Synthesis and NMR spectroscopic analysis of acylated pentasaccharide fragments of mycobacterial arabinogalactan†
Chunjuan
Liu
,
Michele R.
Richards
and
Todd L.
Lowary
*
Alberta Ingenuity Centre for Carbohydrate Science and Department of Chemistry, The University of Alberta, Gunning-Lemieux Chemistry Centre, Edmonton, Alberta, Canada. E-mail: tlowary@ualberta.ca; Fax: 780-492-7705; Tel: 780-492-1861
First published on 21st October 2010
Abstract
The mycolyl–arabinogalactan (mAG) complex, a large glycolipid composed of arabinofuranose and galactofuranose monosaccharides and mycolic acid lipids, provides mycobacteria with substantial protection from their environment. It has been proposed that the presence of flexible furanose rings in the mAG facilitates the packing of the hydrophobic mycolic acids, forming a dense protective barrier of low permeability. In a previous article, we probed this “flexible scaffold hypothesis” through the synthesis and NMR analysis of di- and trisaccharide fragments of the mAG acylated with linear fatty acids. However, we saw few conformational changes due to the presence of the acyl chains. We proposed that branched pentasaccharide glycolipids 5–8 might exhibit larger changes due to the presence of more acyl chains, and studies with these compounds are described here. The carbohydrate portion of 5–8 was synthesized in a 1 + 2 + 2 manner. First, the monosaccharide diol was treated with an excess of appropriately protected thioglycoside donor to give a trisaccharide, which, following selective deprotection to a diol, was converted to the pentasaccharide in a one-pot glycosylation. The resulting differentially protected pentasaccharide 20 gave glycolipids 5–8 upon removal of the protecting groups at the primary positions, acylation, and hydrogenolysis. The conformations of 5–8 were probed using NMR spectroscopy, and chemical shift selective filtering 1D-TOCSY spectra allowed for the determination of all the ring coupling constants. It was found that the addition of four fatty acids to the parent pentasaccharide had little effect on the conformation of the compounds in solution.
Introduction
Mycobacterial diseases, including tuberculosis and leprosy, remain important health concerns given their widespread occurrence worldwide1 and the emergence of strains resistant to front-line antibiotics used to treat these infections.2 Mycobacteria possess an unusual, densely packed, and impermeable cell wall structure that both prevents the passage of antibiotics into the organism and allows it to evade the immune system of the infected host.3 The largest structural component of the cell wall is the mycolyl–arabinogalactan (mAG) complex, which is composed of a polysaccharide esterified at the non-reducing terminus with long chain lipids termed mycolic acids (vide infra).4 The dense packing of the mycolic acids at the outer region of the cell wall complex leads to the impermeability of the structure.In a previous paper,5 we reported the synthesis and conformational analysis of di- and trisaccharide arabinofuranose-containing glycolipids (e.g., 1 and 2, Fig. 1), which were designed as model compounds for portions of the nonreducing end of the mAG complex (3, Fig. 2).3,4 Motivated by the unusual structure of this glycan, which is composed nearly entirely of furanose rings, this study was undertaken to probe the effect of acylation on the conformation of the arabinofuranose rings in these glycolipids. It has been proposed that the inherent flexibility of the furanoside rings in the structure facilitates tight packing of the mycolic acids of mAG leading to the impermeable barrier that protects the organism from its environment.6,7 Testing of this “flexible scaffold hypothesis” is important as it may lead to an understanding of the factors that stabilize the mycobacterial cell wall macrostructure. Such knowledge may lead to improved therapies for the treatment of mycobacterial diseases through identification of key residues in the structures whose biosynthesis could be targeted via novel inhibitors. We postulated that if this hypothesis is correct, then the conformations of oligosaccharide fragments of the mAG lacking acylation should differ from those in which this modification is present.
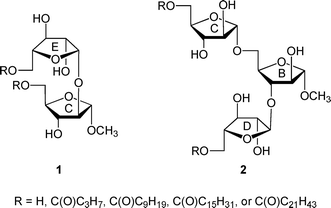 | ||
| Fig. 1 Model di- and trisaccharide glycolipids 1 and 2. | ||
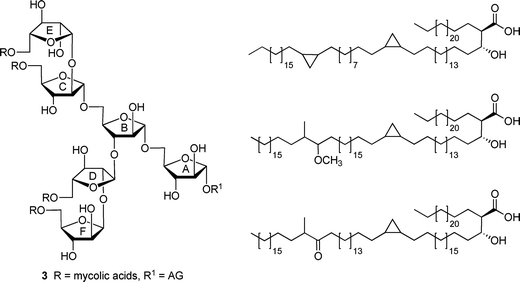 | ||
| Fig. 2 (left) Hexasaccharide motif (3) found at the non-reducing end of the mycolyl–arabinogalactan (mAG) complex. (right) Examples of mycolic acids found in Mycobacterium tuberculosis. | ||
The results of the investigations with these di- and trisaccharides indicated that acylation with linear, long-chain fatty acids did not substantially influence the conformation of the five-membered rings in these structures nor rotation about the C4–C5 bond, which is immediately adjacent to the site of acylation. In addition, on the basis of 1H and 13C NMR chemical shift data, acylation did not lead to significant differences in the conformation about the glycosidic linkages in 1 and 2.5 These results led us to postulate that a more pronounced effect may be observed in glycolipids with structures more similar to the non-reducing terminus of the mAG; i.e., those that possess a larger number of monosaccharide residues and acyl chains, or those that are functionalized with branched acyl groups that more closely resemble the mycolic acids. In this paper, we extend our investigations to a series of pentasaccharide glycolipids (Fig. 3) that are models of the terminal branched structure of mAG (3). These investigations, when coupled to those with more complicated analogues (e.g., those with lipids incorporating β-hydroxy groups and branching) will allow us to address, in a systematic fashion, the effect of various lipid structural motifs on carbohydrate conformation in these glycolipids. We describe here the synthesis of target acylated pentasaccharides 5–8 and conformational analysis of these compounds.
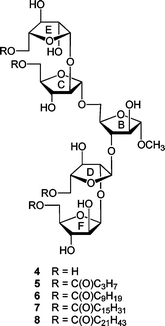 | ||
| Fig. 3 Compounds 4–8 are the target pentasaccharides with the rings labeled to facilitate comparison with 3. | ||
Results and discussion
Preparation of pentasaccharides 4 and 9
The unacylated parent pentasaccharide (4) was synthesized as described previously.8 The route used for the synthesis of 5–8 required a different protecting group strategy to allow for the installation of the acyl chains. For this purpose, we chose as the key intermediate pentasaccharide 9 (Scheme 1), which could be acylated and then debenzylated leading to the target glycolipids. Shown in Scheme 1 is the retrosynthetic analysis of 9. We envisioned a convergent strategy in which the key steps would be two glycosylation reactions of mono- or trisaccharide diols with an excess of a suitably protected donors. Thus, treatment of 139 with an excess of 12 and subsequent debenzoylation would give trisaccharide diol 11, which could be converted by coupling with an excess of 1010,11 and removal of the p-methoxybenzyl ethers. As described earlier for the preparation of acylated disaccharides such as 1,5 the key issue in the preparation of 9 was the stereoselective introduction of the two β-arabinofuranoside motifs. The use of 10 follows our earlier work in which the O-5 protecting group was shown to influence the β-selectivity in the glycosylations leading to disaccharide 1.5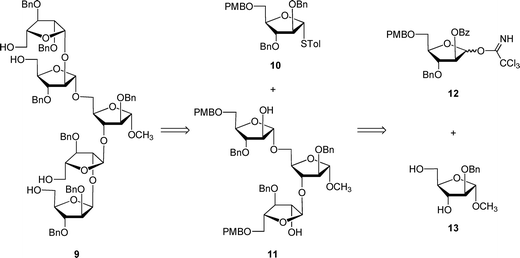 | ||
| Scheme 1 Retrosynthesis of protected pentasaccharide 9. | ||
The preparation of trichloroacetimidate 12 is shown in Scheme 2. First, the known crystalline 1,2,5-orthoester 1512 was prepared in four steps13,14 from the easily accessed methyl glycoside 14.15 Subsequent benzylation and ring-opening by treatment with allyl alcohol afforded intermediate 16 in 72% over six steps from 14. Installation of the PMB ether at the O-5 position of 16 was achieved by reaction with the p-methoxybenzyl trichloroacetimidate and lanthanum triflate16 to give fully protected 17. These mild conditions allowed the protection of the alcohol in the presence of the base-sensitive benzoate ester. Deprotection of the crude allyl ether with palladium chloride and methanol gave the reducing sugar 18 in a 1.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 α:β ratio in 84% yield over two steps, which was converted to trichloroacetimidate 12in situ. One of the attractive features of this route is that many of the intermediates do not need extensive purification; only two of the compounds – 16 and 18 – were purified by chromatography. Next, the target pentasaccharide 9 could be assembled by coupling the building blocks together as illustrated in Scheme 3. After generating trichloroacetimidate 12in situ, the donor was immediately reacted with diol 139 and TMSOTf at 0 °C, which gave an 84% yield of trisaccharide 19. Conversion of 19 into the diol 11 was achieved under the usual sodium methoxide conditions, which provided the product in 82% yield. With 11 in hand, it was coupled with excess donor 1010,11 to form the pentasaccharide in 86% yield. As expected, the product was a mixture of stereoisomers, in which the ratio of desired isomer to the others was 5.9:1. Although the desired product was the major one, separation by chromatography was difficult. We also tried other donors; however, none improved stereoselectivity or purification. In particular, the use of thioglycosides with a benzoate ester, TMDMS ether, or allyl ether at O-5 gave poorer β-selectivity than the 5-O-PMB ether donor 10.5 Analogous protecting group effects have been observed in other studies on stereoselective arabinofuranosylation.11,17
:1 α:β ratio in 84% yield over two steps, which was converted to trichloroacetimidate 12in situ. One of the attractive features of this route is that many of the intermediates do not need extensive purification; only two of the compounds – 16 and 18 – were purified by chromatography. Next, the target pentasaccharide 9 could be assembled by coupling the building blocks together as illustrated in Scheme 3. After generating trichloroacetimidate 12in situ, the donor was immediately reacted with diol 139 and TMSOTf at 0 °C, which gave an 84% yield of trisaccharide 19. Conversion of 19 into the diol 11 was achieved under the usual sodium methoxide conditions, which provided the product in 82% yield. With 11 in hand, it was coupled with excess donor 1010,11 to form the pentasaccharide in 86% yield. As expected, the product was a mixture of stereoisomers, in which the ratio of desired isomer to the others was 5.9:1. Although the desired product was the major one, separation by chromatography was difficult. We also tried other donors; however, none improved stereoselectivity or purification. In particular, the use of thioglycosides with a benzoate ester, TMDMS ether, or allyl ether at O-5 gave poorer β-selectivity than the 5-O-PMB ether donor 10.5 Analogous protecting group effects have been observed in other studies on stereoselective arabinofuranosylation.11,17
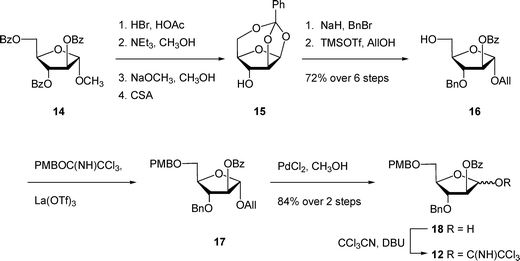 | ||
| Scheme 2 Synthesis of trichloroacetimidate 12. | ||
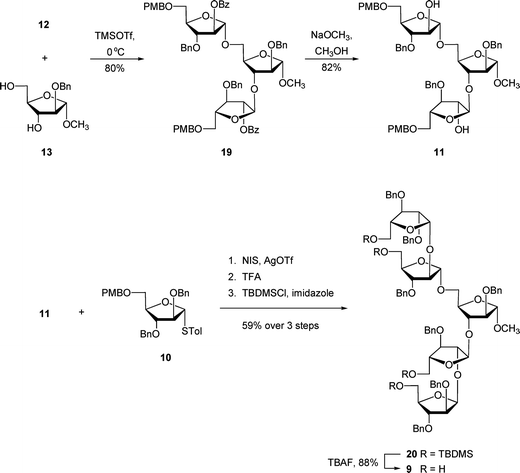 | ||
| Scheme 3 Synthesis of protected pentasaccharide 9. | ||
We postulated that the manipulation of the protecting groups at the four primary positions from PMB ethers to TBDMS would be helpful for the separation. Hence, installation of the TBDMS ethers proceeded in 59% yield over three steps including the glycosylation, treatment with TFA to remove the PMB ethers, and then protection as silyl ethers. At this stage, the desired isomer 20 could be readily separated from the other isomers. The removal of TBDMS ethers was done by treatment of TBAF in THF, which provided 9 in 88% yield.
