Guanidiniocarbonyl-pyrrole-aryl conjugates as nucleic acid sensors: switch of binding mode and spectroscopic responses by introducing additional binding sites into the linker†
Kathrin
Gröger
a,
Domagoj
Baretić
b,
Ivo
Piantanida
*b,
Marko
Marjanović
c,
Marijeta
Kralj
c,
Marina
Grabar
d,
Sanja
Tomić
d and
Carsten
Schmuck
*a
aInstitute for Organic Chemistry, University of Duisburg-Essen, Universitässtrasse 7, 45141, Essen, Germany. E-mail: carsten.schmuck@uni-due.de; Fax: (+) 201 183 4259
bDivision of Organic Chemistry and Biochemistry, “Ruđer Bošković” Institute, P. O. Box 180, HR-10002, Zagreb, Croatia. E-mail: Ivo.Piantanida@irb.hr
cDivision of Molecular Medicine, “Ruđer Bošković” Institute, Zagreb, Croatia
dLaboratory for Chemical and Biological Crystallography, “Ruđer Bošković” Institute, P. O. Box 180, HR-10002, Zagreb, Croatia
First published on 15th November 2010
Abstract
Two novel guanidiniocarbonyl pyrrole–pyrene conjugates 3 and 4 as spectroscopic probes for ds-polynucleotides were synthesized and their interaction with different ds-DNAs/RNAs studied. Compared to a previously reported first set of conjugates (1 and 2) the significant extension and increased rigidity of the central part of the structure resulted in a switch of DNA binding mode from intercalative (previously studied derivatives 1 and 2 with a nonbinding and flexible linker) to minor groove binding of the two novel guanidiniocarbonyl-pyrrole–pyrene conjugates 3 and 4. These two compounds interact strongly with ds-DNAs, but only weakly with ds-RNA. The newly incorporated heterocyclic moieties within the central part of the structure of 3 and 4 were able to control by steric and hydrogen-bonding effects the alignment of the molecules within various, structurally different forms of DNA minor grooves, whereby even small differences in the position of the attached pyrene within the groove were reflected in different fluorimetric responses. In addition, 3 and 4 revealed intriguing in vitro selectivity among various human tumour cell lines.
Introduction
Small molecules targeting DNA/RNA have attracted significant scientific interest due to the medicinal, biochemical and biological implications of such molecular recognition events.1 Most often these small molecules rely on one dominant non-covalent binding mode for their interaction with double stranded (ds) DNA/RNA (e.g. intercalation, minor or major groove binding, external electrostatic binding).2 However, the combination of different binding modes within one molecule as well as additional steric and structural features controlling three-dimensional recognition drew some attention over the last few years with the aim of developing new more selective drugs or biochemical markers as well as research tools on a molecular level. For instance, methods for sequence-specific detection of ds-DNA and ds-RNA are becoming increasingly useful and important as diagnostic and imaging tools, among which fluorescent techniques are representing about 60% of the detection enabling technologies used in molecular biology and medicine.3 However, the design and synthesis of novel small molecules with multiple DNA/RNA-binding features within a structurally well defined architecture is still highly demanding. Within the last two years we have in this context developed guanidiniocarbonyl-pyrrole-aryl hybrid compounds in which a guanidiniocarbonyl pyrrole cation was connected via a flexible linker to an aromatic moiety.4,5 Systematic studies of a series of such compounds were undertaken with the intention to a) determine the minimal size of the aryl-moiety necessary for efficient “anchoring” of the small molecule within ds-DNA/RNA by intercalation; b) to define structural properties (length, flexibility) of the linker between the guanidiniocarbonyl-pyrrole cation and the aryl group, which allows efficient binding of the cation to DNA/RNA after intercalation of the aromatic unit and c) to find an appropriate fluorescence probe to report the binding event in a selective manner. Up till now, we determined that for our compounds the minimal aryl-moiety required for efficient intercalation was acridine and a butyl linker provided sufficient length and flexibility to allow simultaneous intercalation of the aryl unit and groove binding of the cation.Even more efficient for intercalation was a pyrene, and thus the two hybrid compounds 1 and 2 revealed selective and even sequence specific fluorescence responses to several different DNA or RNA sequences.5 While the aliphatic, flexible linker in 1 was inert with respect to DNA/RNA interactions, the positive charge of the lysine side chain in the linker of a second generation compound 2 contributed significantly to DNA/RNA binding.4,5 This was attributed to additional charge interactions with the nucleic acid backbone. This binding mode was also supported by preliminary molecular modelling studies of a 2/poly dAdT-poly dAdT complex which hinted to the importance of anchoring the pyrene by intercalation into DNA in order to allow the accommodation of the guanidiniocarbonyl pyrrole moiety within the DNA minor groove. However, inspired by the previously noted RNA-specific fluorescence change of 1, which was caused by pyrene-excimer formation within the RNA groove,4 we have now designed and prepared two novel pyrenyl-guanidiniocarbonyl pyrrole hybrid compounds 3 and 4 (Scheme 1). In these compounds the linker between pyrene and the guanidinio-pyrrole moiety was significantly modified by introducing additional aromatic moieties next to the pyrene (a methyl imidazole in 3 or a pyrrole-methyl imidazole unit in 4). These heterocycles themselves are able to interact with DNA/RNA as shown by Dervan and coworkers.6 However, these building blocks also significantly rigidified the linker. Most likely a simultaneous intercalation of the pyrene into DNA/RNA and interactions of the linker within DNA/RNA grooves will not be possible. It was therefore expected that a competition between those two binding modes would occur and perhaps might be highly sensitive to the nucleic acid basepair composition and secondary structure.
