First total synthesis of (−)- and (+)-6-O-desmethylantofine†
Meng
Wu
,
Ling
Li
,
Bo
Su
,
Zhihui
Liu
and
Qingmin
Wang
*
State Key Laboratory of Elemento-Organic Chemistry, Research Institute of Elemento-Organic Chemistry, Nankai University, Tianjin, 300071, People's Republic of China. E-mail: wang98h@263.net; Fax: +86-22-23499842
First published on 4th November 2010
Abstract
The first total synthesis of (−)-6-O-desmethylantofine (A) and its unnatural enantiomer (+)-6-O-desmethylantofine (B) is described. The synthetic route is efficient and practical with easily available glutamic acid dimethyl ester hydrochloride as the chiral material under mild conditions.
Introduction
In 1965, Haznagy isolated a compound from C. vincetoxicum and named it C-1.1 In 1969, Wiegrebe also isolated C-1 from C. vincetoxicum and confirmed its configuration as (−)-6-O-desmethylantofine (A)2 (Fig. 1).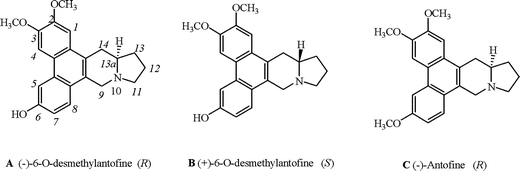 | ||
| Fig. 1 Chemical structures of compounds A, B and C. | ||
(−)-Antofine (C) (Fig. 1) is known for its extremely potent inhibition of cancer cell growth. Antofine has IC50 values in the low nanomolar range, comparable to that of clinically employed cytotoxic drugs.3 As an analogue of (−)-antofine (C), (−)-6-O-desmethylantofine (A) has attracted more and more attention in recent years. There were many reports about in vitro cytotoxic activity of (−)-6-O-desmethylantofine (A) against the KB cancer cell lines,3a,4 with IC50 values lower than that of (−)-antofine (C). Cytotoxicity significantly increased for A relative to C, indicating that the hydroxy group at C6 is of importance.
We have previously reported that (−)-antofine (C), isolated from Cynanchum komarovii, possessed excellent antiviral activity against tobacco mosaic virus (TMV).5 (−)-6-O-Desmethylantofine (A) was also isolated from the same plant for the first time and was found to exhibit better anti-TMV activity than (−)-antofine (C).6
Up to now, (−)-6-O-desmethylantofine (A) can only be obtained by isolation. However, its natural abundance is very low (about 0.003% of the extraction of the plant mentioned above),6 which restricts our research on the antiviral agent. Thus, an efficient and general approach to prepare 6-O-desmethylantofine would be very desirable. Due to the hydroxyl group at C6, the synthesis of 6-O-desmethylantofine is difficult. Only a few syntheses of (±)-6-O-desmethylantofine have been reported.4b,7 However, optically pure (−)-6-O-desmethylantofine (A) or its unnatural enantiomer (+)-6-O-desmethylantofine (B) has not been reported. Herein, we report the first total synthesis of (−)-6-O-desmethylantofine (A) and its enantiomer B.
