzymes, particularly the glycosyltransferases. They are normally derived from sugar 1-phosphates and NTPs catalyzed by sugar 1-phosphate nucleotidyltransferases, also referred to as sugar nucleotide pyrophosphorylases (EC 2.7.7.-). In bacteria and plants, sugar 1-phosphate nucleotidyltransferases are involved in the biosynthesis of starch, peptidoglycans, and lipopolysaccharides, and their participation in natural product biosynthesis has been fully appreciated. In addition, many genes encoding sugar 1-phosphate nucleotidyltransferases have been found in natural products biosynthetic gene clusters, including those of the C7N-aminocyclitol natural products, such as acarbose,11validamycin,16 and salbostatin.27 However, their roles in the biosynthesis of these compounds had not been carefully studied.Whereas VldB, a ValB homologue (99% identity) from another validamycin producing strain, was reported to serve as a glucose 1-phosphate uridylyltransferase,13 results from our in vitro and in vivo studies suggest otherwise. First, ValB, while able to recognize glucose 1-phosphate as substrate, efficiently converts valienol 1-phosphate to its corresponding nucleotidyl derivatives. Interestingly, the 1-epi- and the 1,7-diphosphate-analogues are not preferred substrates. Secondly, ValB most efficiently utilizes ATP and GTP, instead of UTP, as nucleotidyl donor. This, in fact, is consistent with the relatively high sequence identity between ValB (also VldB) and the glucose 1-phosphate adenylyltransferases (GlgC). Furthermore, inactivation of valB in S. hygroscopicus 5008 abolished the production of both validoxylamine A and validamycin A, which provided compelling evidence for the critical role of ValB in validoxylamine A biosynthesis, and not in the conversion of validoxylamine A to validamycin A. In other words, ValB is most likely responsible for the synthesis of ADP- or GDP-valienol, setting the stage for a coupling reaction leading to the bis-cyclitol core unit formation. In addition, our previous study has shown that the glycosyltransferase ValG, which catalyzes the conversion of validoxylamine A to validamycin A, most efficiently uses UDP-glucose, not ADP- or GDP-glucose, as sugar donor.16,17 Therefore, as ValB prefers ATP and GTP as nucleotidyl donor, it is less likely that this enzyme plays a critical role in supplying UDP-glucose for ValG reaction.
Viewed together, the present study not only clarifies the catalytic function and role of ValB in validamycin A biosynthesis but also provides insight into the pathway directly upstream of valienol 1-phosphate (Scheme 4). It now can be postulated that the conversion of valienone 7-phosphate, the product of the cyclitol kinase ValC,28 to valienol 1-phosphate would involve a stereoselective reduction of C-1 followed by phosphate transfer from C-7 to C-1. The latter step may be catalyzed by a phosphohexomutase, possibly ValO.
 | ||
| Scheme 4 Biosynthetic pathway to NDP-valienol. Conversion of valienone 7-phosphate to valienol 1-phosphate requires stereoselective reduction of C-1 ketone and transfer of the phosphate group. | ||
As a valienol 1-phosphate nucleotidyltransferase, ValB represents the first member of the unsaturated carbasugar nucleotidyltransferase family involved in natural product biosynthesis. Other members of this class of enzymes may include AcbR, GacR, and SalF.11,14,27 Although AcbR, GacR, and SalF have been annotated as putative 1-epi-valienol 1,7-diphosphate adenylyltransferases, their natural substrate may very likely be the same as that of ValB. Therefore, careful characterization of those enzymes is warranted to reveal their actual role in acarbose and salbostatin biosynthesis.