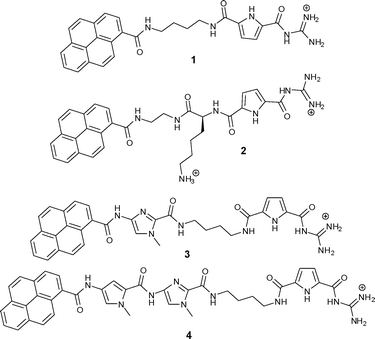 | ||
| Scheme 1 Two previously studied (1 and 2) and two novel (3 and 4) guanidiniocarbonyl-pyrrole-aryl conjugates with different linkers. | ||
Results and discussion
Synthesis
The syntheses of the novel hybrid compounds 3 and 4 were achieved by two analogous procedures as shown in Schemes 2 and 3. Compounds 6 and 14 which are necessary for both syntheses were prepared analogous to the method of Dervan et al.7 and compound 12 was available according to a procedure reported earlier by us.8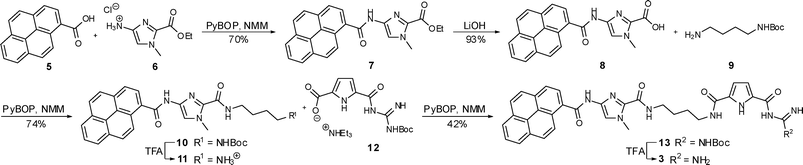 | ||
| Scheme 2 Synthesis of compound 3. | ||
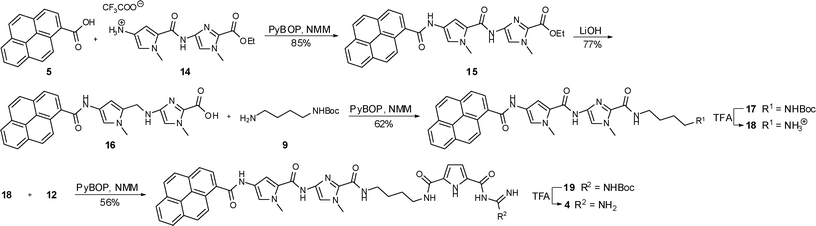 | ||
| Scheme 3 Synthesis of compound 4. | ||
The synthesis of compound 3 (Scheme 2) started with a standard coupling reaction between pyrene carboxylic acid 5 and the imidazole salt 6 by PyBOP activation. After the removal of the ethyl ester in 7 by basic hydrolysis the free acid 8 was activated with PyBOP and then reacted with N-Boc-protected 1,4-diaminobutane 9 to the protected compound 10. Subsequently the Boc protecting group was removed from 10 by the use of trifluoroacetic acid (TFA) yielding the ammonium salt 11, which was afterwards linked to the carboxylate 12 in a PyBOP activated coupling reaction. The desired end product 3 could then be obtained after a final deprotection using TFA. The preparation of compound 4 is shown in Scheme 3. The starting materials were an imidazole-pyrrole salt 14 and again pyrene carboxylic acid 5 which were coupled with the aid of PyBOP to obtain compound 15. After a basic hydrolysis of the ethyl ester in 15 the PyBOP activated free acid 16 was treated with 9 to yield protected compound 17. Boc-removal under acidic conditions (TFA) and a subsequent PyBOP mediated coupling reaction between compound 18 and ammonium salt 12 led to protected compound 19. Finally TFA was used to remove the protecting group and the end product 4 could be isolated.
Physico-chemical and spectroscopic properties of aqueous solutions of 3 and 4
Compounds 3 and 4 were well soluble in water within a 10−4 mol dm−3 range and the absorbencies were found to be proportional to the concentration up to c = 5 × 10−5 mol dm−3 indicating that there is no significant intermolecular stacking which should give rise to hypochromicity effects. Absorption maxima and corresponding molar extinction coefficients (ε) are given in Table 1. In buffer at pH 7 and pH 5 UV/Vis spectra were identical to pure water. Heating of the aqueous solutions of either compound up to 90 °C did not yield any significant changes in the UV/Vis spectra proving the chemical stability of the compounds under these conditions.| λ max/nm (ε × 103/dm3 mol−1 cm−1) | |
|---|---|
| a Previous results.4,5 b Determined at pH 5 (sodium citrate buffer, I = 0.03 mol dm−3) due to poor solubility at pH 7. | |
| 1 a | 242 (24.2); 276 (38.1); 303 (28.1); 342 (20.4) |
| 2 a | 231 (58.17); 242 (48.2); 276 (33.7); 307 (28.6); 344 (18.2); 377 (1.8) |
| 3 b | 295 (29.9); 355 (22.5) |
| 4 b | 295 (21); 355 (17.8) |
According to the comparison of the absorption maxima (Table 1) with the previously prepared compounds 1 and 2,4,5 the maximum at λ = 295 nm could be attributed to the guanidiniocarbonyl pyrrole moiety, while the maximum at λ = 355 nm belongs to pyrene. A more detailed comparison of the data presented in Table 1 indicates more simplified UV/Vis spectra for 3 and 4 compared to 1 and 2, most likely due to the overlapping of absorption bands of the guanidiniocarbonyl pyrrole cation with the absorption bands of the pyrrole and imidazole units incorporated into the linker.
Both 3 and 4 showed fluorescence emission, linearly dependent on the concentration of the compound in water up to c = 5.0 × 10−6 mol dm−3 (Fig. 1). At higher concentrations increase of fluorescence emission became non-proportional due to the inner filter effects. The fluorescence of 3 or 4 was weakly dependent on pH in the range pH = 3–8, most likely due to the reversible protonation and deprotonation of the guanidiniocarbonyl pyrrole moiety. This pH-dependent spectral change allowed us to estimate a pKa ≈ 5.5, indicating that at pH 7, 3 and 4 are less protonated than at pH 5.
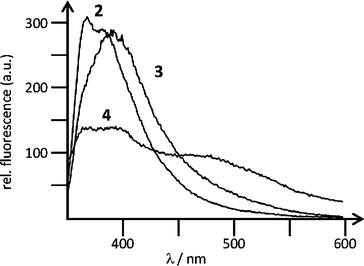 | ||
| Fig. 1 Fluorescence emission spectra of 2 (λexc = 344 nm, c = 1.0 × 10−6 mol dm−3), 3 (λexc = 355 nm, c = 5.0 × 10−7 mol dm−3), 4 (λexc = 355 nm, c = 2.0 × 10−6 mol dm−3), collected under the same instrument setup, at pH 5 (sodium citrate buffer, I = 0.03 mol dm−3). | ||
Interactions with polynucleotides in water
Both 3 and 4 precipitated at pH 7 upon addition of any DNA and RNA studied at higher concentrations, which was not the case at pH 5 due to the better solubility of 3 and 4 caused by protonation of the guanidiniocarbonyl pyrrole moiety. Therefore all further experiments were done at pH 5.| 1 e | 2 e | 3 | 4 | |
|---|---|---|---|---|
| a Error in ΔTm: ±0.5 °C. b r = [compound]/[polynucleotide]. c Biphasic transitions: the first transition at Tm = 30.3 °C is attributed to denaturation of poly rA-poly rU and the second transition at Tm = 85.8 °C is attributed to denaturation of poly rAH+-poly rAH+ since poly rA at pH = 5 is mostly protonated and forms ds-polynucleotide.9,10 d Not determined. e Previous results.4,5 | ||||
| ct-DNA | +7.2 | +9.7 | +4.0 | +7.0 |
| poly dA - poly dT | d | +11.7 | +4.2 | +7.3 |
| poly rA - poly rUc | −1.5/0 | 0 | 0 | 0 |
| poly dAdT - poly dAdT | poly dGdC - poly dGdC | poly dA - poly dT | poly rA - poly rU | |
|---|---|---|---|---|
| a Emission change; I = I(complex)/I(compd). b I(complex) obtained from Scatchard analysis of titration data. c I(complex) estimated from titration data since Scatchard analysis was not possible due to small changes, changes in opposite directions or linear change abruptly ending at well-defined intensity. | ||||