Results and discussion
Our synthetic route is shown in Scheme 1. Perkin condensation of 3,4-dimethoxybenzaldehyde (1) and 4-hydroxyphenylacetic acid (2) could easily afford (E)-2-(4-acetoxyphenyl)-3-(3,4-dimethoxyphenyl)acrylic acid (3), which was converted to its corresponding ester 4 with methanol in the presence of concentrated sulfuric acid. Remarkably, acetyl deprotection and esterification of 3 was accomplished in one pot to afford 4.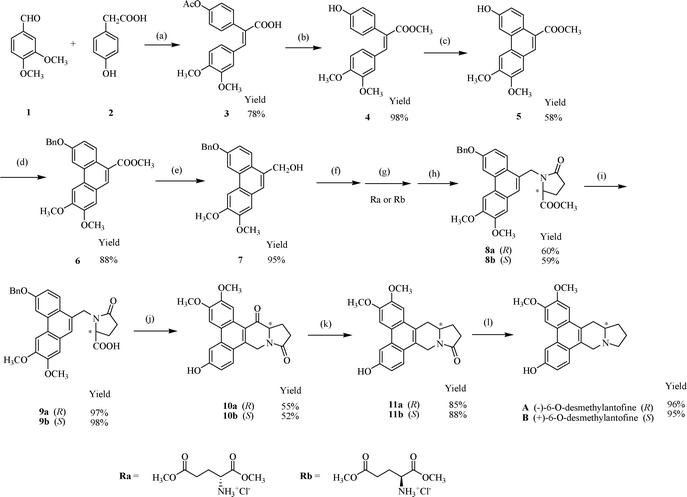 | ||
| Scheme 1 Synthesis of (−)-6-O-desmethylantofine (A) and (+)-6-O-desmethylantofine (B). Reagents and conditions: (a) Ac2O/Et3N; (b) CH3OH/concd. H2SO4; (c) FeCl3/CH2Cl2; (d) BnBr/K2CO3/CH3COCH3; (e) LiAlH4/THF; (f) PBr3/CH2Cl2; (g) Ra or Rb/K2CO3/DMF; (h) AcOH/MeOH; (i) KOH/MeOH/dioxane; (j) (COCl)2/CH2Cl2, SnCl4/CH2Cl2 (k) NaBH4/EtOH, Et3SiH/CF3COOH/CH2Cl2 (l) LiAlH4/THF. | ||
To the best of our knowledge, intramolecular oxidative coupling using oxidative coupling reagents such as thallium(III) trifluoroacetate (TTFA),8 lead(IV) tetraacetate (Pb(OAc)4),9 vanadium oxytrifluoride (VOF3),10 iron(III) chloride (FeCl3)11 and manganese(IV) dioxides (MnO2)12 is the most convenient way to construct phenanthrene ring system. To test the coupling of ester 4, we tried many oxidative coupling reagents such as VOF3, FeCl3 and MnO2. The reaction using FeCl3 gave the best yield (58%), whereas no oxidative coupling product 5 was found when using VOF3 or MnO2. The coupling of ester 4 is much more complicated than that of polymethoxy substituted 2,3-diphenylacrylate.11b,12 Then we protected the hydroxyl using a series of protecting groups such as acetyl, benzyl and methoxymethyl before coupling, but only to make the coupling reaction more complex.
Ester 5 was converted to its corresponding alcohol using lithium aluminium hydride, but the solubility of the corresponding alcohol was so poor that it was difficult to be purified, so it was impossible to be used in the following route. To improve the solubility in the following route, it is necessary to protect the hydroxy group. We introduced a benzyl group to compound 5 using benzyl bromide to afford 6.
Ester 6 was easily converted to corresponding alcohol 7 by lithium aluminium hydride. 7 was treated with phosphorus tribromide and then reacted with D-glutamic acid dimethyl ester hydrochloride followed by cyclization in warm methanol and acetic acid to afford lactam ester 8a successfully. The yield of the combined three steps from 7 to 8a is high (60%). Ester 8a was easily converted to the corresponding acid 9a with potassium hydroxide.
The benzyl derivative was stable in strong base (KOH) and strong acid (HCl) from 8a to 9a. But interestingly, using SnCl4 as Lewis acid, deprotection of benzyl group and intramolecular Friedel–Crafts acylation occurred in one pot to afford 10a from 9a.
10a was treated with sodium borohydride and then immediately converted to methylene using triethylsilane and trifluoroacetic acid to give 11a. At last, 11a was reduced with lithium aluminium hydride to afford (−)-6-O-desmethylantofine (A). The overall yield is 9.68%. By the same procedure, we got (+)-6-O-desmethylantofine (B) using L-glutamic acid dimethyl ester hydrochloride in an overall yield of 9.32%. The enantiomeric excesses of A and B were determined. The ee values were 90% and 91% respectively.
