Chiral inorganic nanoparticles: origin, optical properties and bioapplications
Yunsheng
Xia
ab,
Yunlong
Zhou
a and
Zhiyong
Tang
*a
aLaboratory for Nanomaterials, National Center for Nanoscience and Technology, Beijing, 100190, China. E-mail: zytang@nanoctr.cn
bCollege of Chemistry and Materials Science, Anhui Normal University, Wuhu, 241000, China
First published on 7th February 2011
Abstract
Chirality of inorganic nanoparticles (NPs) is an emerging and hot topic in nanoresearch in the past several years. Many novel and interesting properties of chiral NPs have been explored and studied, which highlight their importance in both fundamental research and potential applications. This review summarizes recent progress in the study of origins, optical properties and bioapplications of chiral NPs, and future developments in this research area are also discussed.
 Yunsheng Xia | Yunsheng Xia is currently a doctoral candidate in Physical Chemistry at the National Center for Nanoscience and Technology China. He received his BS degree from the department of Applied Chemistry, Qingdao University of Science and Technology in 2000, and MS degree from the College of Chemistry and Materials Science, Anhui Normal University in 2007. He is also an associate professor at the College of Chemistry and Materials Science, Anhui Normal University. His current research interests include formation mechanism, optical properties and sensing applications of nanoparticle superstructures. |
 Yunlong Zhou | Dr Yunlong Zhou joined Prof. Zhiyong Tang's group in 2007 and received his Ph.D. degree in physical chemistry from the National Center for Nanoscience and Technology China in 2010. He received his BS degree from department of chemistry, Langfang Normal University in 2003, and received his MS degree from the College of Chemistry and Environmental Science, Hebei University in 2006. Now he is a postdoctoral research fellow in Prof. Kotov's group. His current research interests include synthesis, self-assembly and properties of inorganic nanocrystals. |
 Zhiyong Tang | Zhiyong Tang obtained his Ph.D. degree in 1999 from the Chinese Academy of Sciences. After then, he went to the Swiss Federal Institute of Technology Zurich and the University of Michigan for his postdoctoral research. In November of 2006, he joined the National Center for Nanoscience and Technology (NCNST) China and took a full professor position. Zhiyong Tang's current research interests are focused on fabrication and application of chiral inorganic nanoparticles as well as nanoparticle superstructures. |
1 Background
Inorganic nanoparticles (NPs) with diameters ranging from 1 to 100 nm possess unique size- or shape-dependent physical and chemical properties different from their bulk materials. In semiconductors, the spatial confinement of electronic motion to a length scale, which is comparable to or smaller than the length of the electron Bohr radius, gives rise to size-dependent optical and electrical properties of NPs.1–7 While for noble metals, as their sizes are reduced to tens of nanometers, a strong optical absorption band, also called surface plasmon resonance (SPR) absorption, appears due to the collective oscillation of free electrons in the conduction band on the surface of NPs.8–13 The multifaceted characters of inorganic NPs offer a wide range of research and development possibilities in diverse areas of photonics, electronics, catalysis, and biology.14–20 On the other hand, the concept of chirality has inspired major advances in physics, chemistry, and life sciences.21–23 A chiral molecule is a type of molecule that lacks an internal plane of symmetry and has a non-superposable mirror image, which preferentially absorbs or affects the polarization of light and exhibits an optical circular dichroism (CD) effect. It is worth mentioning that selection of chiral molecules is critical for disease diagnosis and therapy, for example, chiral molecular drugs shared one-third of all drug sales worldwide in 2000.24Although chirality at molecular level (like amino acid, sugar, and synthetic molecules) is exclusively understood, the investigation of nanoscale chirality is scarce. It can be expected that combination of NPs and chiral molecules will bring new opportunities not only in optics but also in biology. The best model to perform the above study is chemically-synthesized NPs, where the organic stabilizers surrounding the inorganic cores can be designed with different types of chiral organic molecules. Here, three questions must be answered by the scientists who are involved in the study of chiral NPs. First, does the introduction of chiral stabilizers change the structures of inorganic cores of NPs and lead to production of chirality in inorganic NPs? Second, can the electronic states of chiral molecules and inorganic NPs interact with each other and generate new optical properties? Third, do chiral NPs have potential biological applications that differ from conventional chiral molecules? Recently, increasing attention has been focused on answering the above questions, and some significant results are obtained. In this review, we will summarize the state-of-the-art progress in the origin, optical properties, and bioapplications of chiral NPs. Before starting the summary, the optical properties of noble metal and semiconductor NPs will be briefly introduced, considering that they will be intensively mentioned in this review. At the end of this review, the prospects of chiral NPs will be also discussed. It should be stressed that this is not a comprehensive review but rather discusses key developments and applications, and we apologize for possible oversights of some significant contributions.