| 3 | b0.5 | b0.7 | 1 | 1 |
| 4 | 1 | c0.9 | b1.3 | c1.1 |
![Fluorimetric titrations of 3 (left column) and 4 (right column) (c(3, 4) = 5.0 × 10−7 mol dm−3, λexc = 355 nm) with various ds-polynucleotides at ratio r[compound]/[polynucleotide] = 0–0.01. Done at pH 5 (sodium citrate buffer, I = 0.03 M).](/image/article/2011/OB/c0ob00103a/c0ob00103a-f2.gif) | ||
| Fig. 2 Fluorimetric titrations of 3 (left column) and 4 (right column) (c(3, 4) = 5.0 × 10−7 mol dm−3, λexc = 355 nm) with various ds-polynucleotides at ratio r[compound]/[polynucleotide] = 0–0.01. Done at pH 5 (sodium citrate buffer, I = 0.03 M). | ||
The titration data with measurable fluorescence changes were processed by means of the Scatchard equation11 using non-linear curve fitting procedures to obtain the binding constants and ratio n[bound compound]/[polynucleotide] (Table 4). Similar binding constants (logKs = 5–6) suggest roughly comparative affinity of 3 and 4 toward most ds-DNAs, which also agrees well with the previously determined affinities of their close analogues 1 and 2.4,5
| dAdT-dAdT logKs (n) | dGdC-dGdC logKs (n) | dAdT logKs (n) | rA-rU logKs (n) | |
|---|---|---|---|---|
| a Titration data were processed using the Scatchard equation, accuracy of obtained n ± 10–30%, consequently log Ks values vary in the same order of magnitude. b Too small changes or accurate calculation. | ||||
| 2 | 6.5 (0.3) | 6.3 (0.2) | b | 5.4 (0.2) |
| 3 | 6.0 (0.2) | 5.5 (0.2) | b | b |
| 4 | b | b | 6.9 (0.1) | b |
![Changes in the CD spectra of various polynucleotides (c = 1.0 × 10−5 mol dm−3) upon addition of 3 (left column) or 4 (right column) at ratios r = [compound]/[polynucleotide] at pH 5.0 (sodium citrate buffer, I = 0.03 mol dm−3).](/image/article/2011/OB/c0ob00103a/c0ob00103a-f3.gif) | ||
| Fig. 3 Changes in the CD spectra of various polynucleotides (c = 1.0 × 10−5 mol dm−3) upon addition of 3 (left column) or 4 (right column) at ratios r = [compound]/[polynucleotide] at pH 5.0 (sodium citrate buffer, I = 0.03 mol dm−3). | ||
Opposite to ds-DNA, addition of any compound resulted in only minor changes in the positive bands of the ds-RNA polynucleotide (poly rA-poly rU, data not shown). The absence of any ICD band in all CD titrations with ds-RNA excludes a uniform orientation of 3 and 4 with respect to the chiral axis of ds-RNA.12,14 The changes in the CD spectra were almost proportional to the ratio r, thus showing no saturation of binding sites even at an excess of compound over ds-RNA. These results suggest non-specific agglomeration of 3 and 4 along the ds-RNA helix caused by hydrophobic and electrostatic interactions.
Molecular modelling
The sensitivity of the fluorescence and ICD bands of 3 and 4 on various secondary structures of ds-DNA/RNA polynucleotides could be attributed to a fine interplay between rather rigid and structurally demanding compounds and differences in binding sites within the DNA/RNA studied. Since experimental results suggested that 3 and 4 bind within the DNA minor groove in contrast to the combined intercalative–minor groove binding of previously studied compound 2, we performed molecular modelling studies of complexes of 2, 3 and 4 bound to the well defined β-helical structure of poly dAdT-poly dAdT.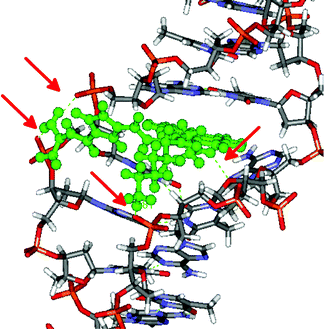 | ||
| Fig. 4 The 2/poly dAdT-poly dAdT complex obtained as a result of 12.5 ns of MD simulations. Red arrows indicate hydrogen bonds. | ||
Apparently the intercalative binding of pyrene within ds-DNA was additionally stabilized by several hydrogen bonds within the DNA minor groove and to the phosphate backbone. The obtained structure of the 2/poly dAdT-poly dAdT complex is in a good agreement with the previously published modelling studies of its close analogue 1 with DNA.4,5 The additional electrostatic interaction of positively charged amine of 2 with the DNA correlates nicely to its stronger binding and thermal stabilization effect of 2 in comparison to 1.5
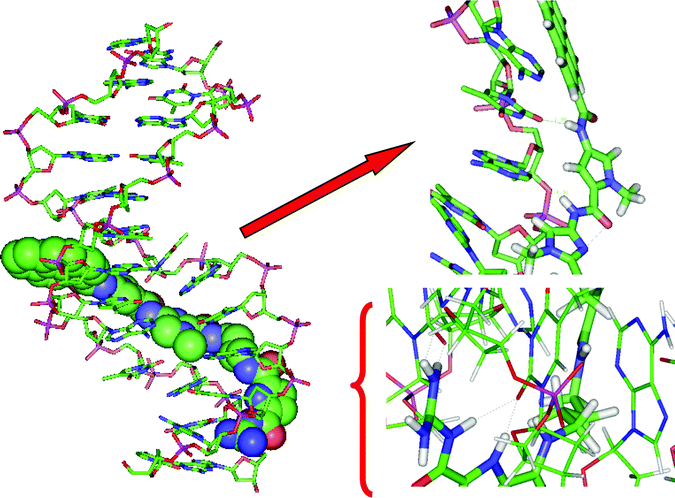 | ||
| Fig. 5 Molecular modelling of 4/poly dAdT-poly dAdT complex, left: complete structure, right: enlarged segments. Dotted grey lines indicate hydrogen bonds. | ||
During 10.5 ns MD simulations, 3 and 4 remained stable in the poly dAdT-poly dAdT minor groove. In the final structure of the complexes, after 10.5 ns of MD simulations, there are several intermolecular hydrogen bonds: four in the 3/poly dAdT-poly dAdT complex and five in the 4/poly dAdT-poly dAdT complex. In the 3/poly dAdT-poly dAdT complex the NH from the pyrrole ring of the guanidiniocarbonyl pyrrole tail forms hydrogen bonds with two neighbouring bases, namely with carbonyl oxygens of two thymine bases; the same oxygens are hydrogen bonded to the amine group from the amide bond between the pyrene moiety and the terminal guanidine. Besides, in the final structure the terminal NH2 group of 3 has established NH-π interactions with the neighbouring base (guanine). The final 4/poly dAdT-poly dAdT complex is stabilized by a hydrogen bond between the amide bond attached to the pyrene moiety of 4 and the neighbouring base (thymine carbonyl oxygen), by two hydrogen bonds between the terminal guanidine and the neighbouring DNA bases, adenine and thymine, combined with one additional hydrogen bond between NH from the terminal amide bond and the neighbouring base (thymine). In addition, pyrene forms twisted “edge-to-face” aromatic stacking interactions with the neighbouring adenine. In both complexes the carbonyl oxygen atoms of the linkers are pointing into the water. Apparently binding of 3 and 4 into the minor groove of poly dAdT-poly dAdT is additionally stabilized by the electrostatic interactions between phosphate groups of the polynucleotide and the positive side of the amide dipoles.