Conclusion
Optically pure (−)-6-O-desmethylantofine (A) and its enantiomer (+)-6-O-desmethylantofine (B) were first synthesized efficiently from cheap materials under mild conditions and their ee values is up to 90%. Intramolecular Friedel–Crafts acylation and deprotection of benzyl group of compounds 9a and 9b was successfully achieved in one pot using SnCl4. The chemistry described here provided a practical synthetic method of optically pure phenanthroindolizidine alkaloids for biochemical and pharmaceutical studies.Experimental
The melting points were determined with an X-4 binocular microscope melting-point apparatus (Beijing Tech Instruments Co., Beijing, China) and were uncorrected. 1H NMR spectra were obtained by using Bruker AV 400 spectrometer. Chemical shifts (δ) were given in parts per million (ppm) and were measured downfield from internal tetramethylsilane. 13C NMR spectra were recorded by using Bruker AV 400 (100 MHz) and CDCl3 or DMSO-d6 as a solvent. Chemical shifts (δ) are reported in parts per million using the solvent peak. High-resolution mass spectra were obtained with an FT-ICR MS spectrometer (Ionspec, 7.0 T). The enantiomeric excesses of A and B were determined by HPLC with a Chiralcel AD-H column using Agilent 1100 instrument. Optical rotations were recorded with a Perkin–Elmer 341 MC polarimeter. All anhydrous solvents were dried and purified by standard techniques just before use.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 7.23 (d, J = 7.6 Hz, 2 H, Ar–H), 7.17 (d, J = 7.6 Hz, 2 H, Ar–H), 6.87 (m, 2 H, Ar–H), 6.41 (s, 1 H, Ar–H), 3.72 (s, 3 H, OMe), 3.30 (s, 3 H, OMe), 2.22 (s, 3 H, COCH3); 13C NMR (100 MHz, DMSO) δ 169.2, 168.3, 149.8, 149.7, 147.8, 139.5, 134.3, 130.7, 129.6, 126.6, 125.2, 122.1, 111.9, 111.1, 55.3, 54.5, 20.8. HRMS (ESI): calcd. for C19H18O6 [M − H]+ 341.1031; found 341.1031.
CH), 7.10 (d, J = 8.4 Hz, 2 H, Ar–H), 6.83–6.85 (m, 3 H, Ar–H), 6.73 (d, J = 8.4 Hz, 1 H, Ar–H), 6.48 (d, J = 1.6 Hz, 1 H, Ar–H), 3.84 (s, 3 H, OMe), 3.79 (s, 3 H, OMe), 3.46 (s, 3 H, OMe); 13C NMR (100 MHz, CDCl3) δ 169.2, 155.6, 149.9, 148.0, 140.6, 131.2, 129.5, 128.0, 127.5, 125.5, 115.9, 112.4, 110.4, 55.7, 55.1, 52.4. HRMS (ESI): calcd. for C18H18O5 [M + Na]+ 337.1046; found 337.1041
CH), 7.23 (d, J = 7.6 Hz, 2 H, Ar–H), 7.17 (d, J = 7.6 Hz, 2 H, Ar–H), 6.87 (m, 2 H, Ar–H), 6.41 (s, 1 H, Ar–H), 3.72 (s, 3 H, OMe), 3.30 (s, 3 H, OMe), 2.22 (s, 3 H, COCH3); 13C NMR (100 MHz, DMSO) δ 169.2, 168.3, 149.8, 149.7, 147.8, 139.5, 134.3, 130.7, 129.6, 126.6, 125.2, 122.1, 111.9, 111.1, 55.3, 54.5, 20.8. HRMS (ESI): calcd. for C19H18O6 [M − H]+ 341.1031; found 341.1031.