2 Optical properties of NPs
2.1 Nobel metal NPs
Noble metal NPs, especially gold and silver NPs, are frequently studied because they exhibit strong SPR absorption in the visible wavelength range. Visible light in resonance with the surface plasmon causes free electrons in noble metal NPs to oscillate. As the wave front of the light passes, the electron density in the particle is polarized to one surface and oscillates in resonance with the light's frequency, causing a standing oscillation. The resonance condition is determined from absorption and scattering spectroscopy and is found to be dependent on the shapes, sizes, inter-distances of NPs, as well as dielectric constants of both the metal and the surrounding materials.132.2 Semiconductor NPs
The most important characteristics of semiconductor NPs are size-dependent properties, which arise from quantum confinement effects. The electron and hole in semiconductor materials can interact through columbic forces and form a Wannier exciton, a state that is very similar to a hydrogen atom.2 The exciton has a finite size within the crystal defined by the Bohr exciton diameter, which ranges from ∼2 to ∼50 nm depending on the nature of materials.4 If the size of a semiconductor NP is smaller than the size of the exciton, the charge carriers become spatially confined, which raises their energy. So, semiconductor NPs with dimensions smaller than Bohr exciton diameter show size-dependent absorption and fluorescence spectra with discrete electronic transitions.3 Origin of chiral NPs
3.1 Origin of single chiral clusters
The early study on chiral NPs was mainly focused on metal or semiconductor clusters. As an example, Au28 clusters were prepared by using chiral glutathione as stabilizers.25 Interestingly, the prepared Au clusters showed chiral optical property and their CD signals fell in the same region of the absorption bands (from ultraviolet (UV) to the near-infrared region). Obviously, this CD signal did not arise from glutathione, since it was only active in UV region. A plausible explanation was that the Au clusters preferentially formed lower symmetry or even chiral structures in order to minimize the total energy of clusters.26 This work opened up the door toward the study of the chiral properties at nanometer scale. Afterwards, both experimental and calculation studies on the origin of optical activity of gold clusters (0.5 to 2 nm) have been reported in succession.27–49 Generally, chirality elucidation of metal clusters can be classified into three models as follows: 1) the intrinsically chiral metallic core model; 2) the dissymmetric field model; and 3) the chiral footprint model. The difference in the three models is to what extent the enantiomeric structure of the chiral ligands is transmitted to the atomic structure of the NPs. Though these three models have been partially supported by individual experimental results, a comprehensive understanding of the chiral origin of metal clusters is still highly desirable. The readers can refer to an excellent review to obtain further information on the recent work on metal clusters.503.2 Origin of single chiral NPs
Very recently, more attention has been paid to the chirality origin of particles that have lager sizes compared with clusters, namely, semiconductor and noble metal NPs with sizes ranging from several to tens of nanometers. It should be emphasized that the change from clusters to NPs is not simply different in size. For instance, the cores of the clusters might be inherently chiral, because small-sized clusters are easily susceptible to surrounding environment and thus produce remarkable structural change. On the other hand, chiral stabilizers can only affect surface structures of large-sized NPs, because crystalline nature of inorganic cores inside NPs is difficult to deform for production of chiral structures due to large mechanic stress.Up to now, a few models have been