Conclusion
The present study provides compelling evidence for the critical role of ValB in validamycin biosynthesis. ValB catalyzes the conversion of the unsaturated carbasugar phosphate valienol 1-phosphate to NDP-valienol. This enzyme requires Mg2+, Mn2+, or Co2+ for its activity and utilizes the purine-based nucleotidyltriphosphates ATP and GTP more efficiently than the pyrimidine-based NTPs (CTP, dTTP, and UTP) as nucleotidyl donor. ValB represents the first characterized unsaturated carbasugar nucleotidyltransferase involved in natural products biosynthesis. Members of this class of enzymes may include AcbR/GacR, and SalF from the acarbose and the salbostatin pathways, respectively.11,14,27 Results of the present study not only expand our understanding of the biosynthesis of carbasugar-derived natural products, but may also facilitate the development of new tools for chemoenzymatic synthesis of carbohydrate mimetics.Experimental
General
All chemical reactions were performed under an argon or nitrogen atmosphere employing oven-dried glassware. Analytical thin-layer chromatography (TLC) was performed using silica gel plates (60 Å) with a fluorescent indicator (254 nm), which were visualized with a UV lamp and ceric ammonium molybdate (CAM) solution. Chromatographic purification of products was performed on silica gel (60 Å, 72–230 mesh) column. Optical rotations were measured on a Jasco P1010 polarimeter (100 mm cell was used) at the sodium D line. Proton NMR spectra were recorded on Bruker 300 or 400 MHz spectrometers. Proton chemical shifts are reported in ppm (δ) relative to the residual solvent signals as the internal standard (CDCl3: δH 7.26; D2O: δH 4.79). Multiplicities in the 1H NMR spectra are described as follows: s = singlet, br s = broad singlet, d = doublet, br d = broad doublet, t = triplet, q = quartet, m = multiplet; coupling constants are reported in Hz. Carbon NMR spectra were recorded on Bruker 300 (75 MHz) or 400 (100 MHz) spectrometers with complete proton decoupling. Carbon chemical shifts are reported in ppm (δ) relative to the residual solvent signals as the internal standard (CDCl3: δC 77.16), or with sodium 2,2-dimethylsilapentane-5-sulfonate (DSS) (δ 0.0) as an external standard. Phosphorus NMR spectra were recorded on a Bruker 300 (112 MHz) spectrometer with complete proton decoupling. Phosphorus chemical shifts are reported in ppm (δ) relative to an 85% H3PO4 (δ 0.0) external standard. Low-resolution electrospray ionization (ESI) mass spectra were recorded on a ThermoFinnigan liquid chromatograph-ion trap mass spectrometer, and High-resolution electrospray mass spectra were recorded on Waters/Micromass LCT spectrometer. Ion-exchange column chromatography was carried out on Dowex 1 × 8 (Aldrich) and size exclusion chromatography was done on Sephadex LH-20 (Pharmacia). Restriction enzymes, T4DNA ligase and Taq polymerase were purchased from various companies (New England Biolabs, Takara, MBI Fermentas, and Toyobo). Synthesis of oligonucleotide primers and DNA sequencing of PCR products were performed by Shanghai Sangon and Invitrogen Co., Ltd. Gel Recovery Kit (Tiangen) was used for DNA purification from agarose gels. TSBY liquid medium (per litre: TSB 30 g, yeast extract 10 g, sucrose 103 g [pH 7.2]) was used for the growth of mycelia and the isolation of total DNA. SFM medium (2% agar, 2% mannitol, 2% soybean powder [pH 7.2]) was used for sporulation and conjugation. YMG liquid medium (0.4% yeast extract, 1% malt extract, 0.4% glucose [pH 7.3]) was used for the fermentation of S. hygroscopicus 5008 and its derivatives. For Streptomyces, apramycin and thiostrepton were used at 30 μg ml−1 and 25 μg ml−1, respectively, both in SFM agar and in liquid media. E. coli strains were cultured as described elsewhere.29Bacterial strains and plasmids
Bacterial strains and plasmids used in this study are listed in Table S1.†S. hygroscopicus 5008, the wild-type producer of validamycin, was used for validamycin isolation, bioassay, and generation of mutant strains. E. coliDH10B was used as the cloning host, and E. coliBW25113 was used as the host for λ-Red-mediated recombination.30 pMD-T vector (Takara) was used for the cloning of PCR amplified fragments. pHZ1358 (an E. coli and Streptomyces shuttle vector) was used for gene replacement in 5008.31 pJTU96832 was used as a transitional cloning vector. pPM927 was used for mutant complementation.33Construction of pJTU702 for targeted replacement of a 936 bp DNA fragment internal to valB