A combination of 1H and 13C NMR spectroscopy was used to establish the structure of 9. The 1H NMR spectrum indicated the presence of two β-arabinofuranosyl anomeric hydrogens at 5.04 ppm (doublet, J = 4.6 Hz) and 4.97 ppm (doublet, J = 4.6 Hz) that were correlated (HMQC) to the resonances in 13C NMR spectrum at 100.4 and 100.1 ppm, respectively. In addition, three signals of the α-linked residues appeared at 5.10 ppm (doublet, J = 1.4 Hz), 5.07 ppm (apparent singlet), and 4.91 ppm (doublet, J = 1.5 Hz), which could be correlated to three resonances in the 13C NMR spectrum at 106.0, 105.9, 106.8 ppm, respectively.
Synthesis of target glycolipids 5–8
As was reported for the di- and trisaccharide analogs,5 we chose fatty acids or fatty acyl chlorides that were commercially available for our initial acylation reactions. Butyryl chloride, decanoyl chloride, palmitoyl chloride, and behenic acid (due to the lack of commercial availability of behenoyl chloride) were used in the esterification reaction. Each member of this homologous series differs from the next by six carbon atoms.The coupling reactions were carried out by treatment of tetraol 9 with an acid chloride and pyridine, or by reaction with behenic acid in the presence of N,N′-dicyclohexylcarbodiimide (DCC). Acylated compounds 21–24 were deprotected using palladium hydroxide on carbon under a hydrogen atmosphere to give the target compounds 5–8 in 25%–75% yield over two steps, as shown in Scheme 4. In these hydrogenation reactions a 1:1 mixture of dichloromethane and methanol was used as the solvent system, and the concentration was kept low (below 0.01 M) to minimize partially deprotected intermediates from forming micelles. We envisioned that the formation of such structures would hinder complete cleavage of the benzyl ethers. In the case of the preparation of 8, the yield of the hydrogenolysis was nevertheless low, which may be due to the adsorption of the amphiphilic compound onto the carbon-supported catalyst.
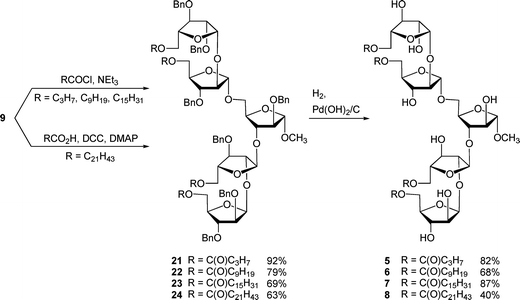 | ||
| Scheme 4 Synthesis of target glycolipids 5–8. | ||
NMR analysis for the measurement of coupling constants.
Because the acylated compounds 5–8 were not soluble in water or water–methanol mixtures, NMR spectra of 4–8 were acquired using a solution of target compound in CD3OD or 1:1 CDCl3–CD3OD, the choice being dictated by solubility of the compound. The assignment process was similar to that described for the related di- and trisaccharide derivatives,5 using the “structural reporter group” concept.18 The α- and β-arabinofuranosides have distinct and well-resolved signals in the anomeric region of the 1H NMR spectrum, and the stereochemistry of glycosidic linkages can easily be determined from the 3JH-1,H-2 magnitude. For the α-isomer, 3JH-1,H-2 = 0–2 Hz; whereas for β-isomer, 3JH-1,H-2 = 3–5 Hz.19
To overcome the spectral overlap of the ring protons in the pentasaccharides, the variable time version of the gradient-enhanced chemical shift selective filtering (ge-CSSF) technique8,20,21 was used; 1D-ge-CSSF-TOCSY and 1D-ge-CSSF-DPFGSE-NOESY spectra were used to provide coupling constants and ring assignments, respectively. In addition, to confirm the magnitudes of all coupling constants, all of the 1D 1H NMR spectra were simulated using the program WinDNMR22 to extract accurate 2JH,H, 3JH,H, or 4JH,H values. An example of the 1D-ge-CSSF-TOCSY is shown for 5 in Fig. 4, while the remainder can be found in the Supporting Information, together with representative examples of the 1D-ge-CSSF-DPFGSE-NOESY spectra. Additional details for obtaining the CSSF spectra and analyzing the data be found in the Supporting Information. The coupling constants for the target glycolipids are shown in Table 1, and their 1H and 13C NMR chemical shifts can also be found in the Supporting Information. Assignment of H-5R and H-5S was based on comparison with previous literature,23,24 and 2JH-5R,H-5S was assumed to be negative.25 No broadening of signals was observed at the concentrations used for the NMR measurements, suggesting that the compounds are not aggregating.
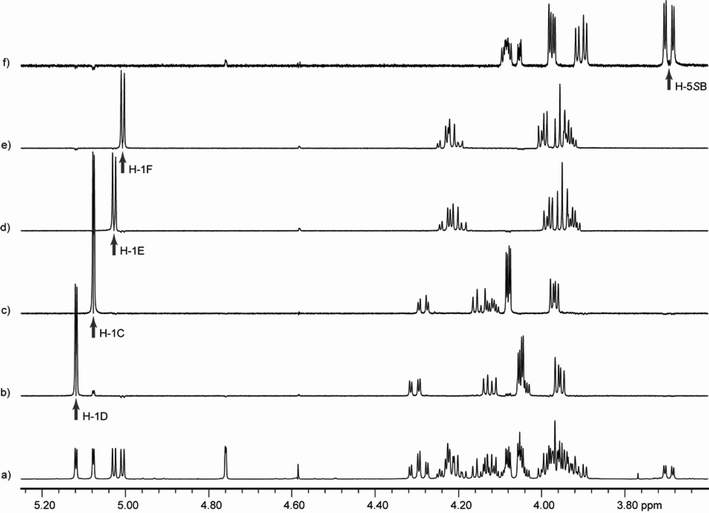 | ||
| Fig. 4 Chemical shift selective filtering TOCSY spectra for 5. The spectra were recorded at 300 K on a 600 MHz spectrometer with 1:1 CD3OD–CDCl3 as a solvent. Spectrum a is an expansion of the 1D 1H spectrum; b through f show 1D-ge-CSSF-TOCSY20,21 spectra of each ring with an arrow indicating the irradiation frequency. The mixing time (mix) and frequency difference (Δ) between the irradiated peak and its nearest neighbor that were used to each spectrum are also indicated: b) ring D, selectively irradiated at 5.12 ppm, mix = 0.2 s, Δ = 3.1 Hz; c) ring C, selectively irradiated at 5.08 ppm, mix = 0.2 s, Δ = 11.6 Hz; d) ring E, selectively irradiated at 5.03 ppm, mix = 0.16 s, Δ = 3.3 Hz; e) ring F, selectively irradiated at 5.01 ppm, mix = 0.16 s, Δ = 3.3 Hz; f) ring B, selectively irradiated at 3.69 ppm, mix = 0.12 s, Δ = 1.4 Hz. | ||
| Ring | Coupling | 4 b | 5 b | 5 c | 6 c | 7 c | 8 c |
|---|---|---|---|---|---|---|---|
|
a Couplings derived from 1D-ge-CSSF-TOCSY20,21 spectra and simulations in WinDNMR.22
b CD3OD as solvent.
c 1:1 CD3OD–CDCl3 as solvent.
|
|||||||
| B | 3 J H-1,H-2 | 1.5 | 1.5 | 1.5 | 1.6 | 1.5 | 1.5 |
| 4 J H-1,H-3 | 0.6 | 0.6 | 0.5 | 0.7 | 0.6 | 0.6 | |
| 3 J H-2,H-3 | 3.0 | 3.1 | 2.9 | 2.8 | 2.9 | 3.0 | |
| 3 J H-3,H-4 | 6.2 | 6.2 | 6.0 | 6.0 | 6.0 | 6.2 | |
| 3 J H-4,H-5R | 5.0 | 4.8 | 4.7 | 4.6 | 4.4 | 4.4 | |
| 3 J H-4,H5S | 3.1 | 3.0 | 2.8 | 2.9 | 2.9 | 2.8 | |
| 2 J H-5R,H5S | −11.3 | −11.3 | −11.3 | −11.4 | −11.4 | −11.5 | |
| C | 3 J H-1,H-2 | 2.2 | 1.9 | 2.2 | 2.0 | 2.0 | 2.0 |
| 4 J H-1,H-3 | 0.0 | 0.5 | 0.0 | 0.0 | 0.0 | 0.0 | |
| 3 J H-2,H-3 | 4.9 | 4.4 | 4.4 | 4.4 | 4.4 | 4.2 | |
| 3 J H-3,H-4 | 7.8 | 7.3 | 6.8 | 6.8 | 6.8 | 6.6 | |
| 3 J H-4,H5R | 2.7 | 3.3 | 3.1 | 3.1 | 3.2 | 3.2 | |
| 3 J H-4,H-5S | 5.0 | 6.0 | 6.1 | 6.2 | 6.2 | 6.3 | |
| 2 J H-5R,H5S | −12.2 | −11.8 | −11.6 | −11.5 | −11.5 | −11.4 | |
| D | 3 J H-1,H-2 | 2.4 | 2.1 | 2.4 | 2.5 | 2.4 | 2.4 |
| 4 J H-1,H-3 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | |
| 3 J H-2,H-3 | 5.2 | 4.5 | 5.0 | 5.1 | 5.0 | 5.0 | |
| 3 J H-3,H-4 | 7.9 | 7.8 | 7.8 | 7.9 | 7.9 | 7.8 | |
| 3 J H-4,H5R | 2.8 | 3.0 | 3.1 | 3.2 | 3.2 | 3.3 | |
| 3 J H-4,H-5S | 5.1 | 5.7 | 6.1 | 5.9 | 5.9 | 5.9 | |
| 2 J H-5R,H5S | −12.3 | −12.0 | −11.9 | −12.0 | −12.0 | −12.0 | |
| E | 3 J H-1,H-2 | 4.4 | 4.2 | 4.5 | 4.5 | 4.9 | 4.5 |
| 4 J H-1,H-3 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | |
| 3 J H-2,H-3 | 7.9 | 8.2 | 7.6 | 8.1 | 8.2 | 7.7 | |
| 3 J H-3,H-4 | 6.5 | 7.2 | 6.9 | 7.2 | 7.2 | 6.9 | |
| 3 J H-4,H5R | 3.2 | 4.0 | 3.1 | 3.4 | 3.4 | 3.1 | |
| 3 J H-4,H-5S | 6.4 | 7.2 | 7.2 | 7.2 | 7.2 | 7.2 | |
| 2 J H-5R,H5S | −11.9 | −1.7 | −11.7 | −11.8 | −11.7 | −11.8 | |
| F | 3 J H-1,H-2 | 4.4 | 4.2 | 4.5 | 4.6 | 4.7 | 4.6 |
| 4 J H-1,H-3 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | |
| 3 J H-2,H-3 | 8.0 | 8.3 | 7.7 | 8.2 | 8.3 | 7.9 | |
| 3 J H-3,H-4 | 6.6 | 7.3 | 6.9 | 6.8 | 6.8 | 6.9 | |
| 3 J H-4,H5R | 3.2 | 3.8 | 3.2 | 3.3 | 3.3 | 3.2 | |
| 3 J H-4,H-5S | 6.4 | 7.4 | 7.4 | 7.3 | 7.3 | 7.4 | |
| 2 J H-5R,H5S | −12.0 | −11.7 | −1.7 | −11.7 | −11.7 | −11.9 | |
As can be seen from the data presented in Table 1, the values for 3JH-1,H-2, 4JH-1,H-3, 3JH-2,H-3, and 3JH-3,H-4 did not change significantly upon acylation of O-5, indicating that addition of a lipid chain had little effect on the ring conformation, a finding that was also observed with the smaller di- and trisaccharide fragments. In addition, all of the coupling constants for 5 were minimally affected by changing the solvent from CD3OD to 1:1 CD3OD–CDCl3, clearly demonstrating that the solvent did not affect ring conformation. The NOE signals for 4 and 5 are also not significantly different from each other or from the hexasaccharide 3 taken in D2O,8 indicating the conformation about the glycosidic linkages is not affected by solvent or by the presence of O-5 acyl groups. For rings C, D, E, and F, values for 3JH-4,H-5R and 3JH-4,H-5S appeared to change more significantly upon acylation, thus we analyzed the rotamer populations about the C-4–C-5 bond (Fig. 5), as reported previously for the acylated di- and tri-saccharides. Table 2 shows the rotamer populations for rings C, D, E, and F in 4–8, and full details of this analysis can be found in the Supporting Information.