proposed for interpretation of chirality of NPs, based on the combination of experiment observations and calculation results. For example, D-penicillamine- and L-penicillamine-capped CdS NPs were explored to possess chiral responses from 200 nm to 380 nm with corresponding mirror images, and scarcely any CD signals were observed for rac-penicillamine capped CdS NPs (Fig. 1A).51 As comparison, CD signals of free chiral penicillamine or Cd-penicillamine complex appeared at the wavelength of shorter than 280 nm. So the observed CD signals in the range of 200 nm – 380 nm should be attributed to chiral NPs themselves rather than ligands. Density functional theory (DFT) was then employed to study the chirality origin of the CdS NPs. The calculation was based on the cluster of [Cd19S17H14(Pen)6(PenH)3]3+, which possessed three identical (100) faces and each face was bound three penicillamine ligands (Fig. 1B and 1C). The calculation result demonstrated that penicillamine ligands bound via N and S to one surface-Cd, and introduced chirality via additional binding of carboxylate with a neighboring surface-Cd. The interaction between ligand and cluster was stronger than that among ligands, so the ligands packed into helical bands on the NP surface and strongly distorted the outermost Cd atoms of the NPs, leading to formation of an enantiomeric structure of the surface layers. As a result, the CD signals at the wavelength of longer than 280 nm were believed to result from near-surface Cd atoms that were enantiomerically distorted by penicillamine ligands. It was also noted that similar geometrical distortion was rare for CdS inside the NP cores.52
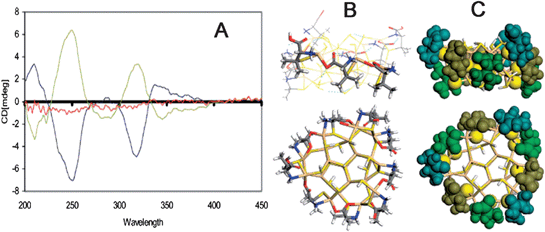 | ||
| Fig. 1 (A) CD spectra of the D- (blue line), L- (yellow line) and rac-penicillamine (red line) stabilized CdS NPs. Side and top views of a (100) face of the optimized model of CdS NPs, (B) stick representation with PenH-Pen-Pen bonding pattern along one face, (C) space-filling representation with non-S parts (colored differently) of each ligand. Note that D-penicillamine ligands form fragments of a left-handed helix around the NP core. The images were taken from ref. 51 and 52 with permission. | ||
Analogously to chiroselective adsorption of biomolecules onto the natural mineral surfaces, which influences biominerization process and induces formation of chiral crystals with different structures, chiral stabilizers can influence the growth rate of semiconductor NPs, i.e., the CdTe NPs showed different growth rate when D-cysteine and L-cysteine were used as the stabilizers, respectively (Fig. 2A and 2B).53 Moreover, such chiral cysteine stabilized CdTe NPs produced obvious CD signals in the wavelength range of 280 nm – 400 nm (Fig. 2C). Although the traditional theory of chiral space group could give perfect structural information on bulk chiral crystallization, it was not suitable to explain the difference in the growth mode of chiral NPs because the surface atoms of NPs with ultra-small size were entirely different from the atoms of bulk materials. The surface atoms, which were more sensitive to stress and other external factors, were more similar to the atoms in organic molecules. Theoretically, the atoms in CdTe NPs were arranged as tetrahedrons, and chirality might occur in apexes of NPs that were most susceptible to chemical modification and substitution (Fig. 2D and 2E). Furthermore, the quantum mechanical calculations revealed that the (D- or L-) cysteine could result in favorable formation of different chiral surface structure on the NPs, and the discrepancy in binding energy between chiral cysteine and chiral NP surface led to the difference in the growth kinetics of CdTe NPs (Fig. 2B). More interestingly, based on the above calculations, it was theoretically deduced that chiral NPs could be obtained even when using racemic compounds as the stabilizers, if the synthesis conditions were carefully controlled.53