A 5.8 kb BamHI fragment from pHZ2229 was cloned into the BamHI site of pHZ1358 to generate pJTU700. Using the gel-purified 1384-bp EcoRI/HindIII fragment from pIJ773 as template,30 a 1.4 kb disruption cassette was obtained by PCR amplification with the primers ValB-PCR-F (5′-ATGGGGCCGCTGGGACGCGGCAGGCTCAAGCCGCTGGTGattccggggatccgtcgacc-3′, nucleotides of pIJ773 are in lowercase) and ValB-PCR-R (5′- GATGGCCGACGCCAGGTGTGTTCCGTCCGGTACCTGGGCtgtaggctggagctgcttc -3′, nucleotides of pIJ773 are in lowercase). Amplification was performed with Taq DNA polymerase in a 50 μl reaction with 100 ng template DNA, 10 mM dNTPs, 50 pmol each primer, and 5% DMSO in a thermocycler (BioRad). After initial denaturation at 94 °C for 2 min, 10 cycles were performed with denaturation at 94 °C for 45 s, annealing at 50 °C for 45 s, and extension at 72 °C for 90 s, followed by 15 cycles with the annealing temperature increased to 55 °C. A last elongation step was done at 72 °C for 5 min. This fragment was used to replace the 936 bp DNA internal to valB in pJTU700 through the λ-Red-mediated recombination. The resulting plasmid pJTU702 was used for targeted replacement of a 936 bp DNA fragment internal to valB with the 1.4 kb oriT-aac(3)IV cassette in the wild-type strain 5008. Oligonucleotide primers ValB-Det-F (5′-TGCTCCACCTGCCTGTA-3′) and ValB-Det-R (5′-CACCTCTTCCCGCTCC-3′) were used for the confirmation of the valB mutant. PCR amplification was performed with Taq DNA polymerase under similar conditions. The PCR product was analyzed by gel electrophoresis and enzymatic digestion.Construction of pJTU918 for the complementation of ZYR-2 with full-length cloned valB
A 1.1-kb NdeI/EcoRI fragment from the expression plasmid pJTU707 harboring a full-length valB was ligated to the NdeI/EcoRI-digested pJTU968 to give pJTU915. Then the 1.4 kb MfeI/EcoRI fragment, containing PermE* promoter and valB, was cleaved from pJTU915 and ligated into EcoRI-digested pPM927 to give pJTU918. The plasmid was introduced into ZYR-2 through conjugation as previously described.18 The thiostrepton-resistant exconjugants were selected.HPLC analysis of validamycin A
For HPLC analysis, the strains were cultured in 50 mL YMG liquid medium in 250 mL baffled flasks at 37 °C and 220 rpm for 6 days. The fermentation broth was acidified with oxalic acid and applied to a Dowex 50 W column (25 mL) as previously reported.34 The extracts were loaded onto Agilent ZORBAX SB-C18 column (5 μm, 4.6 × 250 mm) for HPLC analysis (Waters 2690). The mobile phase (5 mM sodium phosphate buffer–methanol, 98![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, v/v) was applied with the flow rate of 0.6 mL min−1 at room temperature. The eluate was monitored at 210 nm with Waters 996 photodiode array detector and the data was analyzed with a Waters Millennium Chromatography Manager.
:2, v/v) was applied with the flow rate of 0.6 mL min−1 at room temperature. The eluate was monitored at 210 nm with Waters 996 photodiode array detector and the data was analyzed with a Waters Millennium Chromatography Manager.
Synthesis of valienol 1-phosphate, 1-epi-valienol 1-phosphate, valienol 1,7-diphosphate, and 1-epi-valienol 1,7-diphosphate
:1) yielded the title compound (963 mg, 88%); [α]23D +43.2 (c 1.0 in CHCl3); δH(300 MHz; CDCl3) 3.62 (1 H, ddd, J 2.1, 3.3, 9.8), 3.92 (1 H, d, J 13.3), 4.12 (1 H, dd, J 7.4, 9.7), 4.18 (2 H, bd, J 11.2), 4.46 (2 H, dd, J 11.9, 14.6), 4.64–4.68 (1 H, m), 4.70 (2 H, d, J 11.3), 4.83 (1 H, d, J 11.0), 4.88 (1 H, d, J 11.5), 4.96 (1 H, d, J 11.0), 4.99–5.11 (4 H, m), 5.18–5.23 (1 H, m), 5.89 (1 H, dd, J 1.1, 5.7), 7.23–7.41 (30 H, m); δC(75 MHz; CDCl3) 69.23 (d, Jc-p 4.8), 69.30 (d, Jc-p 4.8), 70.0, 71.2 (d, Jc-p 5.9), 72.4, 72.9, 74.6, 75.2, 78.3 (d, Jc-p 5.1), 79.8, 80.3, 121.3 (d, Jc-p 2.8), 127.74, 127.76, 127.79, 127.82, 127.85, 128.0, 128.31, 128.34, 128.46, 128.49, 128.55, 128.63, 136.1 (d, Jc-p 7.4), 136.2 (d, Jc-p 7.7), 138.01, 138.05, 138.5, 138.7, 143.1; m/z (ESI) 819.3073 ([M + Na]+. C49H49O8PNa requires 819.3063).