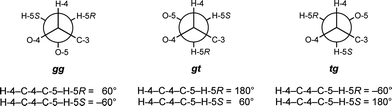 | ||
| Fig. 5 Rotamers about the C-4–C-5 bond for pentasaccharides 5–8. | ||
| Ring | Rotamer | 4 a | 5 a | 5 b | 6 b | 7 b | 8 b |
|---|---|---|---|---|---|---|---|
|
a CD3OD as solvent.
b 1:1 CD3OD–CDCl3 as solvent.
|
|||||||
| C | gg | 53% | 53% | 54% | 52% | 51% | 49% |
| gt | 47% | 43% | 44% | 45% | 45% | 46% | |
| tg | 0% | 4% | 2% | 3% | 4% | 5% | |
| D | gg | 51% | 64% | 54% | 57% | 57% | 55% |
| gt | 48% | 39% | 44% | 41% | 41% | 41% | |
| tg | 1% | −3% | 2% | 2% | 2% | 4% | |
| E | gg | 32% | 18% | 33% | 28% | 28% | 33% |
| gt | 61% | 57% | 57% | 57% | 57% | 57% | |
| tg | 7% | 25% | 10% | 15% | 15% | 10% | |
| F | gg | 32% | 17% | 27% | 28% | 28% | 27% |
| gt | 61% | 60% | 60% | 59% | 59% | 60% | |
| tg | 7% | 23% | 13% | 14% | 14% | 13% | |
For the α-linked residues of the unacylated pentasaccharide 4, rings C and D, both rotamers with O-4 and O-5 gauche to one another are about equally populated to the exclusion of the rotamer with these atoms trans to each other: 53% gg: 47% gt: 0% tg for ring C and 51% gg: 48% gt: 1% tg for ring D. These results are consistent with the gauche effect,26 which would be expected to favor the gt and gg rotamers about this bond. Acylation has little effect on the conformation of the C-4–C-5 bond for rings C and D of 5–8. For these compounds, the average populations for ring C are 52 ± 2% gg: 44 ± 1% gt: 4 ± 1% tg, and for ring D are 57 ± 3% gg: 41 ± 2% gt: 1 ± 2% tg.
For the β-linked rings of 4, residues E and F, the C-4–C-5 rotamer populations are identical, and the gt rotamer is more populated than the gg rotamer: 32% gg: 61% gt: 7% tg. Unlike the α-linked rings, acylation at O-5 seems to have an effect on the conformation; the gg and gt populations both decrease, while the tg population doubles. For compounds 5–8, the average rotamer distributions for ring E is 28 ± 6% gg: 57 ± 0% gt: 15 ± 6% tg, and the averages for ring F are 25 ± 4% gg: 60 ± 1% gt: 15 ± 4% tg. The increase in the tg conformation is somewhat surprising, but upon closer inspection, it appears that this rotamer may allow for a hydrogen bond to form from the hydroxyl at C-3 to the carbonyl oxygen, which had also been observed in the di- and trisaccharide.5
Conclusions
In conclusion, we have synthesized the differentially protected, branched pentasaccharide 20 from the non-reducing end of mycobacterial mAG complex, which allowed for selective functionalization at the O-5 positions. Highlights of the synthesis included the preparation of a trichloroacetimidate arabinofuranose donor 12 that required minimal purification. The use of this donor allowed the α-linkages to be synthesized in a one-pot protocol in 84% yield and excellent selectivity. Both β-(1→2)-linkages were also synthesized in one pot in high yield and good selectivity through the use of a thioglycoside donor (10) with a PMB group on O-5.The proton–proton coupling constants of the target glycolipids were determined through the use of chemical shift selective filtering 1D-TOCSY spectra and the aid of simulations. Acylation at O-5 did not alter the ring conformation but did change the rotamer distribution for the β-linked rings E and F, possibly to accommodate an intramolecular hydrogen bond between the carbonyl group of acyl group and the hydroxyl group at C-3. When these results are compared to those reported for the analogous di- and tri-saccharide structures, it appears that the presence of a linear acyl chain does not substantially influence the conformation of the molecule. We conclude that lipids with branched acyl chains may have more of an effect on ring conformation than the linear lipid chains we have chosen to study to date.
Experimental
General procedure for hydrogenolysis leading to 5–8
To a solution of starting material (0.01 mmol) in CH2Cl2 (1 mL) and CH3OH (1 mL), 20% Pd(OH)2–C (0.15–0.20 fold by weight) was added at room temperature. The reaction was stirred under a positive pressure of hydrogen for 12 h. The resulting mixture was filtered through Celite and concentrated. The crude residue was purified on Iatrobeads with CH2Cl2–CH3OH as the eluent.General procedure for acylation using carboxylic acid chlorides leading to 21–23
To a solution of the alcohol (0.06 mmol) in CH2Cl2 (2 mL) and pyridine (0.05 mL), the carboxylic acid chloride (0.15 mmol) was added at 0 °C. The reaction was kept stirring for 2 h and quenched by the addition of CH3OH. The resulting solution was concentrated and the residue was purified by chromatography (4:1 hexanes–EtOAc) to obtain the acylated compound.
Methyl 5-O-butyryl-β-D-arabinofuranosyl-(1→2)-5-O-butyryl-α-D-arabinofuranosyl-(1→3)-[5-O-butyryl-β-D-arabinofuranosyl-(1→2)-5-O-butyryl-α-D-arabinofuranosyl-(1→5)]-α-D-arabinofuranoside (5)
Isolated 82% as a colorless oil: Rf 0.13 (10:1 CH2Cl2–CH3OH); [α]D +33.1 (c 0.9, CHCl3);1H NMR (500 MHz, CDCl3–CD3OD, 1:1, δH) 5.12 (d, 1H, J1D,2D = 2.4 Hz, H-1D), 5.07 (d, 1 H, J1C,2C = 1.9 Hz, H-1C), 5.03 (d, 1 H, J1F,2F = 4.4 Hz, H-1F), 5.00 (d, 1 H, J1E,2E = 4.4 Hz, H-1E), 4.76 (d, 1 H, J1B,2B = 1.2 Hz, H-1B), 4.33–4.26 (m, 2 H, H-5Ea, H-5Fa), 4.26–4.02 (m, 12 H, H-5Fb, H-5Eb, H-2B, H-2C, H-2D, H-5Da, H-5Ca, H-5Db, H-5Cb, H-3F, H-3E, H-4B), 4.02–3.88 (m, 10 H, H-2F, H-2E, H-3D, H-3C, H-3B, H-4D, H-4C, H-4E, H-4F, H-5Ba), 3.70 (dd, 1 H, J4B,5Bb = 3.0 Hz, J5Ba,5Bb = 11.4 Hz, H-5Bb), 3.27 (s, 3 H, OCH3), 2.34–2.30 (m, 8 H, acyl CH2 × 4), 1.67–1.58 (m, 8 H, acyl CH2 × 4), 0.96–0.90 (m, 12 H, acyl CH3 × 4); 13C NMR (125 MHz, CDCl3, δC) 175.2 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O × 2), 175.1(1) (CO), 175.0(0) (CO), 110.5 (C-1B), 107.6 (C-1C), 107.2 (C-1D), 102.9 (C-1E), 102.8 (C-1F), 89.7 (C-2C), 89.4 (C-2D), 84.6 (C-3B), 82.6 (C-2B), 82.0 (C-4B), 81.6 (C-3E), 81.5 (C-3F), 81.4(4) (C-4D), 81.3(8) (C-4C), 78.6 (C-4E, C-4F), 77.4 (C-2E), 77.2 (C-2F), 76.8(1) (C-3D), 76.7(6) (C-3C), 67.9 (C-5B), 66.9 (C-5E, C-5F), 64.7 (C-5D), 64.6 (C-5C), 55.3 (OCH3), 36.8(4) (acyl CH2), 36.8(3) (acyl CH2), 19.4(5) (acyl CH2), 19.4(1) (acyl CH2), 14.0(3) (acyl CH3), 13.9(8) (acyl CH3). ESIMS: m/z calcd for [C42H68O25Na]+: 995.3942. Found: 995.3957.
O × 2), 175.1(1) (CO), 175.0(0) (CO), 110.5 (C-1B), 107.6 (C-1C), 107.2 (C-1D), 102.9 (C-1E), 102.8 (C-1F), 89.7 (C-2C), 89.4 (C-2D), 84.6 (C-3B), 82.6 (C-2B), 82.0 (C-4B), 81.6 (C-3E), 81.5 (C-3F), 81.4(4) (C-4D), 81.3(8) (C-4C), 78.6 (C-4E, C-4F), 77.4 (C-2E), 77.2 (C-2F), 76.8(1) (C-3D), 76.7(6) (C-3C), 67.9 (C-5B), 66.9 (C-5E, C-5F), 64.7 (C-5D), 64.6 (C-5C), 55.3 (OCH3), 36.8(4) (acyl CH2), 36.8(3) (acyl CH2), 19.4(5) (acyl CH2), 19.4(1) (acyl CH2), 14.0(3) (acyl CH3), 13.9(8) (acyl CH3). ESIMS: m/z calcd for [C42H68O25Na]+: 995.3942. Found: 995.3957.
Methyl 5-O-decanoyl-β-D-arabinofuranosyl-(1→2)-5-O-decanoyl-α-D-arabinofuranosyl-(1→3)-[5-O-decanoyl-β-D-arabinofuranosyl-(1→2)-5-O-decanoyl-α-D-arabinofuranosyl-(1→5)]-α-D-arabinofuranoside (6)
Isolated 68% as a colorless oil: Rf 0.22 (10:1 CH2Cl2–CH3OH); [α]D +34.7 (c 0.2, CHCl3);1H NMR (500 MHz, CDCl3–CD3OD, 1:1, δH) 5.12 (d, 1H, J1D,2D = 2.5 Hz, H-1D), 5.08 (d, 1 H, J1C,2C = 2.0 Hz, H-1C), 5.03 (d, 1 H, J1F,2F = 4.4 Hz, H-1F), 5.00 (d, 1 H, J1E,2E = 4.6 Hz, H-1E), 4.76 (d, 1 H, J1B,2B = 1.2 Hz, H-1B), 4.32–4.02 (m, 14 H, H-5Ea, H-5Fa, H-5Fb, H-5Eb, H-2B, H-2C, H-2D, H-5Da, H-5Ca, H-5Db, H-5Cb, H-3F, H-3E, H-4B), 4.00–3.88 (m, 10 H, H-2F, H-2E, H-3D, H-3C, H-3B, H-4D, H-4C, H-4E, H-4F, H-5Ba), 3.69 (dd, 1 H, J4B,5Bb = 2.8 Hz, J5Ba,5Bb = 11.4 Hz, H-5Bb), 3.34 (s, 3 H, OCH3), 2.36–2.28 (m, 8 H, CH2 × 4), 1.62–1.54 (m, 8 H, acyl CH2 × 4), 1.34–1.16 (m, 48 H, acyl CH2 × 24), 0.90–0.80 (m, 12 H, acyl CH3 × 4); 13C NMR (125 MHz, CDCl3, δC) 175.6(5) (CO), 175.6(4) (CO), 175.5(0) (CO × 2), 110.5 (C-1B), 107.5 (C-1C), 107.2 (C-1D), 103.3 (C-1E), 103.2 (C-1F), 90.2 (C-2C), 89.8 (C-2D), 84.9 (C-3B), 82.9 (C-2B), 81.9 (C-4B), 81.7 (C-3E), 81.5 (C-3F), 81.4(4) (C-4D), 81.1 (C-4C), 78.5(3) (C-4E), 78.4(7) (C-4F), 77.3 (C-2E), 77.1 (C-2F), 76.7(8) (C-3D), 76.7(6) (C-3C), 67.8 (C-5B), 67.2 (C-5E), 67.0 (C-5F), 64.9 (C-5D), 64.7 (C-5C), 56.0 (OCH3), 35.3(1) (acyl CH2), 35.2(9) (acyl CH2), 35.2(7) (acyl CH2), 33.1(5) (acyl CH2), 33.1(4) (acyl CH2), 30.7(4) (acyl CH2), 30.7(3) (acyl CH2), 30.7(0) (acyl CH2), 30.6(2) (acyl CH2), 30.5(9) (acyl CH2), 30.5(6) (acyl CH2), 30.5(5) (acyl CH2), 30.5(4) (acyl CH2), 30.4(4) (acyl CH2), 30.4(1) (acyl CH2), 30.4(0) (acyl CH2), 26.1(5) (acyl CH2), 26.1(2) (acyl CH2), 23.9 (acyl CH2), 15.0 (acyl CH3). ESIMS: m/z calcd for [C66H116O25Na]+: 1331.7698. Found: 1331.7692.