Biological activity
Compounds 3 and 4 were screened for their potential antiproliferative effects on a panel of 5 human cell lines, which were derived from different cancer types including HeLa (cervical carcinoma), MCF-7 (breast carcinoma), SW620 (colon carcinoma), MiaPaCa-2 (pancreatic carcinoma), and H460 (lung carcinoma) (Table 5). Both 3 and 4 showed similar antiproliferative effects against the majority of cell lines similar to the previously studied 2. A significant difference was observed on the SW 620 cell line, on which 3 and 4 revealed antiproliferative activity more than an order of magnitude higher compared to the effect of 2. It is difficult to speculate about the mechanisms underlying this selectivity based just on the preliminary antiproliferative data presented here. However, a pronounced sensitivity of SW 620 cells is certainly an encouraging result for further more detailed biological studies, since this cell line represents a highly metastatic cell line, derived from a lymph node metastasis of a primary colon adenocarcinoma.Conclusions
In this work the flexible and inert aliphatic linker of 2 was extended and rigidified by one (3) or two (4) heterocyclic moieties able to further interact with DNA and RNA by forming additional hydrogen bonds. Such an extension had profound impact on the DNA/RNA binding properties of the compounds. Both 3 and 4 interacted strongly with ds-DNAs, but only weakly with ds-RNA. Observed ICD bands at λ = 310 nm reveal that the guanidiniocarbonyl-pyrrole moiety of 3 and 4 is positioned within the minor groove of all ds-DNAs analogous to previously studied derivative 2.4 However, in contrast to the negative ICD bands at >330 nm attributed to intercalation of the pyrene of 2 into ds-DNA,53 and 4 with most ds-DNAs yielded positive ICD bands attributed to the pyrene moiety (λ = 330–350 nm), strongly indicating the positioning of pyrene within the minor groove of DNA double helix. Furthermore, positioning of 3 and 4 within the DNA minor groove was observed to be strongly dependent on the structural properties of the minor groove. This could explain the absence of any ICD band of the pyrene moiety of 3 and 4 for poly dA-poly dT, which is characterized by a peculiar, twisted helix and much narrower minor groove16 in comparison to the more common β-helix of the other ds-DNAs.10 Molecular modelling studies performed on poly dAdT-poly dAdT/3 and 4 complexes revealed in more detail the nice structural fitting of complete molecules into the minor groove of the classical DNA β-helix, and in addition supported strong binding revealing the presence of at least 4–5 hydrogen bonds between 3 and 4 and DNA. Moreover, molecular modelling results revealed for both 3 and 4 possible “edge-to-face” aromatic interactions between pyrene and the DNA base pairs, which could be correlated to the different fluorimetric responses of 3 and 4 which depend on the structure of the DNA minor groove.The most prominent consequence of the switch of the DNA binding mode from pyrene intercalation (previously studied compounds 1, 2) to pyrene minor groove binding (3 and 4), could be observed in the fluorimetric response. Namely, intercalating compounds either showed similar fluorimetric change for all ds-DNAs (1)4 or fluorescence of 2 depended strongly on basepair composition due to different effects of aromatic stacking interactions between 2-pyrene and A-T (fluorescence increase) vs. G-C basepairs (fluorescence quenching).5 However, 3 and 4 reveal different fluorimetric response patterns in comparison to intercalative compounds, which is not sensitive to the DNA basepair composition but depends strongly on the structural features of the DNA minor groove. By ICD results, both 3 and 4 show similar patterns for all studied polynucleotides, however, ICD effects reflect only the binding position of parts of a molecule but do not describe in detail exact positioning as well as the surroundings of the bound molecule. However, fluorescence of pyrene is extremely polarity-sensitive and therefore it is extensively employed to characterize microheterogeneous systems. Consequently, small differences in the positioning of pyrene caused by structural differences in linker length and rigidity between 3 and 4 result in a fluorescence change of 3 only for the β-helical DNA minor groove but not for the peculiar deep and narrow minor groove of poly dA-poly dT,16 while longer and more rigid molecule 4 reveals the opposite fluorescence response. Although the observed fluorimetric specificity is not accompanied by selective affinity, nevertheless it is to our knowledge unique fluorimetric sensing and therefore 3 and 4 could be considered as promising lead compounds for a further development of minor groove binders selective toward minor structural differences of various ds-DNA, especially those related to changes in A/T tracts.17
The antiproliferative effects of 3 and 4 are strongly dependent on the type of tumor cell line and span almost two orders of magnitude (from 2 μM–100 μM concentrations). Although observed activity cannot compete with the strongest drugs (active in nM range), nevertheless it points out that 3 and 4 efficiently cross the cell membrane and induce considerable biological effects. Thus, future modifications of the pyrene-guanidiniocarbonyl-pyrrole linkers of 3 and 4 by introduction of more groups, which could yield higher affinity, could result in more potent or selective compounds. Moreover, intriguing selectivity among different cell lines supports further detailed biological studies.
Experimental
Synthesis
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O); HR-MS (EI) calcd. for C24H19N3O3˙+ [M]˙+: 397.14209, found: 397.14218.
O); HR-MS (EI) calcd. for C24H19N3O3˙+ [M]˙+: 397.14209, found: 397.14218.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 25 ml) and stirred for 5.5 h at room temperature. Afterwards the solution was acidified with 5% hydrochloric acid to pH = 1 and immediately a solid precipitated. It was filtered off and lyophilised from water (15 ml) yielding 8 (728 mg, 1.97 mmol, 93%) as a slightly yellow solid. Mp > 230 °C; IR (KBr-pellet): νmax/cm−1 = 2923 (w), 2361 (s), 2339 (s), 1657 (m), 1560 (s), 1544 (m), 1075 (m), 464 (s); 1H NMR (400 MHz, [D6]DMSO, 25 °C): δH = 4.02 (s, 3H, N–CH3), 7.87 (s, 1H, imidazole-CH), 8.14 (t, 1H, J = 7.64 Hz, pyrene-CH), 8.23–8.32 (m, 4H, pyrene-CH), 8.33–8.41 (m, 3H, pyrene-CH), 8.54 (d, 1H, J = 9.29 Hz, pyrene-CH), 11.36 (s, 1H, NH); 13C NMR (100 MHz, [D6]DMSO, 25 °C): δC = 35.6 (N–CH3), 115.5 (imidazole-CH), 123.6, 123.8 (2 × pyrene-Cq), 124.4, 124.5, 125.7, 125.9, 126.1, 126.6, 127.2 (7 × pyrene-CH), 128.1 (pyrene-Cq), 128.3, 128.5 (2 × pyrene-CH), 130.1, 130.5, 130.7, 131.9 (4 × pyrene-Cq), 132.1, 137.4 (2 × imidazole-Cq), 160.1, 166.4 (2 × CO); HR-MS (pos. ESI) calcd. for C21H16N3O+ [M − CO2 + H]+: 326.12879, found: 326.12783.