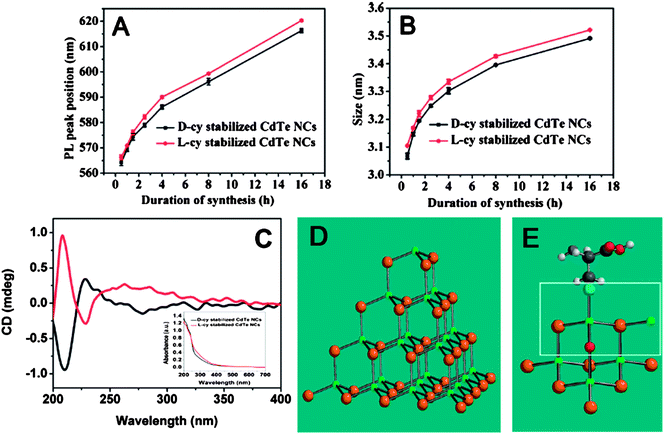 | ||
| Fig. 2 Temporal evolution of photoluminescence peak (A) and particle size (B). (C) CD spectra of L-cysteine- (red line) and D-cysteine stabilized CdTe NPs (black line) after 16 h of synthesis. Insets present the corresponding UV-vis spectra. (D) Ideal tetrahedron of CdTe NPs used in calculations and (E) model of the chiral tetrahedral apex; Cd (green); Te (brown); O (red); S (cyanine). The images were taken from ref. 53 with permission. | ||
Finally, the chiral surface structures of NPs were found to have high stability even after chiral stabilizers were removed. L- and D-cysteinemethylester stabilized CdTe NPs were synthesized, and subsequently the chiral ligands were exchanged by achiral 1-dodecanethiol. The analytical result suggested that almost all (>92%) chiral molecules were exchanged with 1-dodecanethiol. Surprisingly, both post-treated CdTe NPs gave symmetrical mirror CD profiles at the wavelength of above 260 nm, which were identical to those of the original enantiomeric cysteinemethylester-capped CdTe NPs. Furthermore, the chiral memory was robust enough to be preserved for at least one year under ambient conditions.54
3.3 Origin of chiral NP assemblies
In addition to single NPs, the concept of tetrahedron is also applied to explain chirality of NP assemblies. Gold NPs at the tips of the discrete tetrahedrons were constructed by using DNA as scaffolds (Fig. 3).55 This hybrid assembly was obtained by gel electrophoresis separation and confirmed in situ by small-angle X-ray scattering measurements. Impressively, chiral enantiomers were intentionally achieved when four different sized NPs were employed as building blocks in the assembly (Fig. 3).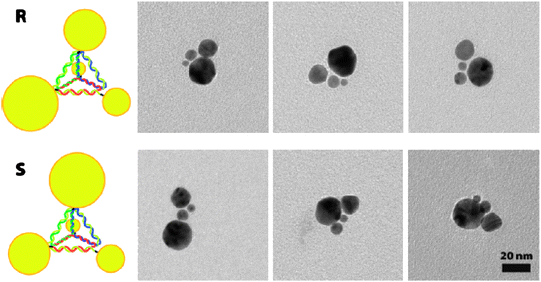 | ||
| Fig. 3 Examples of chiral tetrahedron. Top row, R; bottom row, S. The images were taken from ref. 55 with permission. | ||
Almost at the same time, similar hybrid assembly made of gold NPs and DNA was separately obtained by using polymerase chain reaction (PCR) technique.56 The assembly system was controlled by the density of the primer (DNA) on the surface of gold NPs and the number of PCR cycles. The greater number of primers coating NPs and the larger number of PCR cycles gave rise to the greater number of NPs in the self-assembly system (Fig. 4A–4C). Interestingly, a new chiral signal at about 650 nm was observed for the assembly, and this signal showed a gradual red shift and became weaker with the increase of PCR cycles (Fig. 4D). This chiral signal was contributed by the SPR band of Au NP assembly, in which NPs with varying sizes and shapes organized into chiral tetrahedron in three-dimensional space (Fig. 4C).
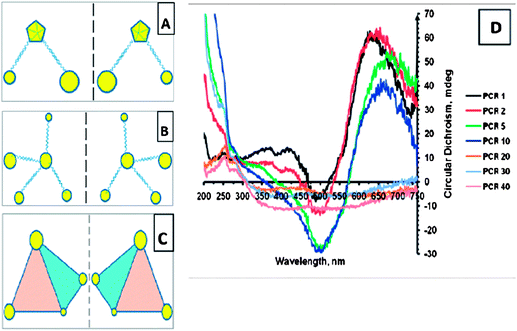 | ||
| Fig. 4 (A-C) Schematics of chiral NP superstructures. (D) CD spectra of products of PCR for increasing number of cycles. The images were taken from ref. 56 with permission. | ||
4 Optical properties of chiral NPs
4.1 Optical properties of single chiral NPs
Early study on chiral NPs was mostly focused on investigation of their origin of optical activity, and less attention was paid to the optical property although some CD spectra were illustrated. Recent theoretical calculations revealed that optical coupling between inorganic NPs and chiral molecules could occur via Coulomb interactions (dipole and multipole) (Fig. 5A).57 Two mechanisms were identified for the interactions of a chiral molecule with a metal NP. The first mechanism was the plasmon-induced change in the electromagnetic field inside the chiral molecule (Fig. 5B). The second one was the optical absorption of the NP-molecule complex due to the chiral currents inside the metal NP induced by the dipole of the chiral molecule. The second mechanism could be used to explain why new CD bands occurred at the plasmon frequency, if a chiral molecule and a metal NP approached each other. Fig. 5C demonstrated the calculated CD spectrum of complex based on a silver NP and a short α-helix (chiral molecule), and new CD peak appeared at the plasmon frequency of silver NP. In addition to metal NPs, this theory was possibly used for explaining the interaction of semiconductor NPs with chiral molecules, since the calculation was based on similar Coulomb interactions.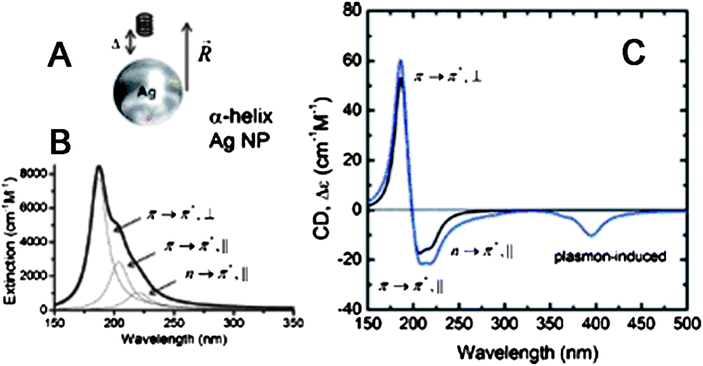 | ||