Methyl 5-O-palmitoyl-β-D-arabinofuranosyl-(1→2)-5-O-palmitoyl-α-D-arabinofuranosyl-(1→3)-[5-O-palmitoyl-β-D-arabinofuranosyl-(1→2)-5-O-palmitoyl-α-D-arabinofuranosyl-(1→5)]-α-D-arabinofuranoside (7)
Isolated 87% as a colorless oil: Rf 0.27 (1:2 hexane–EtOAc); [α]D +25.6 (c 0.5, CHCl3);1H NMR (500 MHz, CDCl3–CD3OD, 1:1, δH) 5.12 (d, 1H, J1D,2D = 2.4 Hz, H-1D), 5.08 (d, 1 H, J1C,2C = 1.9 Hz, H-1C), 5.02 (d, 1 H, J1F,2F = 4.4 Hz, H-1F), 5.00 (d, 1 H, J1E,2E = 4.5 Hz, H-1E), 4.76 (d, 1 H, J1B,2B = 1.2 Hz, H-1B), 4.32–4.02 (m, 14 H, H-5Ea, H-5Fa, H-5Fb, H-5Eb, H-2B, H-2C, H-2D, H-5Da, H-5Ca, H-5Db, H-5Cb, H-3F, H-3E, H-4B), 4.02–3.88 (m, 10 H, H-2F, H-2E, H-3D, H-3C, H-3B, H-4D, H-4C, H-4E, H-4F, H-5Ba), 3.70 (dd, 1 H, J4B,5Bb = 2.8 Hz, J5Ba,5Bb = 11.4 Hz, H-5Bb), 3.34 (s, 3 H, OCH3), 2.38–2.26 (m, 8 H, acyl CH2 × 4), 1.66–1.52 (m, 8 H, acyl CH2 × 4), 1.40–1.06 (m, 96 H, acyl CH2 × 48), 0.90–0.80 (m, 12 H, acyl CH3 × 4); 13C NMR (125 MHz, CDCl3, δC) 175.6(5) (CO), 175.6(3) (CO), 175.5(3) (CO), 175.4(9) (CO), 110.5 (C-1B), 107.5 (C-1C), 107.2 (C-1D), 103.4 (C-1E), 103.3 (C-1F), 90.3 (C-2C), 89.9 (C-2D), 84.8 (C-3B), 82.9 (C-2B), 81.9 (C-4B), 81.7 (C-3E), 81.5 (C-3F), 81.4(4) (C-4D), 81.1 (C-4C), 78.5(3) (C-4E), 78.4(7) (C-4F), 77.3 (C-2E), 77.1 (C-2F), 76.7(5) (C-3D), 76.7(3) (C-3C), 67.7 (C-5B), 67.1 (C-5E), 67.0 (C-5F), 65.0 (C-5D), 64.7 (C-5C), 56.0 (OCH3), 35.3(1) (acyl CH2), 35.2(9) (acyl CH2), 33.2 (acyl CH2), 30.9(6) (acyl CH2), 30.9(3) (acyl CH2), 30.9(2) (acyl CH2), 30.8(3) (acyl CH2), 30.8(1) (acyl CH2), 30.7(8) (acyl CH2), 30.7(7) (acyl CH2), 30.6(6) (acyl CH2), 30.6(3) (acyl CH2), 30.6(0) (acyl CH2), 30.5(9) (acyl CH2), 30.5(1) (acyl CH2), 30.4(8) (acyl CH2), 30.4(5) (acyl CH2), 30.4(3) (acyl CH2), 26.2 (acyl CH2), 26.1 (acyl CH2), 23.9 (acyl CH2), 15.1 (acyl CH3). ESIMS: m/z calcd for ESIMS: m/z calcd for [C90H164O25Na]+: 1648.1459. Found: 1648.1454.
Methyl 5-O-behenoyl-β-D-arabinofuranosyl-(1→2)-5-O-behenoyl-α-D-arabinofuranosyl-(1→3)-[5-O-behenoyl-β-D-arabinofuranosyl-(1→2)-5-O-behenoyl-α-D-arabinofuranosyl-(1→5)]-α-D-arabinofuranoside (8)
Isolated 40% as a colorless oil: Rf 0.36 (1:1 hexane–EtOAc); [α]D +7.2 (c 0.3, CHCl3);1H NMR (500 MHz, CDCl3–CD3OD, 1:1, δH) 5.12 (d, 1H, J1D,2D = 2.2 Hz, H-1D), 5.08 (d, 1 H, J1C,2C = 1.8 Hz, H-1C), 5.03 (d, 1 H, J1F,2F = 4.5 Hz, H-1F), 5.00 (d, 1 H, J1E,2E = 4.4 Hz, H-1E), 4.76 (d, 1 H, J1B,2B = 0.7 Hz, H-1B), 4.32–4.02 (m, 24 H, H-5Ea, H-5Fa, H-5Fb, H-5Eb, H-2B, H-2C, H-2D, H-5Da, H-5Ca, H-5Db, H-5Cb, H-3F, H-3E, H-4B, H-2F, H-2E, H-3D, H-3C, H-3B, H-4D, H-4C, H-4E, H-4F, H-5Ba), 3.70 (dd, 1 H, J4B,5Bb = 2.7 Hz, J5Ba,5Bb = 11.4 Hz, H-5Bb), 3.34 (s, 3 H, OCH3), 2.40–2.24 (m, 8 H, acyl CH2 × 4), 1.70–1.50 (m, 8 H, acyl CH2 × 4), 1.40–1.00 (m, 144 H, acyl CH2 × 72), 0.90–0.76 (m, 12 H, acyl CH3 × 4); 13C NMR (125 MHz, CDCl3, δC) 175.7 (CO), 175.6 (CO), 175.5(3) (CO), 175.4(9) (CO), 110.5 (C-1B), 107.5 (C-1C), 107.3 (C-1D), 103.4 (C-1E), 103.3 (C-1F), 90.3 (C-2C), 89.9 (C-2D), 84.8 (C-3B), 82.9 (C-2B), 81.9 (C-4B), 81.7 (C-3E), 81.5 (C-3F), 81.4(3) (C-4D), 81.1 (C-4C), 78.6 (C-4E), 78.5 (C-4F), 77.3 (C-2E), 77.1 (C-2F), 76.7(2) (C-3D), 76.6(9) (C-3C), 67.7 (C-5B), 67.1 (C-5E), 67.0 (C-5F), 65.0 (C-5D), 64.7 (C-5C), 56.0 (OCH3), 35.3(3) (acyl CH2), 35.3(1) (acyl CH2), 35.2(8) (acyl CH2), 33.2 (acyl CH2), 30.9(8) (acyl CH2), 30.9(4) (acyl CH2), 30.8(6) (acyl CH2), 30.8(4) (acyl CH2), 30.8(1) (acyl CH2), 30.8(0) (acyl CH2), 30.6(4) (acyl CH2), 30.5(0) (acyl CH2), 30.4(7) (acyl CH2), 30.4(5) (acyl CH2), 26.2 (acyl CH2), 26.1 (acyl CH2), 23.9 (acyl CH2), 15.1 (acyl CH3). ESIMS: m/z calcd for [C114H212O25Na]+: 2004.5215. Found: 2004.5210.
Methyl 2,3-di-O-benzyl-β-D-arabinofuranosyl-(1→2)-3-O-benzyl-α-D-arabinofuranosyl-(1→3)-[2,3-di-O-benzyl-β-D-arabinofuranosyl-(1→2)-3-O-benzyl-α-D-arabinofuranosyl-(1→5)]-2-O-benzyl-α-D-arabinofuranoside (9)
To a solution of 20 (189 mg, 0.10 mmol) in THF (6 mL) was added TBAF (0.50 mL, 1.0 M in THF) at rt. The mixture was stirred for 12 h and concentrated. The residue was purified by chromatography (10:1 CH2Cl2–CH3OH) to afford 9 (124 mg, 88%) as a colorless oil: Rf 0.13 (1:3 hexane–EtOAc); [α]D +15.9 (c 0.2, CHCl3);1H NMR (500 MHz, CDCl3, δH) 7.38–7.22 (m, 35 H, Ar), 5.10 (d, 1 H, J1D,2D = 1.4 Hz, H-1D), 5.07 (s, 1 H, H-1C), 5.04 (d, 1 H, J1E,2E = 4.6 Hz, H-1E), 4.97 (d, 1 H, J1F,2F = 4.6 Hz, H-1F), 4.91 (d, 1 H, J1B,2B = 1.5 Hz, H-1B), 4.73–4.67 (m, 2 H, PhCH2O), 4.65–4.61 (m, 2 H, PhCH2O), 4.60–4.42 (m, 10 H, PhCH2O), 4.33–4.29 (m, 2 H, H-2C, H-4E), 4.28–4.18 (m, 4 H, H-2D, H-3F, H-3E, H-3B), 4.13–3.94 (m, 10 H, H-3C, H-3D, H-2E, H-2B, H-2F, H-4B, H-4F, H-4C, H-4D, H-5Ba), 3.80–3.62 (m, 5 H, H-5Bb, H-5Ca, H-5Da, H-5Ea, H-5Fa), 3.60–3.50 (m, 4 H, H-5Db, H-5Cb, H-5Eb, H-5Fb), 3.35 (s, 3 H, OCH3); 13C NMR (125 MHz, CDCl3, δC) 138.0(2) (Ar), 137.9(8) (Ar), 137.8 (Ar), 137.7 (Ar), 137.5(2) (Ar × 2), 137.4(7) (Ar), 128.6 (Ar × 2), 128.5 (Ar × 2), 128.4(3) (Ar × 5), 128.4(2) (Ar × 5), 128.3(9) (Ar × 2), 128.1 (Ar × 2), 128.0 (Ar × 2), 127.9 (Ar), 127.8(4) (Ar), 127.8(2) (Ar), 127.7(9) (Ar), 127.7(8) (Ar), 127.7(6) (Ar × 2), 127.7(0) (Ar × 5), 127.6(8) (Ar × 3), 106.8 (C-1B), 106.0 (C-1D), 105.9 (C-1C), 100.4 (C-1E), 100.1 (C-1F), 88.7 (C-2B), 86.2 (C-2C), 85.8 (C-2D), 84.0 (C-2E, C-2F), 83.1 (C-3E), 83.0 (C-3F), 82.9 (C-3D), 82.6 (C-3C), 82.0(1) (C-4F), 81.9 (7) (C-4D), 80.9 (C-3B), 80.6 (C-4E), 80.5 (C-4C), 80.0 (C-4B), 72.6(7) (PhCH2O), 72.6(6) (PhCH2O), 72.5(8) (PhCH2O), 72.5(2) (PhCH2O), 72.4 (PhCH2O), 72.2 (PhCH2O), 72.1 (PhCH2O), 65.1 (C-5B), 63.5 (C-5C), 63.2 (C-5D), 62.7 (C-5E), 62.6 (C-5F), 54.9 (OCH3). ESIMS: m/z calcd for [C75H86O21Na]+: 1345.5554. Found: 1345.5558.