O); HR-MS (pos. ESI) calcd. for C31H34N5O4+ [M + H]+: 540.26053, found: 540.25907.
O); HR-MS (pos. ESI) calcd. for C26H26N5O2+ [M]+: 440.20810, found: 440.20851.
O); HR-MS (pos. ESI) calcd. for C38H40N9O6+ [M + H]+: 718.30961, found: 718.30996.
:1, 3 ml) and stirred at room temperature for 5 h. After removal of DCM and remaining TFA under reduced pressure, the residual oil was lyophilised from water (3 ml) and purified by flash chromatography (MPLC, RP-18, methanol/water = 7/3, + 0.1% TFA) The desired product 3 (52.9 mg, 72.3 μmol, 60%) was obtained as a yellow solid. Mp 136–138 °C; IR (KBr-pellet): νmax/cm−1 = 2950 (w), 2873 (w), 1702 (s), 1675 (s), 1542 (s), 1474 (m), 1365 (w), 1283 (s), 1200 (s), 1135 (m), 849 (m), 759 (m), 718 (m), 445 (m); 1H NMR (400 MHz, [D6]DMSO, 25 °C): δH = 1.50–1.60 (m, 4H, imidazole-CO-NH-CH2-CH2-CH2-CH2), 3.24–3.33 (m, 4H, imidazole-CO-NH-CH2-CH2-CH2-CH2), 4.02 (s, 3H, N–CH3), 6.84–6.88 (m, 1H, pyrrole-CH), 7.02–7.06 (m, 1H, pyrrole-CH), 7.76 (s, 1H, imidazole-CH), 8.03 (br. t, 1H, J = 6.08 Hz, NH), 8.14 (t, 1H, J = 7.66 Hz, pyrene-CH), 8.17–8.32 (m, 8H, pyrene-CH, NH2 and NH2+), 8.32–8.40 (m, 3H, pyrene-CH), 8.43 (br. t, 1H, J = 5.76 Hz, NH), 8.50 (d, 1H, J = 9.27 Hz, pyrene-CH), 11.02 (br. s, 1H, NH), 11.07 (s, 1H, NH), 12.34 (br. s, 1H, NH);13C NMR (100 MHz, [D6]DMSO, 25 °C): δC = 26.5 (imidazole-CO-NH-CH2-CH2-CH2-CH2), 26.8 (imidazole-CO-NH-CH2-CH2-CH2-CH2), 35.0 (N–CH3), 38.0 (imidazole-CO-NH-CH2-CH2-CH2-CH2), 38.5 (imidazole-CO-NH-CH2-CH2-CH2-CH2), 112.2 (imidazole-CH), 114.7, 115.4 (2 × pyrrole-CH), 123.6, 123.8 (2 × pyrene-Cq), 124.4, 124.4 (2 × pyrene-CH), 125.3 (pyrrole-Cq), 125.6, 125.7, 126.0, 126.6, 127.2 (5 × pyrene-CH), 128.1 (pyrene-Cq), 128.3, 128.5 (2 × pyrene-CH), 130.1, 130.7, 130.7, 131.9 (4 × pyrene-Cq), 132.8 (pyrrole-Cq), 134.4, 136.1 (2 × imidazole-Cq), 155.0 (guanidine-Cq), 158.7, 159.0, 159.6, 166.5 (4 × CO); HR-MS (pos. ESI) calcd. for C33H32N9O4+ [M]+: 618.25718, found: 618.25638; HPLC, LC-18 reversed phase, gradient: methanol/water = 7/3 → methanol/water = 10/0, + 0.1% TFA): 290 nm: retention time 11.40 min, relative area 98.03% → corrected yield: 51.9 mg, 70.9 μmol, 59%; 220 nm: retention time 11.39 min, relative area 87.61% → corrected yield: 46.3 mg, 63.3 μmol, 53%.
O); HR-MS (pos. ESI) calcd. for C30H26N5O4+ [M + H]+: 520.19793, found: 520.19788.
:1, 15 ml) and stirred for 6.5 h at room temperature. Afterwards the solution was acidified with concentrated hydrochloric acid to pH = 1 and immediately a solid precipitated. It was filtered off and lyophilised from water (10 ml) yielding 16 (180 mg, 0.37 mmol, 77%) as a slightly yellow solid. Mp > 230 °C; IR (KBr-pellet): νmax/cm−1 = 2927 (m), 2851 (w), 2361 (s), 2339 (s), 1637 (s), 1543 (s), 1431 (m), 1343 (m), 1264 (m), 1101 (w), 487 (s), 460 (s); 1H NMR (400 MHz, [D6]DMSO, 25 °C): δH = 1.07 (9H, J = 7.20 Hz, CH3, HNEt3+), 2.70–2.87 (m, 6H, CH2, HNEt3+), 3.92 (s, 3H, N–CH3, imidazole), 3.94 (s, 3H, N–CH3, pyrrole), 7.15 (d, 1H, J = 1.67 Hz, pyrrole-CH), 7.36 (s, 1H, imidazole-CH), 7.58 (d, 1H, J = 1.61 Hz, pyrrole-CH), 8.14 (t, 1H, J = 7.65 Hz, pyrene-CH), 8.20–8.32 (m, 4H, pyrene-CH), 8.34–8.41 (m, 3H, pyrene-CH), 8.49 (d, 1H, J = 9.27 Hz, pyrene-CH), 10.56 (s, 1H, NH), 10.72 (s, 1H, NH). 13C NMR (100 MHz, [D6]DMSO, 25 °C): δC = 9.28 (3C, CH3, HNEt3+), 36.3 (N–CH3. imidazole), 36.3 (N–CH3, pyrrole), 45.3 (3C, CH2, HNEt3+), 105.3 (pyrrole-CH), 114.9 (imidazole-CH), 119.4 (pyrrole-CH), 122.3, 122.5 (2 × pyrrole-Cq), 123.7, 123.8 (2 × pyrene-Cq), 124.4, 124.6, 125.3, 125.6, 125.9, 126.6, 127.2 (7 × pyrene-CH), 127.9 (pyrene-Cq), 128.2, 128.3 (2 × pyrene-CH), 130.2, 130.7, 130.8, 131.6 (4 × pyrene-Cq), 131.8, 138.0 (2 × imidazole-Cq), 158.5, 158.5, 166.0 (3 × CO); EI-MSm/z (%) = 447.1 (23.34) [M − CO2]˙+, 351.1 (7.50) [M − C5H6N3O2]˙+, 229.1 (100.00) [M − C11H12N5O3]˙+.