| Fig. 5 (A) A model of interaction between Ag NP and R-helix. (B) Theoretical fit to the extinction of the R-helix. The molar extinction is given per one chromophore (amide group). (C) Calculated CD signals for the R-helix (black line) and for the NP-molecule complex (blue line) shown in (A); αNP = 10 nm, Δ = 7 nm, and R = 17 nm. The images were taken from ref. 57 with permission. | ||
The experimental results proved the optical coupling between chiral molecules and metal NPs described by the model above. Ag NPs was prepared using DNA with 700 base pairs as a template, and the product showed a CD band centered at 425 nm attributed to the plasmon of Ag NPs.58 Similar result was observed for gold NPs. When chiral 1,3-disubstituted diamino calix[4]arene ligands were adsorbed on the surface of gold NPs capped with achiral tetraoctylammonium bromide, a new CD signal appeared in the region of 400–600 nm, which exactly located at the position of SPR band of gold NPs.59 The Cotton effects of gold NPs capped with calixarene enantiomers were mirror images of each other, indicating that they were a chiroptical pair of NP–ligand core-shell systems. Furthermore, remarkable plasmon resonance enhanced absorption and CD peaks were reported by introduction of bimane into L-glutathione stabilized the Ag NP solution.60 The absorption and CD spectra of bimane were enhanced about 70 and 100 times, respectively (Fig. 6A and 6B). Similar results were also observed when they used the mixture of L-glutathione and bimane (L-GSH–bimane) as stabilizers for synthesis of Ag NPs. Such an enhancement effect was attributed to the plasmon-induced electromagnetic enhancement that was similar to other surface-enhanced optical phenomena.61–63
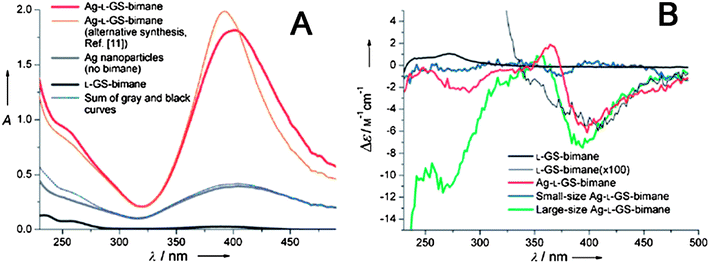 | ||
| Fig. 6 (A) Absorption spectra of different samples. The blue curve is the sum of the separate absorptions of L-GSH-bimane and the Ag NPs. (B) CD spectra of different samples. The small- and large-particle samples were separated from the L-GSH-bimane stabilized Ag NP solution by centrifugation. The thin black curve is the measurement for L-GSH-bimane with Δε multiplied by 100. The images were taken from ref. 60 with permission. | ||
Different from metal NPs, the optical activities of chiral semiconductor NPs are experimentally found to be more complex. For example, both CdS and CdTe NPs exhibited broad CD absorption features in the region of 250–400 nm, as shown in Fig. 1A and 2C.51–54 These CD signals were almost independent on the particle size and composition, so we can conclude that the signals should relate to the trapped states of particle surface rather than the exciton states, although the exact reason of their origin needs to further study.
Besides CD absorption, optical properties of chiral semiconductor NPs were also studied by using circularly polarized luminescence (CPL).64 CdS NPs synthesized within the cavities of ferritins showed left-hands CPL owing to the entire surrounding of helix-rich rhombic dodecahedral proteins. In addition to the direct transition at 498 nm, a strong surface-trapping band at 780 nm was also observed, because CdS NPs synthesized in low temperature easily produced surface defects. Furthermore, the CPL band could be tuned to blue region by laser photobleaching because of gradual decrease of NP sizes. In contrast, no CPL signal was observed for NPs stabilized by short-chain chiral molecules,51 which implied that a three-dimensional chiral confinement was important for production of CPL signals.