Methyl 3-O-benzyl-5-O-p-methoxybenzyl-α-D-arabinofuranosyl-(1→3)-[3-O-benzyl-5-O-p-methoxybenzyl-α-D-arabinofuranosyl-(1→5)]-2-O-benzyl-α-D-arabinofuranoside (11)
To a solution of compound 19 (342 mg, 0.38 mmol) in 1:1 CH3OH–CH2Cl2 (3 mL) a solution of NaOCH3 in CH3OH was added until a pH of 11 was obtained. The reaction mixture was stirred at room temperature for 2 h, neutralized with AcOH, and concentrated. Chromatography of the residue (1:2 hexane–EtOAc) gave 11 (287 mg, 82%) as a colorless syrup: Rf 0.15 (1:1 hexane–EtOAc); [α]D +111.1 (c 0.4, CHCl3);1H NMR (600 MHz, CDCl3, δH) 7.34–7.17 (m, 19 H, Ar), 6.87–6.84 (m, 4 H, Ar), 5.08 (s, 1 H, H-1D), 5.06 (s, 1 H, H-1C), 4.81 (d, 1 H, J1B,2B = 1.3 Hz, H-1B), 4.62–4.49 (m, 6 H, PhCH2O), 4.44–4.38 (m, 4 H, PhCH2O), 4.30–4.28 (m, 2 H, H-3B, H-4C), 4.25 (ddd, 1 H, J3D,4D = J4D,5Da = 2.6 Hz, J4D,5Db = 2.9 Hz, H-4D), 4.16 (ddd, 1 H, J1C,2C = 1.2 Hz, J2C,OH = 9.1 Hz, J2C,3C = 2.1 Hz, H-2C), 4.12–4.08 (m, 2 H, H-4B, H-3D), 3.98–3.94 (m, 2 H, H-2B, H-5Ba), 3.82–3.78 (m, 8 H, H-2D, H-3C, PMBOCH3 × 2), 3.73 (dd, 1 H, J4B,5Bb = 2.7 Hz, J5Ba,5Bb = 11.7 Hz, H-5Bb), 3.58–3.55 (m, 2 H, H-5Ca, H-5Da), 3.46–3.42 (m, 2 H, H-5Cb, H-5Db), 3.36 (d, 1 H, J = 10.2 Hz, OH), 3.35–3.33 (m, 4 H, OH, OCH3); 13C NMR (125 MHz, CDCl3, δC) 159.5 (Ar), 159.4 (Ar), 138.0(3) (Ar), 137.9(5) (Ar), 137.6 (Ar), 129.5 (Ar × 3), 129.4 (Ar), 129.2 (Ar), 128.3(9) (Ar × 3), 128.3(5) (Ar × 3), 127.9 (Ar × 3), 127.7(9) (Ar × 2), 127.7(6) (Ar × 3), 127.6(5) (Ar × 2), 127.6(0) (Ar × 3), 127.5(8) (Ar × 2), 113.9(6) (Ar × 2), 113.9(0) (Ar × 2), 109.2 (C-1C), 108.1 (C-1D), 107.1 (C-1B), 88.5 (C-2B), 85.3 (C-2D), 84.8 (C-3C), 83.7 (C-4D), 82.6 (C-4C), 80.7 (C-4B), 79.5 (C-3B), 78.3 (C-2C), 77.6 (C-3D), 73.4 (PhCH2O), 73.3 (PhCH2O), 72.0 (PhCH2O), 71.9 (PhCH2O), 71.7 (PhCH2O), 69.5 (C-5C, C-5D), 65.8 (C-5B), 55.3 (PMBOCH3 × 2), 54.8 (OCH3). ESIMS: m/z calcd for [C53H62O15Na]+: 961.3981. Found: 961.3978.
Allyl 2-O-benzoyl-3-O-benzyl-α-D-arabinofuranoside (16)
To a solution of 14 (5.00 g, 10.5 mmol) in acetic acid (25 mL) a 33% HBr solution in acetic acid (25 mL) was added. The reaction was stirred for 6 h at room temperature and diluted with CH2Cl2 (200 mL). The clear solution was then washed with cold saturated aqueous NaHCO3 solution (4 × 200 mL) and brine (200 mL), dried (MgSO4) and filtered. To this solution molecular sieves (10 g), triethylamine (6.65 mL, 47.7 mmol) and CH3OH (2 mL, 49.4 mmol) were added. After 12 h stirring, the mixture was concentrated, diluted with CH2Cl2 (200 mL) and washed with water (3 × 200 mL) and brine (200 mL). The solution was dried (MgSO4), filtered and evaporated to yield the orthoester. To a solution of this orthoester in 1:1 CH2Cl2–CH3OH (150 mL) a solution of NaOCH3 in CH3OH (2.5 mL, 1M) was added. The reaction was stirred at 40 °C for 4 h and concentrated. The residue was diluted with CH2Cl2 (200 mL), washed with water (50 mL), dried and concentrated to provide the crude deprotected orthoester. To a solution of the crude orthoester (4.26 g, 7.89 mmol) in CH2Cl2 (79 mL) molecular sieves (5 g) was added. The mixture was stirred for 30 min at room temperature and cooled to 0 °C. To this solution camphorsulfonic acid (26 mg, 0.078 mmol) was added. After TLC indicated the completion of reaction, triethylamine was added. Then the mixture was filtered and concentrated to give the crude 1,2,5-orthoester 15. After the residue was dissolved in DMF (39 mL) 0 °C, NaH (473 mg, 60% dispersion in oil, 11.8 mmol) and BnBr (1.41 mL, 11.8 mmol) were added subsequently. The reaction mixture was stirred for 2 h and the reaction was quenched by the addition of CH3OH. The mixture was diluted with CH2Cl2 (70 mL), washed with water (70 mL) and brine (70 mL), dried and concentrated. After the residue was dissolved in CH2Cl2 (39 mL), molecular sieves (4 g) and allyl alcohol (2.68 mL, 39.5 mmol) were added. The reaction mixture was stirred at room temperature for 30 min and cooled to 0 °C and then a solution of TMSOTf (0.14 mL, 0.79 mmol, 0.1 in CH2Cl2 v/v) was added. The solution was allowed to stir for 10 min and the reaction was quenched by the addition of triethylamine. The mixture was filtered, concentrated and purified by chromatography (2:1 hexane–EtOAc) to give 16 (2.92 g, 72% over 6 steps) as a colorless syrup: Rf 0.21 (2:1 hexane–EtOAc); [α]D +94.5 (c 1.0, CHCl3);1H NMR (500 MHz, CDCl3, δH) 8.06–8.00 (m, 2 H, Ar), 7.62–7.57 (m, 1 H, Ar), 7.48–7.44 (m, 2 H, Ar), 7.38–7.24 (m, 5 H, Ar), 5.99–5.91 (m, 1 H, CH2CHCH2O), 5.43 (d, 1 H, J1,2 = 1.6 Hz, H-2), 5.38–5.33 (m, 1 H, CH2CHCH2O), 5.21–5.20 (m, 2 H, CH2CHCH2O, H-1), 4.84 (d, 1 H, J = 12.1 Hz, PhCH2O), 4.63 (d, 1 H, J = 12.1 Hz, PhCH2O), 4.30–4.26 (m, 2 H, CH2CHCH2O, H-4), 4.13–4.08 (m, 2 H, CH2CHCH2O, H-3), 3.88 (ddd, 1 H, J4,5a = 3.5 Hz, J5a,5b = 12.2 Hz, J5a,OH = 4.5 Hz, H-5a), 3.67 (ddd, 1 H, J4,5b = 3.5 Hz, J5a,5b = 12.2 Hz, J5b,OH = 8.2 Hz, H-5b), 1.97 (dd, 1 H, J5a,OH = 4.5 Hz, J5b,OH = 8.2 Hz, OH); 13C NMR (125 MHz, CDCl3, δC) 165.4 (CO), 137.6 (Ar), 133.9 (Ar), 133.4 (Ar), 129.7 (Ar × 2), 129.4 (Ar), 128.5 (Ar × 2), 128.4 (Ar × 2), 127.9(3) (Ar × 2), 127.8(6) (CH2CH-CH2O), 117.6 (CH2CH–CH2O), 105.2 (C-1), 83.2 (C-4), 82.9 (C-3), 82.2 (C-2), 72.5 (PhCH2O), 68.0 (CH2CH–CH2O), 62.0 (C-5). ESIMS: m/z calcd for [C22H24O6Na]+: 407.1465. Found: 407.1463.
2-O-Benzoyl-3-O-benzyl-5-O-p-methoxybenzyl-D-arabinofuranose (18)
To a solution of PMBOH (0.92 mL, 7.36 mmol) in CH2Cl2 (36 mL), DBU (0.22 mL, 1.47 mol) and CCl3CN (3.69 mL, 36.79 mmol) were added. The solution was stirred at room temperature for 2 h, passed though a short silica plug, and concentrated. The resulting residue and 17 (1.41 g, 3.68 mmol) were dissolved in toluene (42 mL) and La(OTf)3 (50 mg, 0.085 mmol) was added. The reaction mixture was stirred for 5 h, the reaction was quenched by the addition of few drops of acetic acid, and the solution was concentrated. To a solution of this residue in CH3OH (65 mL), PdCl2 (131 mg, 0.74 mmol) was added. The reaction mixture was stirred for 5 h, neutralized by the addition of triethylamine, and filtered through Celite. The filtrate was concentrated and the residue was purified by chromatography (6:1 hexane–EtOAc) to give the reducing sugar 18 (1.42 g, 84% over two steps) in a 1.8:1 α:β ratio as colorless oil: Rf 0.26 (3:1 hexane–EtOAc); [α]D −11.7 (c 0.6, CHCl3); ESIMS: m/z calcd for [C27H28O7Na]+: 487.1727. Found: 487.1726. α isomer: 1H NMR (500 MHz, CDCl3, δH) 7.96–7.92 (m, 2 H, Ar), 7.46–7.40 (m, 3 H, Ar), 7.36–7.28 (m, 5 H, Ar), 7.18–7.14 (m, 2 H, Ar), 6.81–6.77 (m, 2 H, Ar), 5.51 (dd, 1 H, J1,2 = 0.8 Hz, J1,OH = 7.5 Hz, H-1), 5.37 (d, 1 H, J1,2 = 0.8 Hz, H-2), 4.81 (d, 1 H, J = 12.1 Hz, PhCH2O), 4.68 (d, 1 H, J = 12.1 Hz, PhCH2O), 4.56–4.52 (m, 1 H, H-4), 4.50–4.42 (m, 2 H, PhCH2O), 4.05 (ddd, 1 H, J1,3 = 1.1 Hz, J2,3 = 1.0 Hz, J3,4 = 3.5 Hz, H-3), 3.77 (s, 3 H, PMBOCH3), 3.60–3.50 (m, 2 H, H-5a, H-5b), 3.37 (d, 1 H, J2,OH = 7.5 Hz, OH); 13C NMR (125 MHz, CDCl3, δC) 165.4 (CO), 159.2 (Ar), 137.1 (Ar), 133.4 (Ar), 129.9 (Ar), 129.8 (Ar), 129.3 (Ar × 2), 129.2 (Ar), 128.4(9) (Ar × 2), 128.4(7) (Ar × 3), 128.0 (Ar × 3), 113.7 (Ar × 2), 101.1 (C-1), 82.8 (C-3), 82.7 (C-4), 80.7 (C-2), 73.0 (PhCH2O), 72.3 (PhCH2O), 69.3 (C-5), 55.2 (OCH3). β isomer: 1H NMR (500 MHz, CDCl3, δH) 8.02–7.98 (m, 2 H, Ar), 7.61–7.56 (m, 3 H, Ar), 7.36–7.28 (m, 5 H, Ar), 7.25–7.22 (m, 2 H, Ar), 6.88–6.85 (m, 2 H, Ar), 5.61 (dd, 1 H, J1,2 = 4.7 Hz, J1,OH = 10.3 Hz, H-1), 5.28 (dd, 1 H, J1,2 = 4.7 Hz, J2,3 = 5.4 Hz, H-2), 4.69 (d, 1 H, J = 11.9 Hz, PhCH2O), 4.58–4.52 (m, 2 H, PhCH2O), 4.50–4.42 (m, 2 H, PhCH2O, H-3), 4.24 (ddd, 1 H, J3,4 = 3.0 Hz, J4,5a = 3.1 Hz, J4,5b = 4.6 Hz, H-4), 3.95 (d, 1 H, J2,OH = 10.3 Hz, OH), 3.79 (s, 3 H, PMBOCH3), 3.66 (dd, 1 H, J4,5a = 3.1 Hz, J5a,5b = 10.3 Hz, H-5a), 3.60–3.50 (m, 1 H, H-5b); 13C NMR (125 MHz, CDCl3, δC) 165.7 (CO), 159.5 (Ar), 137.5 (Ar), 133.3 (Ar), 129.7 (Ar × 4), 129.6 (Ar × 2), 129.5 (Ar), 129.0 (Ar), 128.4 (Ar × 2), 127.9 (Ar), 127.8 (Ar × 2), 114.0 (Ar × 2), 95.8 (C-1), 80.8 (C-3, C-4), 80.2 (C-2), 73.5 (PhCH2O), 72.1 (PhCH2O), 70.1 (C-5), 55.2 (OCH3).