O); HR-MS (pos. ESI) calcd. for C37H40N7O5+ [M + H]+: 662.30854, found: 662.30724.
O, CF3COO−), 158.6, 158.7, 166.0 (3 × CO); HR-MS (pos. ESI) calcd. for C32H32N7O3+ [M]+: 562.25611, found: 562.25612.
O); HR-MS (pos. ESI) calcd. for C44H46N11O7+ [M + H]+: 840.35762, found: 840.35605.
O); HR-MS (pos. ESI) calcd. for C39H38N11O5+ [M]+: 740.30519, found: 740.30519; HPLC, LC-18 reversed phase, isocratic: methanol, + 0.1% TFA: 290 nm: retention time 13.26 min, relative area 97.37% → corrected yield: 50.4 mg, 59.0 μmol, 97%; 220 nm: retention time 13.27 min, relative area 95.21% → corrected yield: 49.3 mg, 57.7 μmol, 95%.
:1, 25 ml) and stirred for 5.5 h at room temperature. Afterwards the solution was acidified with 5% hydrochloric acid to pH = 1 and immediately a solid precipitated. It was filtered off and lyophilised from water (15 ml) yielding 8 (728 mg, 1.97 mmol, 93%) as a slightly yellow solid. Mp > 230 °C; IR (KBr-pellet): νmax/cm−1 = 2923 (w), 2361 (s), 2339 (s), 1657 (m), 1560 (s), 1544 (m), 1075 (m), 464 (s); 1H NMR (400 MHz, [D6]DMSO, 25 °C): δH = 4.02 (s, 3H, N–CH3), 7.87 (s, 1H, imidazole-CH), 8.14 (t, 1H, J = 7.64 Hz, pyrene-CH), 8.23–8.32 (m, 4H, pyrene-CH), 8.33–8.41 (m, 3H, pyrene-CH), 8.54 (d, 1H, J = 9.29 Hz, pyrene-CH), 11.36 (s, 1H, NH); 13C NMR (100 MHz, [D6]DMSO, 25 °C): δC = 35.6 (N–CH3), 115.5 (imidazole-CH), 123.6, 123.8 (2 × pyrene-Cq), 124.4, 124.5, 125.7, 125.9, 126.1, 126.6, 127.2 (7 × pyrene-CH), 128.1 (pyrene-Cq), 128.3, 128.5 (2 × pyrene-CH), 130.1, 130.5, 130.7, 131.9 (4 × pyrene-Cq), 132.1, 137.4 (2 × imidazole-Cq), 160.1, 166.4 (2 × CO); HR-MS (pos. ESI) calcd. for C21H16N3O+ [M − CO2 + H]+: 326.12879, found: 326.12783.
O); HR-MS (pos. ESI) calcd. for C31H34N5O4+ [M + H]+: 540.26053, found: 540.25907.
O); HR-MS (pos. ESI) calcd. for C26H26N5O2+ [M]+: 440.20810, found: 440.20851.
O); HR-MS (pos. ESI) calcd. for C38H40N9O6+ [M + H]+: 718.30961, found: 718.30996.
:1, 3 ml) and stirred at room temperature for 5 h. After removal of DCM and remaining TFA under reduced pressure, the residual oil was lyophilised from water (3 ml) and purified by flash chromatography (MPLC, RP-18, methanol/water = 7/3, + 0.1% TFA) The desired product 3 (52.9 mg, 72.3 μmol, 60%) was obtained as a yellow solid. Mp 136–138 °C; IR (KBr-pellet): νmax/cm−1 = 2950 (w), 2873 (w), 1702 (s), 1675 (s), 1542 (s), 1474 (m), 1365 (w), 1283 (s), 1200 (s), 1135 (m), 849 (m), 759 (m), 718 (m), 445 (m); 1H NMR (400 MHz, [D6]DMSO, 25 °C): δH = 1.50–1.60 (m, 4H, imidazole-CO-NH-CH2-CH2-CH2-CH2), 3.24–3.33 (m, 4H, imidazole-CO-NH-CH2-CH2-CH2-CH2), 4.02 (s, 3H, N–CH3), 6.84–6.88 (m, 1H, pyrrole-CH), 7.02–7.06 (m, 1H, pyrrole-CH), 7.76 (s, 1H, imidazole-CH), 8.03 (br. t, 1H, J = 6.08 Hz, NH), 8.14 (t, 1H, J = 7.66 Hz, pyrene-CH), 8.17–8.32 (m, 8H, pyrene-CH, NH2 and NH2+), 8.32–8.40 (m, 3H, pyrene-CH), 8.43 (br. t, 1H, J = 5.76 Hz, NH), 8.50 (d, 1H, J = 9.27 Hz, pyrene-CH), 11.02 (br. s, 1H, NH), 11.07 (s, 1H, NH), 12.34 (br. s, 1H, NH);13C NMR (100 MHz, [D6]DMSO, 25 °C): δC = 26.5 (imidazole-CO-NH-CH2-CH2-CH2-CH2), 26.8 (imidazole-CO-NH-CH2-CH2-CH2-CH2), 35.0 (N–CH3), 38.0 (imidazole-CO-NH-CH2-CH2-CH2-CH2), 38.5 (imidazole-CO-NH-CH2-CH2-CH2-CH2), 112.2 (imidazole-CH), 114.7, 115.4 (2 × pyrrole-CH), 123.6, 123.8 (2 × pyrene-Cq), 124.4, 124.4 (2 × pyrene-CH), 125.3 (pyrrole-Cq), 125.6, 125.7, 126.0, 126.6, 127.2 (5 × pyrene-CH), 128.1 (pyrene-Cq), 128.3, 128.5 (2 × pyrene-CH), 130.1, 130.7, 130.7, 131.9 (4 × pyrene-Cq), 132.8 (pyrrole-Cq), 134.4, 136.1 (2 × imidazole-Cq), 155.0 (guanidine-Cq), 158.7, 159.0, 159.6, 166.5 (4 × CO); HR-MS (pos. ESI) calcd. for C33H32N9O4+ [M]+: 618.25718, found: 618.25638; HPLC, LC-18 reversed phase, gradient: methanol/water = 7/3 → methanol/water = 10/0, + 0.1% TFA): 290 nm: retention time 11.40 min, relative area 98.03% → corrected yield: 51.9 mg, 70.9 μmol, 59%; 220 nm: retention time 11.39 min, relative area 87.61% → corrected yield: 46.3 mg, 63.3 μmol, 53%.