:1; 2 mL), and the reaction mixture was stirred for 2 h at room temperature. The reaction mixture was diluted with water (5 mL) and EtOAc (10 mL), and the organic layer further washed with water (5 mL) and saturated aqueous Na2CO3 solution (3 × 5 mL). The organic layer was dried over MgSO4, filtered and concentrated under reduced vacuum to give a crude sample. The crude sample was dissolved in MeOH (2.5 mL) and NaOMe (0.8 mL of 30% solution in MeOH) was added to the reaction mixture. The reaction mixture was stirred for 4 h at room temperature and quenched with water (2 mL) and saturated aqueous NH4Cl (10 mL). The reaction mixture was diluted with EtOAc (5 mL), and the organic layer further washed with water (5 mL) and brine (5 mL). The organic layer was dried over MgSO4, filtered and concentrated under reduced vacuum. Column chromatography (silica gel, Hex : EtOAc = 1:2) yielded the title compound (51 mg, 62%); [α]24D +2.3 (c 0.23 in CHCl3); δH(300 MHz; CDCl3) 1.73 (1 H, t, J 6.3), 2.58 (1 H, d, J 3.3), 3.60 (1 H, dd, J 3.9, 9.2), 4.05–4.13 (3 H, m), 4.16 (1 H, d, J 6.9), 4.30 (1 H, q, J 4.2), 4.67 (1 H, d, J 11.4), 4.69 (1 H, d, J 11.7), 4.75 (1 H, d, J 11.1), 4.80 (1 H, d, J 11.4), 4.83 (1 H, d, J 11.1), 4.92 (1 h, d, J 11.1 Hz), 5.85 (1 H, dd, J 1.5, 4.8), 7.29–7.39 (15 H, m); δC(75 MHz; CDCl3) 64.0, 65.2, 73.1, 74.2, 74.9, 79.1, 79.28, 79.33, 124.1, 127.90, 128.1, 128.2, 128.6, 128.70, 128.72, 138.1, 138.3, 138.6, 141.7; m/z (ESI) 469.1978 ([M + Na]+. C28H30O5Na requires 469.1991).
:2 and toluene–EtOAc = 3:1) the title compound was obtained (58.1 mg, 66%); [α]23D +26.0 (c 1.0 in CHCl3); δH(300 MHz; CDCl3) 3.47–3.52 (1 H, m), 3.99–4.07 (2 H, m), 4.40 (1 h, dd, J 6.3, 12.6), 4.53–4.61 (3 H, m), 4.64 (1 H, d, J 11.1), 4.77 (1 H, d, J 10.8), 4.82 (1 H, d, J 11.7), 4.91 (1 H, d, J 11.1), 4.94–5.04 (8 H, m), 5.04–5.09 (1 H, m), 5.74 (1 h, d, J 5.4), 7.20–7.39 (35 H, m); δC(75 MHz; CDCl3) 67.0 (d, Jc-p 5.3), 69.30 (d, Jc-p 5.8), 69.36 (d, Jc-p 6.1), 69.51 (d, Jc-p 5.6), 69.56 (d, Jc-p 5.5), 70.8 (d, Jc-p 6.2), 72.5, 74.7, 75.1, 78.1 (d, Jc-p 4.8), 78.8, 80.0, 122.5, 127.75, 127.79, 128.0, 128.06, 128.11, 128.3, 128.4, 128.49, 128.53, 128.58, 128.69, 128.76, 135.79, 135.88, 135.94, 136.04, 136.10, 136.20, 138.0, 138.2, 138.6, 140.5, 140.6; m/z (ESI) 989.3180 ([M + Na]+. C56H56O11P2Na requires 989.3196).