Methyl 2-O-benzoyl-3-O-benzyl-5-O-p-methoxybenzyl-α-D-arabinofuranosyl-(1→3)-[2-O-benzoyl-3-O-benzyl-5-O-p-methoxybenzyl-α-D-arabinofuranosyl-(1→5)]-2-O-benzyl-α-D-arabinofuranoside (19)
To a solution of alcohol 18 (443 mg, 0.95 mmol) and CCl3CN (0.29 mL, 2.86 mmol) in CH2Cl2 (19 mL) was added DBU (0.029 mL, 0.19 mmol). The reaction was stirred at rt for 4 h. The solvent was evaporated and the residue was quickly filtered through a short column of silica gel. The resulting solution was then concentrated to give the imidate derivative 12, which was used immediately in the next step. To a mixture of imidate residue, 13 (101 mg, 0.40 mmol) and 4 Å molecular sieves (700 mg) in CH2Cl2 (10 mL) was added a solution of TMSOTf (86 μL, 0.1 in CH2Cl2 v:v) dropwise at 0 °C. After stirring at this temperature for 10 min, the reaction was quenched by the addition of triethylamine. The solution was then diluted with CH2Cl2 (30 mL) and filtered through Celite. The filtrate was concentrated and purified by chromatography (2:1 hexane–EtOAc) to afford 19 (382 mg, 84%) as a white solid: Rf 0.23 (2:1 hexane–EtOAc); [α]D +68.0 (c 0.6, CHCl3); 1H NMR (500 MHz, CDCl3, δH) 7.98–7.94 (m, 4 H, Ar), 7.60–7.54 (m, 2 H, Ar), 7.42–7.38 (m, 4 H, Ar), 7.32–7.10 (m, 19 H, Ar), 6.77–6.74 (m, 4 H, Ar), 5.46 (d, 1 H, J1C,2C = 1.3 Hz, H-2C), 5.34 (d, 1 H, J1D,2D = 1.1 Hz, H-2D), 5.30 (s, 1 H, H-1C), 5.26 (s, 1 H, H-1D), 4.94 (d, 1 H, J1B,2B = 1.5 Hz, H-1B), 4.80–4.74 (m, 2 H, PhCH2O), 4.60–4.54 (m, 4 H, PhCH2O), 4.48–4.42 (m, 2 H, PhCH2O), 4.42–4.34 (m, 2 H, H-4C, PhCH2O), 4.32–4.28 (m, 2 H, H-3B, H-4D), 4.24–4.20 (m, 1 H, H-4B), 4.06–3.98 (m, 4 H, H-2B, H-5Ba, H-3D, H-3C), 3.82 (dd, 1 H, J4B,5Bb = 2.7 Hz, J5Ba,5Bb = 11.4 Hz, H-5Bb), 3.80–3.72 (m, 6 H, PMBOCH3 × 2), 3.64–3.50 (m, 4 H, H-5Da, H-5Ca, H-5Db, H-5Cb), 3.36 (s, 3 H, OCH3); 13C NMR (125 MHz, CDCl3, δC) 165.3 (CO), 165.2 (CO), 159.1 (Ar × 2), 137.9 (Ar), 137.8 (Ar), 137.7 (Ar), 133.3 (Ar), 133.2 (Ar), 130.2 (Ar), 130.1 (Ar), 129.8(1) (Ar × 2), 129.7(8) (Ar × 2), 129.6 (Ar), 129.5 (Ar), 129.2(4) (Ar × 2), 129.2(0) (Ar × 2), 129.2(0) (Ar × 2), 128.3(9) (Ar × 2), 128.3(8) (Ar × 2), 128.2(9) (Ar × 4), 128.2(4) (Ar × 2), 127.8(4) (Ar × 2), 127.8(2) (Ar × 4), 127.6(4) (Ar), 127.6(1) (Ar), 127.5(3) (Ar), 113.7 (Ar × 4), 107.1 (C-1B), 106.2 (C-1C), 105.6 (C-1D), 88.3 (C-2B), 83.6 (C-3C), 83.2 (C-3D), 82.4 (C-3B), 82.2 (C-4C), 82.1 (C-2D), 81.7 (C-2C), 80.6 (C-4D), 80.1 (C-4B), 73.0(2) (PhCH2O), 73.0(0) (PhCH2O), 72.1 (PhCH2O), 72.0 (PhCH2O), 71.9 (PhCH2O), 69.0 (C-5C), 68.8 (C-5D), 66.0 (C-5B), 55.2(0) (PMBOCH3), 55.1(9) (PMBOCH3), 54.9 (OCH3). ESIMS: m/z calcd for [C67H70O17Na]+: 1169.4505. Found: 1169.4509.
Methyl 2,3-di-O-benzyl-5-O-tert-butyldimethylsilyl-β-D-arabinofuranosyl-(1→2)-3-O-benzyl-5-O-tert-butyldimethylsilyl-α-D-arabinofuranosyl-(1→3)-[2,3-di-O-benzyl-5-O-tert-butyldimethylsilyl-β-D-arabinofuranosyl-(1→2)-3-O-benzyl-5-O-tert-butyldimethylsilyl-α-D-arabinofuranosyl-(1→5)]-2-O-benzyl-α-D-arabinofuranoside (20)
To a mixture of 11 (567 mg, 0.60 mmol), 10 (823 mg, 1.45 mmol), and 4 Å molecular sieves (1.3 g) in CH2Cl2 (20 mL) was added N-iodosuccinimide (352 mg, 1.56 mmol) and AgOTf (16 mg, 0.06 mmol) at −60 °C. The solution was warmed to −40 °C over a period of 1 h and the reaction was quenched by the addition of triethylamine. The solution was then diluted with CH2Cl2 (30 mL) and filtered through Celite. The filtrate was concentrated and purified by chromatography (4:1 hexane–EtOAc) to afford a mixture of pentasaccharides (947 mg, 86%) as a colorless oil. To a solution of this mixture (947 mg, 0.52 mmol) in CH2Cl2 (51 mL) trifluoroacetic acid (1.04 mL) was added dropwise. After TLC indicated the completion of the reaction, the resulting solution was neutralized by the addition of triethylamine. The reaction mixture was then concentrated and the residue purified by chromatography (10:1 CH2Cl2–CH3OH) to give a syrup, that was redissolved in CH2Cl2 (51 mL). TBDMSCl (162 mg, 1.07 mmol), imidazole (244 mg, 3.58 mmol) and DMAP (44 mg, 3.58 mmol) were added and the reaction mixture was stirred at rt overnight before the reaction was quenched by the addition of CH3OH. The reaction mixture was then concentrated and purified by chromatography (6:1 hexane–EtOAc) to afford 20 (645 mg, 59%) over three steps as a colorless oil: Rf 0.26 (4:1 hexane–EtOAc) [α]D −8.9 (c 0.3, CHCl3);1H NMR (500 MHz, CDCl3, δH) 7.34–7.20 (m, 35 H, Ar), 5.14 (d, 1 H, J1E,2E = 4.5 Hz, H-1E), 5.13 (d, 1 H, J1C,2C = 1.0 Hz, H-1C), 5.09 (s, 1 H, H-1D), 5.00 (d, 1 H, J1F,2F = 4.4 Hz, H-1F), 4.86 (d, 1 H, J1B,2B = 1.0 Hz, H-1B), 4.72–4.44 (m, 14 H, PhCH2O), 4.39 (dd, 1 H, J1C,2C = 1.0 Hz, J2C,3C = 3.0 Hz, H-2C), 4.33 (dd, 1 H, J1D,2D = 1.0 Hz, J2D,3D = 2.9 Hz, H-2D), 4.25 (dd, 1 H, J2B,3B = 3.0 Hz, J3B,4B = 6.8 Hz, H-3B), 4.18–4.06 (m, 5 H, H-4B, H-4C, H-3E, H-3F, H-4D), 4.03–3.92 (m, 8 H, H-2F, H-2E, H-2B, H-5Ba, H-3D, H-3C, H-4F, H-4E), 3.79 (dd, 1 H, J4B,5Bb = 2.3 Hz, J5Ba,5Bb = 12.0 Hz, H-5Bb), 3.77–3.66 (m, 8 H, H-5Da, H-5Ca, H-5Ea, H-5Fa, H-5Db, H-5Cb, H-5Eb, H-5Fb), 3.29 (s, 3 H, OCH3), 0.92–0.84 (m, 36 H, t-BuCH3), 0.06–0.00 (m, 24 H, SiCH3); 13C NMR (125 MHz, CDCl3, δC) 138.5 (Ar), 138.4 (Ar), 138.3 (Ar), 138.2 (Ar), 137.9 (Ar), 137.8 (Ar), 137.7 (Ar), 128.4(0) (Ar × 2), 128.3(7) (Ar × 4), 128.3(2) (Ar × 2), 128.2(9) (Ar × 2), 128.2(6) (Ar × 2), 128.2(1) (Ar × 2), 127.9(2) (Ar × 2), 127.8(5) (Ar × 2), 127.7(9) (Ar), 127.7(3) (Ar × 4), 127.6(8) (Ar), 127.6(1) (Ar × 2), 127.5(9) (Ar × 2), 127.5(5) (Ar), 127.4(8) (Ar × 3), 127.4(6) (Ar × 2), 127.3(7) (Ar), 107.0 (C-1B), 106.5 (C-1C), 104.9 (C-1D), 100.3 (C-1E), 100.0 (C-1F), 88.2 (C-2B), 85.9 (C-2C), 85.4 (C-2D), 84.5 (C-3E), 84.2(2) (C-2E), 84.1(7) (C-3F), 84.1(4) (C-3C), 84.1(1) (C-2F), 84.0 (C-3D), 83.2 (C-4D), 82.7 (C-4C), 82.2 (C-4F), 82.1 (C-4E), 81.2 (C-4B), 80.1 (C-3B), 72.3(3) (PhCH2O), 72.2(7) (PhCH2O), 72.2(1) (PhCH2O), 72.1(8) (PhCH2O), 72.0(7) (PhCH2O), 72.0(6) (PhCH2O), 71.7 (PhCH2O), 65.8 (C-5B), 65.5 (C-5C), 65.4 (C-5D), 63.2 (C-5E), 63.1 (C-5F), 54.7 (OCH3), 25.9(8) (t-Butyl CH3 × 4), 26.0 (t-Butyl CH3 × 6), 25.9 (t-Butyl CH3 × 6), 18.4 (t-Butyl C × 2), 18.3 (t-Butyl C × 2), −5.1(8) (Si CH3), −5.2(4) (Si CH3), −5.2(6) (Si CH3 × 2), −5.3(0) (Si CH3 × 2), −5.3(1) (Si CH3 × 2). ESIMS: m/z calcd for [C99H142O21Na]+: 1801.9013. Found: 1801.9016.