CH), 7.10 (d, J = 8.4 Hz, 2 H, Ar–H), 6.83–6.85 (m, 3 H, Ar–H), 6.73 (d, J = 8.4 Hz, 1 H, Ar–H), 6.48 (d, J = 1.6 Hz, 1 H, Ar–H), 3.84 (s, 3 H, OMe), 3.79 (s, 3 H, OMe), 3.46 (s, 3 H, OMe); 13C NMR (100 MHz, CDCl3) δ 169.2, 155.6, 149.9, 148.0, 140.6, 131.2, 129.5, 128.0, 127.5, 125.5, 115.9, 112.4, 110.4, 55.7, 55.1, 52.4. HRMS (ESI): calcd. for C18H18O5 [M + Na]+ 337.1046; found 337.1041
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) to give 5.82 g (58%) of 5 as a white solid. mp 188–190 °C; 1H NMR (400 MHz, DMSO-d6): δ 9.91 (s, 1 H, Ar–OH), 8.66 (d, J = 9.2 Hz, 1 H, Ar–H), 8.26 (s, 1 H, Ar–H), 8.03 (s, 1 H, Ar–H), 7.94 (s, 1 H, Ar–H), 7.59 (s, 1 H, Ar–H), 7.18 (d, J = 8.8 Hz, 1 H, Ar–H), 4.04 (s, 3 H, OMe), 3.93 (s, 3 H, OMe), 3.92 (s, 3 H, OMe); 13C NMR (100 MHz, CDCl3) δ 168.3, 154.1, 150.9, 149.7, 131.9, 129.6, 128.6, 126.5, 125.5, 123.7, 123.2, 116.6, 109.1, 106.4, 103.1, 55.9, 52.1. HRMS (ESI): calcd. for C18H16O5 [M + Na]+ 335.0890; found 335.0889
:10:1, v/v/v) to give 17.28 g (88%) of 6 as a white solid. mp 156–157 °C; 1H NMR (400 MHz, CDCl3): δ 8.94 (d, J = 9.2 Hz, 1 H, Ar–H), 8.31 (s, 1H, Ar–H), 7.97 (d, J = 2.8 Hz, 1 H, Ar–H), 7.78 (s, 1 H, Ar–H), 7.54 (d, J = 7.2 Hz, 2 H, Ar–H), 740–7.44 (m, 2 H, Ar–H), 7.34–7.37 (m, 2 H, Ar–H), 7.26 (s, 1 H, Ar–H), 5.30 (s, 2 H, benzyl-CH2), 4.10 (s, 3 H, OMe), 4.03 (s, 3 H, OMe), 4.01 (s, 3 H, OMe); 13C NMR (100 MHz, CDCl3) δ 168.1, 157.2, 150.9, 149.6, 136.9, 131.6, 129.6, 128.7, 128.4, 128.1, 127.6, 126.7, 125.5, 123.7, 123.3, 116.2, 109.2, 106.0, 103.1, 70.4, 56.0, 55.9, 52.1. HRMS (ESI): calcd. for C25H22O5 [M + Na]+ 425.1359; found 425.1352
:1 then 1:1, v/v) to give 7.49 g (60%) of 8a as a white solid. mp 182–183 °C; [α]20D −56.0 (c 1.0, CH2Cl2); 1H NMR (400 MHz, CDCl3): δ 8.03 (d, J = 8.8 Hz, 1 H, Ar–H), 7.97 (d, J = 2.4 Hz, 1 H, Ar–H), 7.78 (s, 1 H, Ar–H), 7.52–7.54 (m, 2 H, Ar–H), 7.40–7.44 (m, 2 H,Ar–H), 7.34–7.37 (m, 2 H, Ar–H), 7.31 (dd, J = 2.4, 8.8 Hz, 1H, Ar–H), 7.16 (s, 1 H, Ar–H), 5.50 (d, J = 14.4 Hz, 1 H, N–CH2), 5.28 (s, 2 H, benzyl-CH2), 4.40 (d, J = 14.4 Hz, 1 H, N–CH2), 4.09 (s, 3 H, OMe), 4.03 (s, 3 H, OMe), 3.84 (dd, J = 2.8, 9.2 Hz, 1 H, N–CH), 3.58 (s, 3 H, OMe), 2.55–2.64 (m, 1 H, N–CH–CH2), 2.04–2.48 (m, 1 H, NCO–CH2), 2.08–2.18 (m, 1 H, N–CH–CH2), 1.95–2.02 (m, 1 H, NCO–CH2) 13C NMR (100 MHz, CDCl3) δ 174.6, 172.3, 157.4, 149.6, 149.4, 137.0, 131.7, 128.7, 128.1, 127.7, 127.6, 126.6, 126.2, 126.2, 124.6, 124.4, 115.8, 108.2, 106.4, 103.3, 70.4, 58.6, 56.0, 55.9, 52.2, 44.4, 29.8, 22.8. HRMS (ESI): calcd. for C30H29NO6 [M + Na]+ 522.1887; found 522.1883