O); HR-MS (pos. ESI) calcd. for C30H26N5O4+ [M + H]+: 520.19793, found: 520.19788.
:1, 15 ml) and stirred for 6.5 h at room temperature. Afterwards the solution was acidified with concentrated hydrochloric acid to pH = 1 and immediately a solid precipitated. It was filtered off and lyophilised from water (10 ml) yielding 16 (180 mg, 0.37 mmol, 77%) as a slightly yellow solid. Mp > 230 °C; IR (KBr-pellet): νmax/cm−1 = 2927 (m), 2851 (w), 2361 (s), 2339 (s), 1637 (s), 1543 (s), 1431 (m), 1343 (m), 1264 (m), 1101 (w), 487 (s), 460 (s); 1H NMR (400 MHz, [D6]DMSO, 25 °C): δH = 1.07 (9H, J = 7.20 Hz, CH3, HNEt3+), 2.70–2.87 (m, 6H, CH2, HNEt3+), 3.92 (s, 3H, N–CH3, imidazole), 3.94 (s, 3H, N–CH3, pyrrole), 7.15 (d, 1H, J = 1.67 Hz, pyrrole-CH), 7.36 (s, 1H, imidazole-CH), 7.58 (d, 1H, J = 1.61 Hz, pyrrole-CH), 8.14 (t, 1H, J = 7.65 Hz, pyrene-CH), 8.20–8.32 (m, 4H, pyrene-CH), 8.34–8.41 (m, 3H, pyrene-CH), 8.49 (d, 1H, J = 9.27 Hz, pyrene-CH), 10.56 (s, 1H, NH), 10.72 (s, 1H, NH). 13C NMR (100 MHz, [D6]DMSO, 25 °C): δC = 9.28 (3C, CH3, HNEt3+), 36.3 (N–CH3. imidazole), 36.3 (N–CH3, pyrrole), 45.3 (3C, CH2, HNEt3+), 105.3 (pyrrole-CH), 114.9 (imidazole-CH), 119.4 (pyrrole-CH), 122.3, 122.5 (2 × pyrrole-Cq), 123.7, 123.8 (2 × pyrene-Cq), 124.4, 124.6, 125.3, 125.6, 125.9, 126.6, 127.2 (7 × pyrene-CH), 127.9 (pyrene-Cq), 128.2, 128.3 (2 × pyrene-CH), 130.2, 130.7, 130.8, 131.6 (4 × pyrene-Cq), 131.8, 138.0 (2 × imidazole-Cq), 158.5, 158.5, 166.0 (3 × CO); EI-MSm/z (%) = 447.1 (23.34) [M − CO2]˙+, 351.1 (7.50) [M − C5H6N3O2]˙+, 229.1 (100.00) [M − C11H12N5O3]˙+.
O); HR-MS (pos. ESI) calcd. for C37H40N7O5+ [M + H]+: 662.30854, found: 662.30724.
O, CF3COO−), 158.6, 158.7, 166.0 (3 × CO); HR-MS (pos. ESI) calcd. for C32H32N7O3+ [M]+: 562.25611, found: 562.25612.
O); HR-MS (pos. ESI) calcd. for C44H46N11O7+ [M + H]+: 840.35762, found: 840.35605.
O); HR-MS (pos. ESI) calcd. for C39H38N11O5+ [M]+: 740.30519, found: 740.30519; HPLC, LC-18 reversed phase, isocratic: methanol, + 0.1% TFA: 290 nm: retention time 13.26 min, relative area 97.37% → corrected yield: 50.4 mg, 59.0 μmol, 97%; 220 nm: retention time 13.27 min, relative area 95.21% → corrected yield: 49.3 mg, 57.7 μmol, 95%.
Spectroscopic studies
Notes: Polynucleotides were purchased as noted: poly dA-poly dT, poly dAdT-poly dAdT, poly dGdC-poly dGdC, poly rA-poly rU (Sigma), calf thymus (ct)-DNA (Aldrich). Polynucleotides were dissolved in Na-cacodylate buffer, I = 0.05 mol dm−3, pH = 7. Calf thymus (ct)-DNA was additionally sonicated and filtered through a 0.45 μm filter. Polynucleotide concentration was determined spectroscopically as a concentration of phosphates.18The electronic absorption spectra were obtained on a Varian Cary 100 Bio spectrometer, CD spectra were collected on the Jasco J-810 spectrometer and fluorescence spectra were recorded on a Varian Cary Eclipse fluorimeter; all in quartz cuvettes (1 cm). The measurements were performed in aqueous buffer solution (pH = 7 - Na-cacodylate buffer, I = 0.05 mol dm−3, pH = 5 - sodium citrate buffer, I = 0.03 M). Under the experimental conditions used the absorbance and fluorescence intensities of studied compounds were proportional to their concentration. The sample concentration in fluorescence measurements had an optical absorbance below 0.05 at the excitation wavelength. In fluorimetric titrations, an excitation wavelength of λexc > 320 nm was used to avoid inner filter effects caused by absorption of excitation light by added polynucleotide. The binding constant (Ks) and [bound compound]/[polynucleotide phosphate] ratio (n) were calculated according to the Scatchard equation by non-linear least-square fitting, giving excellent correlation coefficients (>0.999) for obtained values for Ks and n. Thermal melting curves for ds-polynucleotides and their complexes with studied compounds were determined as previously described by following the absorption change at 260 nm as a function of temperature.19 The absorbance of studied compound was subtracted from every curve, and the absorbance scale was normalized. Obtained Tm values are the midpoints of the transition curves, determined from the maximum of the first derivative or graphically by a tangent method. Given ΔTm values were calculated subtracting Tm of the free nucleic acid from Tm of the complex. Every ΔTm value reported here was the average of at least two measurements, the error in ΔTm is ± 0.5 °C.
Molecular modelling method
Compound 2 was manually intercalated into the space between two base pairs of double stranded DNA (poly dAdT-poly dAdT) and compounds 3 and 4 were placed into the DNA minor groove. For this purpose we constructed a 13 base pair DNA (poly dAdT-poly dAdT with one dC-dG base pair on the each end for better stability) with the program NUCGEN, a part of the Amber program suit.20 Compounds 2, 3 and 4 were built using the module ‘Builder’ within the program InsightII.21 The ds-DNA-2 complex was built by inserting the pyrene moiety of 2 into the gap made by removing a base pair in the middle of ds-DNA, the DNA backbone was connected in a common way and the complete complex was minimised to assure common bond lengths within ds-DNA. The AMBER ff99 force field of Duan et al.22 and the general AMBER force field GAFF were used to parameterize the substrate-DNA complexes. Each of the complexes was placed in the centre of the octahedral box filled with TIP3P type water molecules, a water buffer of 7 Å was used, and Na+ ions were added to neutralize the systems. The solvated complexes were geometry optimized using the steepest descent and conjugate gradient methods, 2500 steps of each, and equilibrated for 0.5 ns. During equilibration the temperature was gently increased from 0 to 300 K and the volume was kept constant. The equilibrated systems were subjected to 12.5 ns (complex with 2) and 10.5 ns (complexes with 3 and 4) of the productive molecular dynamics (MD) simulation at constant temperature and pressure (300 K, 1 atm). No restraints were used during the productive MD simulations. The time step during the simulation was 1 fs and the temperature was kept constant using Langevin dynamics with a collision frequency of 1 ps−1.Geometry optimisation and molecular dynamics (MD) simulations were accomplished using the AMBER 9 program package. The simulation was accomplished using Periodic Boundary Conditions (PBC). The Particle Mesh Ewald (PME) method was used for calculation of the long range electrostatic interactions. In the direct space the pairwise interactions were calculated within the cutoff-distance of 11 Å.