4.2 Optical properties of chiral NP assemblies
The study on optical properties of chiral NP assemblies is in burgeon stage. Like the model introduced in Part 4.1, the CD signal of the assembly could also be deduced though Coulomb interaction between NPs inside the assembly.65 The calculation result indicated that chiral assembly composed of gold NPs had both strong positive and negative CD bands in the visible wavelength range, and further the CD signals were very sensitive to their geometry and composition. For example, helical NP complexes with N = 4 (N is the number of NPs in assembly) and N = 5 exhibited inverted CD spectra. Actually strong optical activity was experimentally observed in the assembly system of DNA and Au NPs prepared by PCR technique (Fig. 4).The assembly was also prepared by attachment and subsequent growth of gold NPs on the chiral surface of diphenylalanine peptide nanotubes (PNT) (Fig. 7A-7C),66 Obviously, the superstructure made of gold NPs and PNT showed bisignated CD signal with symmetrical mirror images at the plasmon frequency of gold NPs, and its anisotropic factor (g-factor) was about 5 × 10−5. As comparison, no CD signal was observed at the same region before formation of gold NP assemblies, pointing to optical coupling between chiral molecules and NPs.
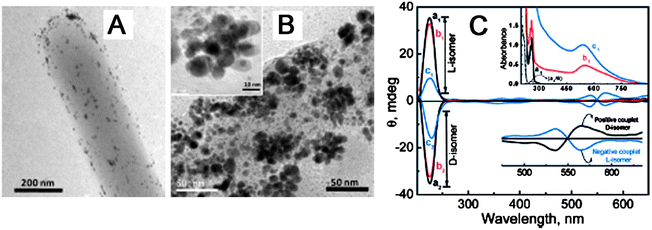 | ||
Fig. 7 (A) TEM images of PNT attachment of Au NPs. (B) Subsequent growth of Au NP assemblies. (C) Absorption (inset) and CD spectra of (a) PNT in water/HFP (99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at pH 10 on subsequent addition of (b) Au NPs and (c) growth of Au NPs. The bottom inset represents CD spectra at the surface plasmon frequency. The images were taken from ref. 66 with permission. :1) at pH 10 on subsequent addition of (b) Au NPs and (c) growth of Au NPs. The bottom inset represents CD spectra at the surface plasmon frequency. The images were taken from ref. 66 with permission. | ||
5 Bioapplications of chiral NPs
Thanks to their unique size effect and optical properties, biological applications of NPs have attracted considerable attention in recent years. Metal (semiconductor) NPs possess strong and tunable light absorption (emission), and remarkable resistance to chemical- and photo-degradation. So they have been considered as alternative materials for biodetection and biolabels.17 On the other hand, chirality is paramount in both structures and functions of bio-organisms. So, it is of great interest to study the biological properties and applications of chiral NPs.Inorganic NPs have shown potential applications in recognition of biomolecular enantiomers. Cyclodextrin-modified CdSe/ZnS core/shell NPs were found to have different fluorescent response to L-/D- amino acids including methionine and tyrosine.67 Although all amino acids could lead to the fluorescent enhancement of cyclodextrin-modified NPs, but the enhancement extent was largely dependent on the chirality of amino acids. The net fluorescence intensity increase of α-cyclodextrin-NPs with L-methionine was 3.4 times greater than that with D-methionine, while β-cyclodextrin-NPs exhibited 7.9 times net fluorescent enhancement in the presence of L-tyrosine than that of D-tyrosine. The difference in fluorescent enhancement of cyclodextrin-modified NPs induced by chiral amino acids was attributed to the difference in the stability constants of cyclodextrin/(amino acid) complexes. Gold NPs also had potential application in chiral recognition of enantiomers. Enantiomeric cysteine was mixed with the citrate-capped gold NPs in aqueous solution, and the rate for homochiral cysteine (LL, or DD) mediated assembly of NPs was explored to be 1 order of magnitude higher than that for the heterochiral mixture.68 The NP-regulated chiral recognition was a result of the creation of a nanoscale environment either favoring or not favoring the preferential configuration of the pairwise zwitterionic dimerization of the enantiomeric cysteines adsorbed on gold NPs. DFT calculation results indicated that the binding energies of the LL (or DD) and DL dimers were 11.2 and 8.5 kcal/mol, respectively, which demonstrated that LL (or DD) dimer was more stable than DL dimer when adsorbed onto Au NPs. Unfortunately, this method can not distinguish between L-cysteine and D-cysteine, since homochiral cysteine leads to similar aggregation of gold NPs. One advancement is the direct use of chiral NPs as substrates to recognize the enantiomers.69 As a example, the fluorescence intensity of L-cysteine stabilized CdSe/ZnS core/shell NPs decayed in the presence of D-carnitine but were not affected by L-carnitine (Fig. 8A). On the contrary, the fluorescence of D-cysteine-modified NPs was only affected by L-carnitine (Fig. 8B). This chiral recognition behavior was interpreted in terms of a “preferential interaction” model between a D-carnitine enantiomer with a chiral L-cysteine-capped NP surface and vice versa. The bond between the carboxyl group of cysteine and the NP surface was broken as cysteine interacted with carnitine, resulting in a dramatic quenching of its fluorescence.