:1, v/v) to give 0.62 g (55%) of 10a as a light-yellow solid. mp 260–261 °C; [α]20D −94.4 (c 0.5, DMF); 1H NMR (400 MHz, DMSO-d6): δ 10.41(s, 1 H, Ar–OH), 8.99 (s, 1 H, Ar–H), 8.14 (d, J = 8.8 Hz, 1 H, Ar–H), 8.02 (d, J = 2.0 Hz, 1 H, Ar–H), 7.95 (s, 1 H, Ar–H), 7.23 (dd, J = 2.0, 8.8 Hz, 1 H, Ar–H), 5.55 (d, J = 18.4 Hz, 1 H, 9-H), 4.78 (d, J = 18.0 Hz, 1 H, 9-H), 4.54–4.57 (m, 1 H,13a-H), 4.02 (s, 3 H, OMe), 3.91 (s, 3 H, OMe), 2.30–2.44 (m, 4 H, 12-H, 13-H); 13C NMR (100 MHz, DMSO-d6) δ 196.1, 172.8, 159.2, 149.6, 148.4, 139.6, 133.9, 127.3, 123.7, 123.3, 120.3, 119.8, 117.5, 107.4, 106.7, 103.5, 60.5, 55.3, 55.1, 39.9, 29.4, 20.4. HRMS (ESI): calcd. for C22H19NO5 [M + Na]+ 400.1155; found 400.1152
:3, v/v and then CH2Cl2–EtOAc, 1:8, v/v) to give 0.34 g (85%) of 11a as a light-yellow solid. mp 221 °C dec; [α]20D −85.6 (c 1, DMF); 1H NMR (400 MHz, CDCl3): δ 7.76 (s, 1 H, Ar–H), 7.74 (s, 1 H, Ar–H), 7.55 (d, J = 8.8 Hz, 1 H, Ar–H), 7.10 (d, J = 8.4 Hz, 1 H, Ar–H), 7.04 (s, 1 H, Ar–H), 6.94 (s, 1 H, OH), 5.30 (d, J = 17.2 Hz, 1 H, 9-H), 4.47 (d, J = 17.2 Hz, 1 H, 9-H), 4.06 (s, 3 H, OMe), 4.01 (s, 3 H,OMe), 3.93–3.94 (m, 1H, 13a-H), 3.30–3.34 (m, 1H, 14-H), 2.53–2.78 (m, 4H, 14-H, 12-H, 13-H), 2.00–2.09 (m, 1H, 13-H); 13C NMR (100 MHz, DMSO-d6) δ 173.1, 155.6, 149.0, 148.2, 130.0, 125.7, 124.0, 123.4, 123.2, 122.8, 121.6, 116.5, 106.5, 104.1, 103.9, 55.4, 55.3, 52.3, 40.1, 32.1, 29.4, 24.4. HRMS (ESI): calcd. for C22H21NO4 [M + H]+ 364.1543; found 364.1540
:15 and 0.1% triethylamine, 254 nm, tR (major) = 22.75 min, tR (minor) = 25.14 min]. 1H NMR (400 MHz, DMSO-d6): δ 9.69 (s,1 H, OH), 7.95 (s, 1 H, Ar–H), 7.92 (s, 1 H, Ar–H), 7.73 (d, J = 8.8 Hz, 1 H, Ar–H), 7.31 (s, 1 H, Ar–H), 7.09 (d, J = 9.2 Hz, 1 H, Ar–H), 4.55 (d, J = 15.2 Hz, 1 H, 9-H), 3.99 (s, 3 H, OMe), 3.94 (s, 3 H, OMe), 3.53 (d, J = 14.8 Hz, 1 H, 9-H), 3.30–3.35 (m, 2 H, 13a-H, 11-H), 2.72–2.78 (m, 1 H, 11-H), 2.32–2.38 (m, 2 H, 14-H), 2.12–2.18 (m, 1 H, 13-H), 1.82–1.90 (m, 2 H, 12-H), 1.59–1.69 (m, 1 H, 13-H); 13C NMR (100 MHz, DMSO-d6) δ 155.3, 149.0, 148.0, 129.9, 126.3, 126.1, 124.4, 124.0, 122.6, 122.2, 116.2, 106.4, 104.0, 103.9, 59.8, 55.4, 55.3, 54.4, 53.2, 32.9, 30.8, 21.1. HRMS (ESI): calcd. for C22H23NO3 [M + H]+ 350.1751; found 350.1753
:15 and 0.1% triethylamine, 254 nm, tR (major) = 25.02 min, tR (minor) = 22.80 min]. other data are the same as those for B.