![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif)
![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif)
![[T with combining low line]](https://www.rsc.org/images/entities/char_0054_0332.gif)
![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) AGACGGAGTGCGTGCC-3′, ValB-R, 5′-ACAGCGCCACCTCTTCCCGCTC-3′) and ligated to pMD T vector (Takara) and sequenced. The confirmed gene was cut out with BamHI and EcoRI and inserted into pRSET B expression vector (Invitrogen). The valBplasmid DNA was introduced into E. coli BL21 Gold(DE3)/pLysS by heat-pulse transformation. The mixture was then plated onto LB agar plates containing ampicillin (100 μg mL−1) and chloramphenicol (25 μg mL−1), and incubated at 37 °C for overnight. Single colonies were transferred into test tubes containing LB medium with the above antibiotics and incubated on a rotary shaker (220 rpm) at 37 °C for overnight. Seed cultures in LB medium (10 mL) containing the antibiotics were inoculated with 100 μL of the test tube culture and were grown at 37 °C for overnight. The production cultures in LBBS medium (50 mL) containing the antibiotics were inoculated with seed cultures (5 mL), and were incubated at 37 °C to an OD600 of 0.60. Isopropyl β-D-1-thiogalactopyranoside (IPTG) (0.2 mM) was then added, and the incubation was continued at 28 °C to an OD600 of 1.04. The cells were harvested by centrifugation at 5000 rpm for 15 min and stored frozen at −80 °C until further use.
AGACGGAGTGCGTGCC-3′, ValB-R, 5′-ACAGCGCCACCTCTTCCCGCTC-3′) and ligated to pMD T vector (Takara) and sequenced. The confirmed gene was cut out with BamHI and EcoRI and inserted into pRSET B expression vector (Invitrogen). The valBplasmid DNA was introduced into E. coli BL21 Gold(DE3)/pLysS by heat-pulse transformation. The mixture was then plated onto LB agar plates containing ampicillin (100 μg mL−1) and chloramphenicol (25 μg mL−1), and incubated at 37 °C for overnight. Single colonies were transferred into test tubes containing LB medium with the above antibiotics and incubated on a rotary shaker (220 rpm) at 37 °C for overnight. Seed cultures in LB medium (10 mL) containing the antibiotics were inoculated with 100 μL of the test tube culture and were grown at 37 °C for overnight. The production cultures in LBBS medium (50 mL) containing the antibiotics were inoculated with seed cultures (5 mL), and were incubated at 37 °C to an OD600 of 0.60. Isopropyl β-D-1-thiogalactopyranoside (IPTG) (0.2 mM) was then added, and the incubation was continued at 28 °C to an OD600 of 1.04. The cells were harvested by centrifugation at 5000 rpm for 15 min and stored frozen at −80 °C until further use.
Enzymatic assays for electrospray mass spectroscopy and TLC analyses were carried out using ten times concentrations of substrates and metal ions. The mixtures were incubated for 1 h at 30 °C and the products were checked by TLC (254 nm) and mass spectroscopy (ESI, negative ion mode).
Acknowledgements
The project described was supported by Award Number R01AI061528 from the National Institute of Allergy and Infectious Diseases and the Herman Frasch Foundation. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or the National Institutes of Health. Work at Shanghai Jiaotong University was supported by grants from the 973 and 863 Programs of the Ministry of Science and Technology, the Natural Science Foundation of China, and Shanghai Leading Academic Discipline Project B203. HX was in part supported by the exchange program from China Scholarship Council and the U.S. National Institutes of Health.References
- P. Sears and C. H. Wong, Science, 2001, 291, 2344–2350 CrossRef CAS.
- C. R. Bertozzi and L. L. Kiessling, Science, 2001, 291, 2357–2364 CrossRef CAS.
- A. Helenius and M. Aebi, Science, 2001, 291, 2364–2369 CrossRef CAS.
- P. M. Rudd, T. Elliott, P. Cresswell, I. A. Wilson and R. A. Dwek, Science, 2001, 291, 2370–2376 CrossRef.
- D. H. Dube and C. R. Bertozzi, Nat. Rev. Drug Discovery, 2005, 4, 477–488 CrossRef CAS.
- S. A. Gruner, E. Locardi, E. Lohof and H. Kessler, Chem. Rev., 2002, 102, 491–514 CrossRef CAS.
- T. Mahmud, Nat. Prod. Rep., 2003, 20, 137–166 RSC.
- T. Mahmud, Curr. Opin. Chem. Biol., 2009, 13, 161–170 CrossRef CAS.
- P. M. Flatt and T. Mahmud, Nat. Prod. Rep., 2007, 24, 358–392 RSC.
- T. Mahmud, S. Lee and H. G. Floss, Chem. Rec., 2001, 1, 300–310 CrossRef CAS.
- U. F. Wehmeier and W. Piepersberg, Appl. Microbiol. Biotechnol., 2004, 63, 613–625 CrossRef CAS.
- D. Singh, M. J. Seo, H. J. Kwon, A. Rajkarnikar, K. R. Kim, S. O. Kim and J. W. Suh, Gene, 2006, 376, 13–23 CrossRef CAS.