Methyl 2,3-di-O-benzyl-5-O-butyryl-β-D-arabinofuranosyl-(1→2)-3-O-benzyl-5-O-butyryl-α-D-arabinofuranosyl-(1→3)-[2,3-di-O-benzyl-5-O-butyryl-β-D-arabinofuranosyl-(1→2)-3-O-benzyl-5-O-butyryl-α-D-arabinofurano-syl-(1→5)]-2-O-benzyl-α-D-arabinofuranoside (21)
Isolated 92% as a colorless oil: Rf 0.19 (4:1 hexane–EtOAc); [α]D +13.7 (c 0.3, CHCl3);1H NMR (500 MHz, CDCl3, δH) 7.37–7.20 (m, 35 H, Ar), 5.13 (s, 1H, H-1C), 5.12 (d, 1 H, J1E,2E = 4.5 Hz, H-1E), 5.07 (s, 1 H, H-1D), 4.96 (d, 1 H, J1F,2F = 4.4 Hz, H-1F), 4.85 (d, 1 H, J1B,2B = 1.0 Hz, H-1B), 4.72–4.64 (m, 4 H, PhCH2O), 4.62–4.44 (m, 9 H, PhCH2O), 4.43 (m, 1 H, PhCH2O), 4.40–4.38 (m, 1 H, H-2C), 4.34 (dd, 1 H, J1D,2D = 1.1 Hz, J2D,3D = 2.9 Hz, H-2D), 4.30–4.04 (m, 16 H, H-3B, H-5Fa, H-5Ea, H-5Fb, H-5Eb, H-4F, H-4E, H-3F, H-3E, H-5Da, H-5Ca, H-5Db, H-5Cb, H-4D, H-4C, H-4B), 4.01–3.92 (m, 6 H, H-2B, H-5Ba, H-2F, H-2E, H-3D, H-3C), 3.78 (dd, 1 H, J4B,5Bb = 2.2 Hz, J5Ba,5Bb = 12.0 Hz, H-5Bb), 3.35 (s, 3 H, OCH3), 2.29–2.14 (m, 8 H, acyl CH2 × 4), 1.64–1.50 (m, 8 H, acyl CH2 × 4), 0.92–0.80 (m, 12 H, acyl CH3 × 4); 13C NMR (125 MHz, CDCl3, δC) 173.3 (CO), 173.1(8) (CO), 173.1(5) (CO × 2), 137.9 (Ar), 137.8 (Ar), 137.7(5) (Ar), 137.7(1) (Ar), 137.5(2) (Ar), 137.5(0) (Ar), 137.4(1) (Ar), 128.5(1) (Ar × 2), 128.4(8) (Ar × 2), 128.4(3) (Ar × 2), 128.4(2) (Ar × 3), 128.4(0) (Ar × 2), 128.3(5) (Ar × 2), 128.3(1) (Ar × 2), 128.1 (Ar), 128.0(0) (Ar × 2), 127.9(6) (Ar × 2), 127.8(4) (Ar), 127.8(0) (Ar), 127.7(8) (Ar), 127.7(4) (Ar × 3), 127.7(0) (Ar × 2), 127.6(7) (Ar × 2), 127.6(3) (Ar × 5), 106.9 (C-1B), 106.6 (C-1C), 105.3 (C-1D), 100.6 (C-1E), 100.3 (C-1F), 88.3 (C-2B), 85.9 (C-2C), 85.6 (C-2D), 84.2 (C-3C), 84.1 (C-3D), 83.7(1) (C-2E), 83.6(9) (C-2F), 82.5(5) (C-3E), 82.4(6) (C-3F), 81.0 (C-4B), 80.4 (C-4F), 80.2 (C-4E), 79.8 (C-3B), 78.9 (C-4C), 78.7 (C-4D), 72.5(1) (PhCH2O), 72.4(7) (PhCH2O), 72.4(2) (PhCH2O), 72.4(1) (PhCH2O), 72.3(0) (PhCH2O), 72.2(8) (PhCH2O), 71.8 (PhCH2O), 66.0 (C-5C), 65.9 (C-5D), 65.5 (C-5B), 63.5(4) (C-5E), 63.4(7) (C-5F), 54.7 (OCH3), 35.8(7) (acyl CH2), 35.8(6) (acyl CH2), 35.8(4) (acyl CH2), 18.2(8) (acyl CH2), 18.2(6) (acyl CH2), 13.6(3) (acyl CH3), 13.6(0) (acyl CH3), 13.5(8) (acyl CH3). ESIMS: m/z calcd for [C91H110O25Na]+: 1625.7228. Found: 1625.7215.
Methyl 2,3-di-O-benzyl-5-O-decanoyl-β-D-arabinofuranosyl-(1→2)-3-O-benzyl-5-O-decanoyl-α-D-arabinofuranosyl-(1→3)-[2,3-di-O-benzyl-5-O-decanoyl-β-D-arabinofuranosyl-(1→2)-3-O-benzyl-5-O-decanoyl-α-D-arabino-furanosyl-(1→5)]-2-O-benzyl-α-D-arabinofuranoside (22)
Isolated 79% as a colorless oil: Rf 0.23 (4:1 hexane–EtOAc); [α]D +12.4 (c 0.3, CHCl3);1H NMR (600 MHz, CDCl3, δH) 7.37–7.20 (m, 35 H, Ar), 5.13 (s, 1H, H-1C), 5.12 (d, 1 H, J1E,2E = 4.4 Hz, H-1E), 5.07 (s, 1 H, H-1D), 4.96 (d, 1 H, J1F,2F = 4.3 Hz, H-1F), 4.85 (d, 1 H, J1B,2B = 0.9 Hz, H-1B), 4.73–4.65 (m, 4 H, PhCH2O), 4.62–4.57 (m, 3 H, PhCH2O), 4.55–4.44 (m, 6 H, PhCH2O), 4.40–4.39 (m, 2 H, PhCH2O, H-2C), 4.34 (dd, 1 H, J1D,2D = 1.1 Hz, J2D,3D = 2.8 Hz, H-2D), 4.30–4.04 (m, 16 H, H-3B, H-5Fa, H-5Ea, H-5Fb, H-5Eb, H-4F, H-4E, H-3F, H-3E, H-5Da, H-5Ca, H-5Db, H-5Cb, H-4D, H-4C, H-4B), 4.02–3.92 (m, 6 H, H-2B, H-5Ba, H-2F, H-2E, H-3D, H-3C), 3.79 (dd, 1 H, J4B,5Bb = 2.2 Hz, J5Ba,5Bb = 12.0 Hz, H-5Bb), 3.30 (s, 3 H, OCH3), 2.28–2.14 (m, 8 H, acyl CH2 × 4), 1.60–1.38 (m, 8 H, acyl CH2 × 4), 1.32–1.18 (m, 48 H, acyl CH2 × 24), 0.92–0.86 (m, 12 H, acyl CH3 × 4); 13C NMR (125 MHz, CDCl3, δC) 173.5 (CO), 173.4 (CO), 173.3 (CO × 2), 137.8(8) (Ar), 137.8(7) (Ar), 137.7(7) (Ar), 137.7(4) (Ar), 137.5(2) (Ar), 137.5(0) (Ar), 137.4 (Ar), 128.5(1) (Ar × 2), 128.4(9) (Ar × 2), 128.4(2) (Ar × 3), 128.4(0) (Ar × 2), 128.3(4) (Ar × 2), 128.3(1) (Ar × 2), 128.0(4) (Ar), 128.0(2) (Ar × 2), 127.9(6) (Ar × 3), 127.8(3) (Ar), 127.8(0) (Ar), 127.7(7) (Ar), 127.7(5) (Ar × 2), 127.7(2) (Ar), 127.6(7) (Ar × 2), 127.6(4) (Ar × 3), 127.6(3) (Ar × 3), 127.6(1) (Ar × 2), 106.9 (C-1B), 106.6 (C-1C), 105.3 (C-1D), 100.6 (C-1E), 100.3 (C-1F), 88.3 (C-2B), 85.8 (C-2C), 85.5 (C-2D), 84.3 (C-3C), 84.2 (C-3D), 83.7(2) (C-2E), 83.7(0) (C-2F), 82.7 (C-3E), 82.6 (C-3F), 81.1 (C-4B), 80.4 (C-4F), 80.3 (C-4E), 79.8 (C-3B), 78.9 (C-4C), 78.7 (C-4D), 72.5(1) (PhCH2O), 72.4(8) (PhCH2O), 72.4(3) (PhCH2O × 2), 72.3 (PhCH2O × 2), 71.8 (PhCH2O), 66.0 (C-5C), 65.9 (C-5D), 65.5 (C-5B), 63.6 (C-5E), 63.5 (C-5F), 54.7 (OCH3), 34.0(3) (acyl CH2), 34.0(1) (acyl CH2), 33.9(9) (acyl CH2), 33.9(6) (acyl CH2), 31.8(8) (acyl CH2), 31.8(5) (acyl CH2), 29.4(7) (acyl CH2), 29.4(1) (acyl CH2), 29.3(3) (acyl CH2), 29.3(2) (acyl CH2), 29.2(8) (acyl CH2), 29.2(5) (acyl CH2), 29.1(8) (acyl CH2), 29.1(3) (acyl CH2), 24.8 (acyl CH2), 22.6 (acyl CH2), 14.1 (acyl CH3). ESIMS: m/z calcd for [C115H158O25Na]+: 1962.0984. Found: 1962.0986.
Methyl 2,3-di-O-benzyl-5-O-palmitoyl-β-D-arabinofuranosyl-(1→2)-3-O-benzyl-5-O-palmitoyl-α-D-arabinofuranosyl-(1→3)-[2,3-di-O-benzyl-5-O-palmitoyl-β-D-arabinofuranosyl-(1→2)-3-O-benzyl-5-O-palmitoyl-α-D-arabinofuranosyl-(1→5)]-2-O-benzyl-α-D-arabinofuranoside (23)
Isolated 69% as a colorless oil: Rf 0.43 (4:1 hexane–EtOAc); [α]D +9.8 (c 0.3, CHCl3);1H NMR (600 MHz, CDCl3, δH) 7.37–7.20 (m, 35 H, Ar), 5.16 (s, 1H, H-1C), 5.14 (d, 1 H, J1E,2E = 4.5 Hz, H-1E), 5.03 (s, 1 H, H-1D), 4.96 (d, 1 H, J1F,2F = 4.4 Hz, H-1F), 4.86 (d, 1 H, J1B,2B = 0.8 Hz, H-1B), 4.72–4.65 (m, 4 H, PhCH2O), 4.61–4.57 (m, 3 H, PhCH2O), 4.55–4.44 (m, 6 H, PhCH2O), 4.42–4.39 (m, 2 H, PhCH2O, H-2C), 4.34 (dd, 1 H, J1D,2D = 1.1 Hz, J2D,3D = 2.8 Hz, H-2D), 4.30–4.04 (m, 16 H, H-3B, H-5Fa, H-5Ea, H-5Fb, H-5Eb, H-4F, H-4E, H-3F, H-3E, H-5Da, H-5Ca, H-5Db, H-5Cb, H-4D, H-4C, H-4B), 4.02–3.93 (m, 6 H, H-2B, H-5Ba, H-2F, H-2E, H-3D, H-3C), 3.79 (dd, 1 H, J4B,5Bb = 2.1 Hz, J5Ba,5Bb = 11.9 Hz, H-5Bb), 3.30 (s, 3 H, OCH3), 2.28–2.14 (m, 8 H, acyl CH2 × 4), 1.58–1.48 (m, 8 H, acyl CH2 × 4), 1.34–1.19 (m, 96 H, acyl CH2 × 48), 0.92–0.87 (m, 12 H, acyl CH3 × 4); 13C NMR (125 MHz, CDCl3, δC) 173.5 (CO), 173.4 (CO), 173.3 (CO × 2), 137.8(7) (Ar), 137.8(6) (Ar), 137.7(5) (Ar), 137.7(2) (Ar), 137.5(1) (Ar), 137.4(8) (Ar), 137.3(9) (Ar), 128.5(2) (Ar × 2), 128.4(9) (Ar × 2), 128.4(3) (Ar × 3), 128.4(0) (Ar × 2), 128.3(5) (Ar × 2), 128.3(1) (Ar × 2), 128.0(5) (Ar), 128.0(3) (Ar × 2), 127.9(8) (Ar × 3), 127.8(4) (Ar), 127.8(1) (Ar), 127.7(7) (Ar), 127.7(6) (Ar × 2), 127.7(3) (Ar), 127.6(8) (Ar × 2), 127.6(5) (Ar × 3), 127.6(3) (Ar × 3), 127.6(2) (Ar × 2), 106.9 (C-1B), 106.6 (C-1C), 105.2 (C-1D), 100.6 (C-1E), 100.3 (C-1F), 88.3 (C-2B), 85.8 (C-2C), 85.4 (C-2D), 84.3 (C-3C), 84.2 (C-3D), 83.7(1) (C-2E), 83.6(9) (C-2F), 82.6(9) (C-3E), 82.5(9) (C-3F), 81.1 (C-4B), 80.4 (C-4F), 80.2 (C-4E), 79.9 (C-3B), 78.9 (C-4C), 78.7 (C-4D), 72.5(0) (PhCH2O), 72.4(9) (PhCH2O), 72.4(3) (PhCH2O × 2), 72.3 (PhCH2O × 2), 71.8 (PhCH2O), 66.0(0) (C-5C), 65.9(4) (C-5D), 65.5 (C-5B), 63.6 (C-5E), 63.5 (C-5F), 54.7 (OCH3), 34.0(3) (acyl CH2), 34.0(1) (acyl CH2), 33.9(8) (acyl CH2), 33.9(6) (acyl CH2), 33.9(3) (acyl CH2), 29.7(1) (acyl CH2), 29.6(8) (acyl CH2), 29.6(4) (acyl CH2), 29.5(6) (acyl CH2), 29.4(9) (acyl CH2), 29.3(7) (acyl CH2), 29.2(8) (acyl CH2), 29.2(1) (acyl CH2), 29.1(5) (acyl CH2), 24.8 (acyl CH2), 22.7 (acyl CH2), 14.1 (acyl CH3). ESIMS: m/z calcd for [C135H206O25Na]+: 2298.4746. Found: 2298.4740.