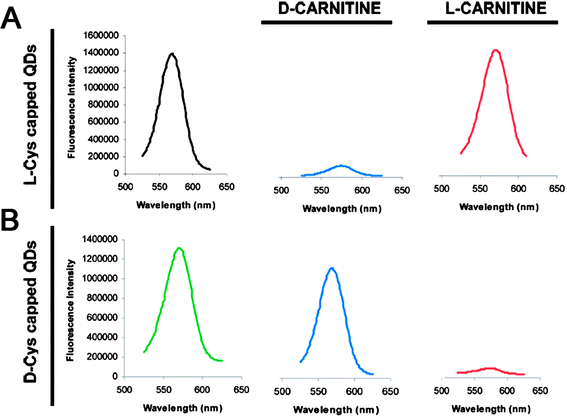 | ||
| Fig. 8 Emission spectra of (A) L-cysteine- and (B) D-cysteine-capped CdSe/ZnS NPs in the presence and absence of D-carnitine or L-carnitine enantiomers. The images were taken from ref. 69 with permission. | ||
In addition to recognition of biomolecular enantiomers, other potential applications of chiral NPs include bioimaging and biotherapy. Before application, the cytotoxicity of chiral NPs must be investigated. Recently, chiral penicillamine stabilized CdS nanotetrapods were proved to produce no cytotoxicity to the NG108-15 cells, and no obvious difference in the cytotoxicity was observed for enantiomer penicillamine stabilized CdS nanotetrapods.70
6 Concluding remarks and prospects
Although big progresses in study of chiral NPs have been made in recent years, it is still in its infancy at present. Further systematic research both in experiments and theories is needed for fundamental understanding and potential applications of chiral NPs.First, the general theory for the chirality origin of NP is desirable. As described in Part 3, two new models on surface structures of NPs have been proposed to interpret the chirality origin of CdS and CdTe NPs. However, these models are based on individual experimental results, and their applicability should be further verified by more experiments. It has been observed that a few experiments about NP chirality are interesting and difficult to be explained by present models. For example, why do CdTe NPs exhibit chiral memory effect54 and how can achiral stabilizers lead to formation of chiral Au NPs?71 So, the chirality origin of inorganic NPs need to be further study, and the exclusive understanding of chiral NPs is necessary. Obviously, this is a big challenge at present, because it is very difficult to characterize in situ the surface states of NPs and meanwhile the complexity of calculation is also an inevitable problem due to the large size of NPs.
Second, deep study of the chiral NPs will help us find new optical properties and potential applications. It is known that chiral signals of most biomolecules are located in the UV region, while chiral signals with tunable wavelength in visible optical regime are important for exploring their applications, such as negative index materials and second-order nonlinear materials. The calculation results demonstrate that CD absorption at longer wavelengths can be obtained through exciton-plasmon interactions or helical gold NP assembly. However, on one hand, this theoretical calculation need to be further verified by experimental data; on the other hand, these CD signals are too weak to be used although 70 times enhancement induced by metal NPs has been obtained.58 Whether it is possible to achieve larger amplification of chiral signals, especially in visible wavelength region, remains an open question. A possible strategy is controllable assembly or aggregation of noble metal NPs. Another key question is if the peak positions of chiral optical absorption could be tuned in visible region upon demand. It is known that the optical properties of semiconductor and noble metal NPs are size- or shape-dependent. So, could their optical activity be tuned by controlling NP sizes or shapes? For instance, most current studies on the chiral semiconductor NPs with different sizes only show broad CD absorption features in the region of 250–400 nm, which originate from their surface trap states. If one can design the chiral semiconductor NPs which optical activities are dominated by their exciton states, a size-dependent CD or CPL spectroscopy will be expected to be obtained.