:1, v/v) to give 5.82 g (58%) of 5 as a white solid. mp 188–190 °C; 1H NMR (400 MHz, DMSO-d6): δ 9.91 (s, 1 H, Ar–OH), 8.66 (d, J = 9.2 Hz, 1 H, Ar–H), 8.26 (s, 1 H, Ar–H), 8.03 (s, 1 H, Ar–H), 7.94 (s, 1 H, Ar–H), 7.59 (s, 1 H, Ar–H), 7.18 (d, J = 8.8 Hz, 1 H, Ar–H), 4.04 (s, 3 H, OMe), 3.93 (s, 3 H, OMe), 3.92 (s, 3 H, OMe); 13C NMR (100 MHz, CDCl3) δ 168.3, 154.1, 150.9, 149.7, 131.9, 129.6, 128.6, 126.5, 125.5, 123.7, 123.2, 116.6, 109.1, 106.4, 103.1, 55.9, 52.1. HRMS (ESI): calcd. for C18H16O5 [M + Na]+ 335.0890; found 335.0889
:10:1, v/v/v) to give 17.28 g (88%) of 6 as a white solid. mp 156–157 °C; 1H NMR (400 MHz, CDCl3): δ 8.94 (d, J = 9.2 Hz, 1 H, Ar–H), 8.31 (s, 1H, Ar–H), 7.97 (d, J = 2.8 Hz, 1 H, Ar–H), 7.78 (s, 1 H, Ar–H), 7.54 (d, J = 7.2 Hz, 2 H, Ar–H), 740–7.44 (m, 2 H, Ar–H), 7.34–7.37 (m, 2 H, Ar–H), 7.26 (s, 1 H, Ar–H), 5.30 (s, 2 H, benzyl-CH2), 4.10 (s, 3 H, OMe), 4.03 (s, 3 H, OMe), 4.01 (s, 3 H, OMe); 13C NMR (100 MHz, CDCl3) δ 168.1, 157.2, 150.9, 149.6, 136.9, 131.6, 129.6, 128.7, 128.4, 128.1, 127.6, 126.7, 125.5, 123.7, 123.3, 116.2, 109.2, 106.0, 103.1, 70.4, 56.0, 55.9, 52.1. HRMS (ESI): calcd. for C25H22O5 [M + Na]+ 425.1359; found 425.1352
:1 then 1:1, v/v) to give 7.49 g (60%) of 8a as a white solid. mp 182–183 °C; [α]20D −56.0 (c 1.0, CH2Cl2); 1H NMR (400 MHz, CDCl3): δ 8.03 (d, J = 8.8 Hz, 1 H, Ar–H), 7.97 (d, J = 2.4 Hz, 1 H, Ar–H), 7.78 (s, 1 H, Ar–H), 7.52–7.54 (m, 2 H, Ar–H), 7.40–7.44 (m, 2 H,Ar–H), 7.34–7.37 (m, 2 H, Ar–H), 7.31 (dd, J = 2.4, 8.8 Hz, 1H, Ar–H), 7.16 (s, 1 H, Ar–H), 5.50 (d, J = 14.4 Hz, 1 H, N–CH2), 5.28 (s, 2 H, benzyl-CH2), 4.40 (d, J = 14.4 Hz, 1 H, N–CH2), 4.09 (s, 3 H, OMe), 4.03 (s, 3 H, OMe), 3.84 (dd, J = 2.8, 9.2 Hz, 1 H, N–CH), 3.58 (s, 3 H, OMe), 2.55–2.64 (m, 1 H, N–CH–CH2), 2.04–2.48 (m, 1 H, NCO–CH2), 2.08–2.18 (m, 1 H, N–CH–CH2), 1.95–2.02 (m, 1 H, NCO–CH2) 13C NMR (100 MHz, CDCl3) δ 174.6, 172.3, 157.4, 149.6, 149.4, 137.0, 131.7, 128.7, 128.1, 127.7, 127.6, 126.6, 126.2, 126.2, 124.6, 124.4, 115.8, 108.2, 106.4, 103.3, 70.4, 58.6, 56.0, 55.9, 52.2, 44.4, 29.8, 22.8. HRMS (ESI): calcd. for C30H29NO6 [M + Na]+ 522.1887; found 522.1883