Methyl 5-O-behenoyl-2,3-di-O-benzyl-β-D-arabinofuranosyl-(1→2)-5-O-behenoyl-3-O-benzyl-α-D-arabinofuranosyl-(1→3)-[5-O-behenoyl-2,3-di-O-benzyl-β-D-arabinofuranosyl-(1→2)-5-O-behenoyl-3-O-benzyl-α-D-arabino-furanosyl-(1→5)]-2-O-benzyl-α-D-arabinofuranoside (24)
To a solution of alcohol (0.10 mmol) in CH2Cl2 (3 mL), behenic acid (131 mg, 0.38 mmol), DCC (79 mg, 0.38 mol) and DCC (5 mg) were added at 0 °C. The reaction was kept stirring for 4 h and quenched by the addition of CH3OH. The resulting solution was concentrated and the residue was purified by chromatography (10:1 hexanes–EtOAc) to obtain a 63% yield of 24 as a white solid: Rf 0.32 (6:1 hexane–EtOAc); [α]D +7.3 (c 0.6, CHCl3);1H NMR (600 MHz, CDCl3, δH) 7.36–7.20 (m, 35 H, Ar), 5.13 (s, 1H, H-1C), 5.11 (d, 1 H, J1E,2E = 4.4 Hz, H-1E), 5.06 (s, 1 H, H-1D), 4.96 (d, 1 H, J1F,2F = 4.4 Hz, H-1F), 4.85 (s, 1 H, H-1B), 4.72–4.64 (m, 4 H, PhCH2O), 4.62–4.56 (m, 3 H, PhCH2O), 4.55–4.43 (m, 6 H, PhCH2O), 4.42–4.38 (m, 2 H, PhCH2O, H-2C), 4.35–4.32 (m, 1 H, H-2D), 4.30–4.04 (m, 16 H, H-3B, H-5Fa, H-5Ea, H-5Fb, H-5Eb, H-4F, H-4E, H-3F, H-3E, H-5Da, H-5Ca, H-5Db, H-5Cb, H-4D, H-4C, H-4B), 4.00–3.92 (m, 6 H, H-2B, H-5Ba, H-2F, H-2E, H-3D, H-3C), 3.78 (dd, 1 H, J4B,5Bb = 2.1 Hz, J5Ba,5Bb = 11.9 Hz, H-5Bb), 3.30 (s, 3 H, OCH3), 2.30–2.14 (m, 8 H, acyl CH2 × 4), 1.60–1.46 (m, 8 H, acyl CH2 × 4), 1.40–1.10 (m, 144 H, acyl CH2 × 72), 0.92–0.84 (m, 12 H, acyl CH3 × 4); 13C NMR (125 MHz, CDCl3, δC) 173.5 (CO), 173.4 (CO), 173.3 (CO × 2), 137.8(8) (Ar), 137.8(7) (Ar), 137.7(7) (Ar), 137.7(4) (Ar), 137.5(3) (Ar), 137.5(0) (Ar), 137.4(1) (Ar), 128.5(1) (Ar × 2), 128.4(8) (Ar × 2), 128.4(2) (Ar × 4), 128.3(9) (Ar × 2), 128.3(4) (Ar × 2), 128.3(0) (Ar × 2), 128.1 (Ar × 2), 128.0 (Ar × 2), 127.8(2) (Ar), 127.7(9) (Ar), 127.7(5) (Ar × 2), 127.7(1) (Ar), 127.6(6) (Ar × 2), 127.6(4) (Ar × 4), 127.6(2) (Ar × 4), 127.6(1) (Ar × 2), 106.9 (C-1B), 106.6 (C-1C), 105.3 (C-1D), 100.6 (C-1E), 100.3 (C-1F), 88.3 (C-2B), 85.8 (C-2C), 85.4 (C-2D), 84.3 (C-3C), 84.2 (C-3D), 83.7(3) (C-2E), 83.7(1) (C-2F), 82.7 (C-3E), 82.6 (C-3F), 81.1 (C-4B), 80.4 (C-4F), 80.3 (C-4E), 79.9 (C-3B), 78.9 (C-4C), 78.7 (C-4D), 72.5(0) (PhCH2O), 72.4(7) (PhCH2O), 72.4(2) (PhCH2O × 2), 72.3 (PhCH2O × 2), 71.8 (PhCH2O), 66.0(0) (C-5C), 65.9 (C-5D), 65.5 (C-5B), 63.6 (C-5E), 63.5 (C-5F), 54.7 (OCH3), 34.0(4) (acyl CH2), 34.0(1) (acyl CH2), 33.9(9) (acyl CH2), 33.9(7) (acyl CH2), 29.7(2) (acyl CH2), 29.6(7) (acyl CH2), 29.5(7) (acyl CH2), 29.5(0) (acyl CH2), 29.3(7) (acyl CH2), 29.2(9) (acyl CH2), 29.2(1) (acyl CH2), 29.1(6) (acyl CH2), 24.8(3) (acyl CH2), 24.8(1) (acyl CH2), 22.7 (acyl CH2), 14.1 (acyl CH3). ESIMS: m/z calcd for [C163H254O25Na]+: 2634.8502. Found: 2634.8496.
Acknowledgements
This work was supported by the Alberta Ingenuity Centre for Carbohydrate Science and the Natural Sciences and Engineering Research Council of Canada. M.R.R. is supported by an AHFMR Ph.D. studentship. We thank Dr Sara Duncan (AstraZeneca, UK) for sending us the pulse sequences for the CSSF NMR experiments.Notes and references
- World Health Organization, Geneva, 2009.
- C. Y. Chiang and W. W. Yew, The International Journal of Tuberculosis and Lung Disease, 2009, 13, 304–311 Search PubMed.
- S. Bhamidi, M. S. Scherman and M. R. McNeil, in Bacterial Polysaccharides: Current Innovations and Future Trends, ed. M. Ullrich, Caister Academic Press, Norfolk, UK, 2009, pp. 39–65 Search PubMed.
- S. Mahapatra, J. Basu, P. J. Brennan and D. C. Crick, in Tuberculosis and the Tubercle Bacillus, ed. S. T. Cole, K. D. Eisenback, D. N. McMurray and W. R. Jacobs, Jr., ASM Press, Washington, D.C., 2005, pp. 275–285 Search PubMed.
- C. Liu, M. R. Richards and T. L. Lowary, J. Org. Chem., 2010, 75, 4992–5007 CrossRef CAS.
- H. Nikaido, S.-H. Kim and E. Y. Rosenberg, Mol. Microbiol., 1993, 8, 1025–1030 CrossRef CAS.
- P. J. Brennan and H. Nikaido, Annu. Rev. Biochem., 1995, 64, 29–63 CrossRef CAS.
- C. Rademacher, G. K. Shoemaker, H. S. Kim, R. B. Zheng, H. Taha, C. Liu, R. C. Nacario, D. C. Schriemer, J. S. Klassen, T. Peters and T. L. Lowary, J. Am. Chem. Soc., 2007, 129, 10489–10502 CrossRef CAS.
- H. Yin, F. W. D'Souza and T. L. Lowary, J. Org. Chem., 2002, 67, 892–903 CrossRef CAS.
- A. Ishiwata, H. Akao, Y. Ito, M. Sunagawa, N. Kusunose and Y. Kashiwazaki, Bioorg. Med. Chem., 2006, 14, 3049–3061 CrossRef CAS.
- A. Ishiwata, H. Akao and Y. Ito, Org. Lett., 2006, 8, 5525–5528 CrossRef CAS.
- T. Bamhaoud, S. Sanchez and J. Prandi, Chem. Commun., 2000, 659–660 RSC.
- K. Marotte, S. Sanchez, T. Bamhaoud and J. Prandi, Eur. J. Org. Chem., 2003, 3587–3598 CrossRef CAS.
- A. Hölemann, B. L. Stocker and P. H. Seeberger, J. Org. Chem., 2006, 71, 8071–8088 CrossRef.
- J. Lu and B. Fraser-Reid, Org. Lett., 2004, 6, 3051–3054 CrossRef CAS.
- A. N. Rai and A. Basu, Tetrahedron Lett., 2003, 44, 2267–2269 CrossRef CAS.
- A. Imamura and T. L. Lowary, Org. Lett., 2010, 12, 3686–3689 CrossRef CAS.
- J. F. G. Vliegenthart, L. Dorland and H. Vanhalbeek, Adv. Carbohydr. Chem. Biochem., 1983, 41, 209–374 CAS.
- K. Mizutani, R. Kasai, M. Nakamura, O. Tanaka and H. Matsuura, Carbohydr. Res., 1989, 185, 27–38 CrossRef CAS.
- P. T. Robinson, T. N. Pham and D. Uhrin, J. Magn. Reson., 2004, 170, 97–103 CrossRef CAS.
- S. J. Duncan, R. Lewis, M. A. Bernstein and P. Sandor, Magn. Reson. Chem., 2007, 45, 283–288 CrossRef CAS.
- H. J. Reich, Journal of Chemical Education Software, 3D2, Madison, WI, USA, 1996 Search PubMed.
- A. S. Serianni and R. Barker, Can. J. Chem., 1979, 57, 3160–3167 CrossRef CAS.
- G. D. Wu, A. S. Serianni and R. Barker, J. Org. Chem., 1983, 48, 1750–1757 CrossRef CAS.
- E. Pretsch, P. Bühlmann and C. Affolter, in Structure and Determination of Organic Compounds, Springer-Verlag, New York, 2000, p. 166 Search PubMed.
- S. Wolfe, Acc. Chem. Res., 1972, 5, 102–111 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental methods, 1D-ge-CSSF-TOCSY and 1D-ge-CSSF-DPFGSE-NOESY spectra used in the conformational analysis; details on the equations used to determine the rotamer populations in Table 2, and 1H and 13C NMR spectra of new compounds. See DOI: 10.1039/c0ob00423e |
| This journal is © The Royal Society of Chemistry 2011 |