Proliferation assays
The growth inhibition activity was assessed as described previously,23 according to the slightly modified procedure of the National Cancer Institute, Developmental Therapeutics Program.24 The cells were inoculated onto standard 96-well microtiter plates on day 0. Test agents were then added in five consecutive 10-fold dilutions and incubated for a further 72 h. Working dilutions were freshly prepared on the day of testing. The solvent (DMSO) was also tested for eventual inhibitory activity by adjusting its concentration to be the same as in working concentrations (maximal concentration of DMSO was 0.25%). After 72 h of incubation, the cell growth rate was evaluated by performing the MTT assay which detects dehydrogenase activity in viable cells. The absorbency (OD, optical density) was measured on a microplate reader at 570 nm. Each test point was performed in quadruplicate in three individual experiments. The results are expressed as IC50, which is the concentration necessary for 50% of inhibition. The IC50 values for each compound are calculated from dose-response curves using linear regression analysis by fitting the test concentrations that give PG values above and below the reference value (i.e. 50%). Each result is a mean value from three separate experiments.Acknowledgements
C.S. thanks the DFG (Deutsche Forschungsgemeinschaft) and the Fonds of the Chemical Industry for ongoing financial support of his work. I.P., S.T. and M.K. thank the Ministry of Science, Education and Sport of Croatia for financial support of this study (Projects 098-0982914-2918, 098-1191344-2860, 098-0982464-2514). We also acknowledge the support from a bilateral funding project between the DAAD and the Ministry of Science, Education and Sport of Croatia.Notes and references
- R. B. Silverman, The Organic Chemistry of Drug Design and Drug Action, Elsevier Academic Press, New York, 2004 Search PubMed.
- M. Demeunynck, C. Bailly, W. D. Wilson, ed. DNA and RNA Binders; Wiley-VCH: Weinheim, 2002 Search PubMed.
- E. Trinquet and G. Mathis, Mol. BioSyst., 2006, 2, 380 RSC.
- L. Hernandez-Folgado, C. Schmuck, S. Tomić and I. Piantanida, Bioorg. Med. Chem. Lett., 2008, 18, 2977–2981 CrossRef CAS.
- L. Hernandez-Folgado, D. Baretić, I. Piantanida, M. Marjanović, M. Kralj, T. Rehm and C. Schmuck, Chem.–Eur. J., 2010, 16, 3036–3056 CrossRef CAS.
- P. B. Dervan, Bioorg. Med. Chem., 2001, 9, 2215–2235 CrossRef CAS.
- E. E. Baird and P. B. Dervan, J. Am. Chem. Soc., 1996, 118, 6141–6146 CrossRef CAS.
- C. Schmuck, V. Bickert, M. Merschky, L. Geiger, D. Rupprecht, J. Dudaczek, P. Wich, T. Rehm and U. Machon, Eur. J. Org. Chem., 2008, 324–329 CrossRef CAS.
- G. Malojčić, I. Piantanida, M. Marinić, M. Žinić, M. Marjanović, M. Kralj, K. Pavelić and i H.-J. Schneider, Org. Biomol. Chem., 2005, 3, 4373–4381 RSC.
- C. R. Cantor and P. R. Schimmel, Biophysical Chemistry, vol. 3. WH Freeman and Co., San Francisco, 1980, 1109–1181 Search PubMed.
- G. Scatchard, Ann. N. Y. Acad. Sci., 1949, 51, 660–672 CrossRef CAS; J. D. McGhee and P. H. von Hippel, J. Mol. Biol., 1976, 103, 679–684 CrossRef.
- A. Rodger, B. Norden, In Circular Dichroism and Linear Dichroism; Oxford University Press: New York, 1997, Chapter 2 Search PubMed.
- N. Berova, K. Nakanishi, R. W. Woody ( 2000) Circular dichroism Principles and Applications, 2nd edn, New YorkWiley-VCH Search PubMed.
- M. Eriksson and B. Nordén, Methods in Enzymology, Vol 340, 2001, Pages 68–98, Table I Search PubMed.
- B. Norden and F. Tjerneld, Biopolymers, 1982, 21, 1713–1734 CrossRef CAS; R. Lyng, A. Rodger and B. Norden, Biopolymers, 1991, 31, 1709–1820 CAS; P. E. Schipper, B. Norden and F. Tjerneld, Chem. Phys. Lett., 1980, 70, 17–21 CrossRef CAS.
- W. D. Wilson, Y.-H. Wang, C. R. Krishnamoorthy and J. C. Smith, Biochemistry, 1985, 24, 3991–3999 CrossRef CAS.
- H. S Koo, H. M Wu and D. M Crothers, Nature, 1986, 320, 501–506 CrossRef CAS; D. MacDonald, K. Herbert, X. ZZhang, T. Polgruto and P. Lu, J. Mol. Biol, 2001, 5, 1081–1098 CrossRef.
- M. Radić Stojković and I. Piantanida, Tetrahedron, 2008, 64, 7807–7814 CrossRef CAS.
- J.-L. Mergny and L. Lacroix, Oligonucleotides, 2003, 13, 515–537 CrossRef CAS.
- http://amber.scripps.edu/ .
- INSIGHTII – Accelerys San Diego 2001–2008. (http://accelrys.com/services/training/life-science/insight-migration.html).
- Y. Duan, C. Wu, S. Chowdhury, M. C. Lee, G. Xiong, W. Zhang, R. Yang, P. Cipelak, R. Luo and T. Lee, A point-charge force field for molecular mechanics simulations of proteins, J. Comput. Chem., 2003, 24, 1999–2012 CrossRef CAS.
- M. Marjanović, M. Kralj, F. Supek, L. Frkanec, I. Piantanida, T. Šmuc and L. Tušek-Bo,žić, J. Med. Chem., 2007, 50, 1007–1018 CrossRef CAS.
- M. R. Boyd and D. P. Kenneth, Drug Dev. Res., 1995, 34, 91–109 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: CD titrations of ct‐DNA with 3 and 4. See DOI: 10.1039/c0ob00103a |
| This journal is © The Royal Society of Chemistry 2011 |