:1, v/v) to give 0.62 g (55%) of 10a as a light-yellow solid. mp 260–261 °C; [α]20D −94.4 (c 0.5, DMF); 1H NMR (400 MHz, DMSO-d6): δ 10.41(s, 1 H, Ar–OH), 8.99 (s, 1 H, Ar–H), 8.14 (d, J = 8.8 Hz, 1 H, Ar–H), 8.02 (d, J = 2.0 Hz, 1 H, Ar–H), 7.95 (s, 1 H, Ar–H), 7.23 (dd, J = 2.0, 8.8 Hz, 1 H, Ar–H), 5.55 (d, J = 18.4 Hz, 1 H, 9-H), 4.78 (d, J = 18.0 Hz, 1 H, 9-H), 4.54–4.57 (m, 1 H,13a-H), 4.02 (s, 3 H, OMe), 3.91 (s, 3 H, OMe), 2.30–2.44 (m, 4 H, 12-H, 13-H); 13C NMR (100 MHz, DMSO-d6) δ 196.1, 172.8, 159.2, 149.6, 148.4, 139.6, 133.9, 127.3, 123.7, 123.3, 120.3, 119.8, 117.5, 107.4, 106.7, 103.5, 60.5, 55.3, 55.1, 39.9, 29.4, 20.4. HRMS (ESI): calcd. for C22H19NO5 [M + Na]+ 400.1155; found 400.1152
:3, v/v and then CH2Cl2–EtOAc, 1:8, v/v) to give 0.34 g (85%) of 11a as a light-yellow solid. mp 221 °C dec; [α]20D −85.6 (c 1, DMF); 1H NMR (400 MHz, CDCl3): δ 7.76 (s, 1 H, Ar–H), 7.74 (s, 1 H, Ar–H), 7.55 (d, J = 8.8 Hz, 1 H, Ar–H), 7.10 (d, J = 8.4 Hz, 1 H, Ar–H), 7.04 (s, 1 H, Ar–H), 6.94 (s, 1 H, OH), 5.30 (d, J = 17.2 Hz, 1 H, 9-H), 4.47 (d, J = 17.2 Hz, 1 H, 9-H), 4.06 (s, 3 H, OMe), 4.01 (s, 3 H,OMe), 3.93–3.94 (m, 1H, 13a-H), 3.30–3.34 (m, 1H, 14-H), 2.53–2.78 (m, 4H, 14-H, 12-H, 13-H), 2.00–2.09 (m, 1H, 13-H); 13C NMR (100 MHz, DMSO-d6) δ 173.1, 155.6, 149.0, 148.2, 130.0, 125.7, 124.0, 123.4, 123.2, 122.8, 121.6, 116.5, 106.5, 104.1, 103.9, 55.4, 55.3, 52.3, 40.1, 32.1, 29.4, 24.4. HRMS (ESI): calcd. for C22H21NO4 [M + H]+ 364.1543; found 364.1540
:15 and 0.1% triethylamine, 254 nm, tR (major) = 22.75 min, tR (minor) = 25.14 min]. 1H NMR (400 MHz, DMSO-d6): δ 9.69 (s,1 H, OH), 7.95 (s, 1 H, Ar–H), 7.92 (s, 1 H, Ar–H), 7.73 (d, J = 8.8 Hz, 1 H, Ar–H), 7.31 (s, 1 H, Ar–H), 7.09 (d, J = 9.2 Hz, 1 H, Ar–H), 4.55 (d, J = 15.2 Hz, 1 H, 9-H), 3.99 (s, 3 H, OMe), 3.94 (s, 3 H, OMe), 3.53 (d, J = 14.8 Hz, 1 H, 9-H), 3.30–3.35 (m, 2 H, 13a-H, 11-H), 2.72–2.78 (m, 1 H, 11-H), 2.32–2.38 (m, 2 H, 14-H), 2.12–2.18 (m, 1 H, 13-H), 1.82–1.90 (m, 2 H, 12-H), 1.59–1.69 (m, 1 H, 13-H); 13C NMR (100 MHz, DMSO-d6) δ 155.3, 149.0, 148.0, 129.9, 126.3, 126.1, 124.4, 124.0, 122.6, 122.2, 116.2, 106.4, 104.0, 103.9, 59.8, 55.4, 55.3, 54.4, 53.2, 32.9, 30.8, 21.1. HRMS (ESI): calcd. for C22H23NO3 [M + H]+ 350.1751; found 350.1753
:15 and 0.1% triethylamine, 254 nm, tR (major) = 25.02 min, tR (minor) = 22.80 min]. other data are the same as those for B.