- M. J. Seo, E. M. Im, D. Singh, A. Rajkarnikar, H. J. Kwon, C. G. Hyun, J. W. Suh, Y. R. Pyun and S. O. Kim, J. Microbiol. Biotechnol., 2006, 16, 1311–1315 CAS.
- Y. Rockser and U. F. Wehmeier, J. Biotechnol., 2009, 140, 114–123 CrossRef CAS.
- K. S. Ko, C. J. Zea and N. L. Pohl, J. Am. Chem. Soc., 2004, 126, 13188–13189 CrossRef CAS.
- L. Bai, L. Li, H. Xu, K. Minagawa, Y. Yu, Y. Zhang, X. Zhou, H. G. Floss, T. Mahmud and Z. Deng, Chem. Biol., 2006, 13, 387–397 CrossRef CAS.
- H. Xu, K. Minagawa, L. Bai, Z. Deng and T. Mahmud, J. Nat. Prod., 2008, 71, 1233–1236 CrossRef.
- Y. Yu, L. Bai, K. Minagawa, X. Jian, L. Li, J. Li, S. Chen, E. Cao, T. Mahmud, H. G. Floss, X. Zhou and Z. Deng, Appl. Environ. Microbiol., 2005, 71, 5066–5076 CrossRef CAS.
- Y. K. Chang, B. Y. Lee, D. J. Kim, G. S. Lee, H. B. Jeon and K. S. Kim, J. Org. Chem., 2005, 70, 3299–3302 CrossRef CAS.
- M. Scholl, S. Ding, C. W. Lee and R. H. Grubbs, Org. Lett., 1999, 1, 953–956 CrossRef CAS.
- K.-L. Yu and B. Fraser-Reid, Tetrahedron Lett., 1988, 29, 979–982 CrossRef CAS.
- T. Mahmud, I. Tornus, E. Egelkrout, E. Wolf, C. Uy, H. G. Floss and S. Lee, J. Am. Chem. Soc., 1999, 121, 6973–6983 CrossRef CAS.
- G. Yang, X. Ding and F. Kong, Tetrahedron Lett., 1997, 38, 6725–6728 CrossRef CAS.
- R. H. Upson, R. P. Haugland, M. N. Malekzadeh and R. P. Haugland, Anal. Biochem., 1996, 243, 41–45 CrossRef CAS.
- C. M. Bejar, X. Jin, M. A. Ballicora and J. Preiss, J. Biol. Chem., 2006, 281, 40473–40484 CrossRef CAS.
- M. Morar, K. Bhullar, D. W. Hughes, M. Junop and G. D. Wright, Structure, 2009, 17, 1649–1659 CrossRef CAS.
- W. S. Choi, X. Wu, Y. H. Choeng, T. Mahmud, B. C. Jeong, S. H. Lee, Y. K. Chang, C. J. Kim and S. K. Hong, Appl. Microbiol. Biotechnol., 2008, 80, 637–645 CrossRef CAS.
- K. Minagawa, Y. Zhang, T. Ito, L. Bai, Z. Deng and T. Mahmud, ChemBioChem, 2007, 8, 632–641 CrossRef CAS.
- J. Sambrook, E. F. Fritsch and T. Maniatis, Molecular Cloning: a Laboratory Manual, 2nd edn, Cold Spring Harbor, Cold Spring Harbor, NY, 1989 Search PubMed.
- B. Gust, G. L. Challis, K. Fowler, T. Kieser and K. F. Chater, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 1541–1546 CrossRef CAS.
- Y. Sun, X. Zhou, J. Liu, K. Bao, G. Zhang, G. Tu, T. Kieser and Z. Deng, Microbiology, 2002, 148, 361–371 CAS.
- H. Xu, Y. Zhang, J. Yang, T. Mahmud, L. Bai and Z. Deng, Chem. Biol., 2009, 16, 567–576 CrossRef CAS.
- T. Smokvina, P. Mazodier, F. Boccard, C. J. Thompson and M. Guerineau, Gene, 1990, 94, 53–59 CrossRef CAS.
- H. Dong, T. Mahmud, I. Tornus, S. Lee and H. G. Floss, J. Am. Chem. Soc., 2001, 123, 2733–2742 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Strains used in the study; NMR spectra. See DOI: 10.1039/c0ob00475h |
| This journal is © The Royal Society of Chemistry 2011 |