Acknowledgements
We are grateful to the National Key Project for Basic Research (2010CB126100) and the National Natural Science Foundation of China (20872072, 30890143) for generous financial support for our programsNotes and references
-
(a) A. Haznagy, K. Szendrei and L. Toth, Pharmazie, 1965, 20(9), 541–548 CAS
; (b) A. Haznagy, L. Toth and K. Szendrei, Pharmazie, 1965, 20, 649–651 CAS
- W. Wiegrebe, L. Faber, H. Brockman and K. U. Bidzikiewicz, Justus Liebigs Ann. Chem., 1969, 721, 154–162 CAS
-
(a) D. Staérk, A. K. Lykkeberg, J. Christensen, B. A. Budnik, F. Abe and J. W. Jaroszewski, J. Nat. Prod., 2002, 65, 1299–1302 CrossRef
-
(a) A. Toribio, A. Bonfils, E. Delannay, E. Prost, D. Harakat, E. Henon, B. Richard, M. Litaudon, J.-M. Nuzillard and J.-H. Renault, Org. Lett., 2006, 8(17), 3825–3828 CrossRef CAS
-
(a) T. Y. An, R. Q. Huang, Z. Yang, D. K. Zhang, G. R. Li, Y. C. Yao and J. Gao, Phytochemistry, 2001, 58, 1267–1269 CrossRef CAS :76069].
-
(a)
Q. M. Wang, K. L. Wang, Z. Q. Huang, Y. X. Liu, H. Li, T. S. Hu, Z. Jin, Z. J. Fan, R. Q. Huang CN 101189968, 2008 [Chem.Abstr. 149:97630];
(b) Z. Q. Huang, Y. X. Liu, Z. J. Fan, Q. M. Wang, G. R. Li, Y. C. Yao, X. S. Yu and R. Q. Huang, Fine Chemical Intermediates (Ch.), 2007, 37, 20–24 Search PubMed
-
(a) W. Wiegrebe, L. Faber and H. Budzikiewicz, Justus Liebigs Ann. Chem., 1970, 733, 125–140 CrossRef CAS
-
(a) E. C. Taylor, J. G. Andrade, G. J. H. Rall and A. Mckillop, J. Am. Chem. Soc., 1980, 102, 6513–6519 CrossRef CAS
- K. S. Feldman and S. M. Ensel, J. Am. Chem. Soc., 1994, 116, 3357–3366 CrossRef CAS
-
(a) B. Halton, A. I. Maidment, D. L. Officer and J. M. Warner, Aust. J. Chem., 1984, 37, 2119–2128 CAS
-
(a) K. L. Wang, M. Y. Lü, A. Yu, X. Q. Zhu and Q. M. Wang, J. Org. Chem., 2009, 74, 935–938 CrossRef CAS
- K. L. Wang, Y. N. Hu, Z. Li, M. Wu, Z. H. Liu, B. Su, A. Yu, Y. Liu and Q. M. Wang, Synthesis, 2009, 7, 1083–1090
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C NMR and HRMS spectra for the compounds 1–7, 8a–11a, A. (To avoid repetition, the NMR and HRMS spectra for 8b–11b, B are not given.) See DOI: 10.1039/c0ob00287a |
| This journal is © The Royal Society of Chemistry 2011 |