Third, more attention should be paid to bio-effects of chiral NPs. It is known chiral (bio-)molecules have been widely used in therapy applications. For example, cis-platinum complexes are well-known as antineoplastic drugs. On the other hand, inorganic NPs have shown great potential in medical diagnostics and cancer therapy. So, an exciting question is whether the scientists can transfer chirality from molecular scale to nanometer scale and extend its bioapplications further.
In summary, chirality of NPs is giving rise to more interests because of its novel properties and potential applications,72 and we can expect a fast development of the corresponding research in the near future.
Acknowledgements
The authors are grateful for financial support from National Natural Science Foundation for Distinguished Youth Scholars of China (21025310, Z.Y.T.), National Natural Science Foundation of China (20973047, Z.Y.T.), the National Research Fund for Fundamental Key Project (2009CB930401, Z.Y.T.), China-Korea Joint Research Project (2010DFA51700, Z.Y.T.), and 100-Talent Program of Chinese Academy of Sciences (Z.Y.T.).References
- L. Brus, J. Phys. Chem., 1986, 90, 2555–2560 CrossRef CAS.
- H. Weller, Angew. Chem., Int. Ed. Engl., 1993, 32, 41–53 CrossRef.
- A. P. Alivisatos, Science, 1996, 271, 933–937 CrossRef CAS.
- A. P. Alivisatos, J. Phys. Chem., 1996, 100, 13226–13239 CrossRef CAS.
- C. B. Murray, C. R. Kagan and M. G. Bawendi, Annu. Rev. Mater. Sci., 2000, 30, 545–610 CrossRef CAS.
- A. M. Smith and S. Nie, Acc. Chem. Res., 2010, 43, 190–200 CrossRef CAS.
- M. D. Regulaclo and M. Y. Han, Acc. Chem. Res., 2010, 43, 621–630 CrossRef.
- R. Jin, Y. W. Cao, E. Hao, G. S. Metraux, G. C. Schatz and C. A. Mirkin, Nature, 2003, 425, 487–490 CrossRef CAS.
- M. C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS.
- S. K. Ghosh and T. Pal, Chem. Rev., 2007, 107, 4797–4862 CrossRef CAS.
- J. Zhao, A. O. Pinchuk, J. M. McMahon, S. Li, L. K. Ausman, A. L. Atkinson and G. C. Schatz, Acc. Chem. Res., 2008, 41, 1710–1720 CrossRef CAS.
- S. E. Skrabalak, J. Chen, Y. Sun, X. Lu, L. Au, C. M. Cobley and Y. Xia, Acc. Chem. Res., 2008, 41, 1587–1595 CrossRef CAS.
- S. Eustis and M. A. El-Sayed, Chem. Soc. Rev., 2006, 35, 209–217 RSC.
- J. M. Caruge, J. E. Halpert, V. Wood, V. Bulović and M. G. Bawendi, Nat. Photonics, 2008, 2, 247–250 CrossRef CAS.
- D. V. Talapin, J. S. Lee, M. V. Kovalenko and E. V. Shevchenko, Chem. Rev., 2010, 110, 389–458 CrossRef CAS.
- C. Burda, X. Chen, R. Narayanan and M. A. El-Sayed, Chem. Rev., 2005, 105, 1025–1102 CrossRef CAS.
- X. Michalet, F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Sundaresan, A. M. Wu, S. S. Gambhir and S. Weiss, Science, 2005, 307, 538–544 CrossRef CAS.
- R. A. Sperling, P. R. Gil, F. Zhang, M. Zanella and W. J. Parak, Chem. Soc. Rev., 2008, 37, 1896–1908 RSC.
- E. Boisselier and D. Astruc, Chem. Soc. Rev., 2009, 38, 1759–1782 RSC.
- N. A. Frey, S. Peng, K. Cheng and S. Sun, Chem. Soc. Rev., 2009, 38, 2532–2542 RSC.
- G. H. Wagniére, On Chirality and the Universal Asymmetry, Verlag Helvetica Chimica Acta, Zürich, and Wiley-VCH, Weinheim, 2007 Search PubMed.
- L. D. Barron, Molecular Light Scattering and Optical Activity, Cambridge University Press, Cambridge, 2004 Search PubMed.
- S. H. Yu, H. Cölfen, K. Tauer and Markus Antonietti, Nat. Mater., 2005, 4, 51–55 CrossRef CAS.
- S. C. Stinson, Chem. Eng. News, 2000, 43, 55–78 CrossRef.
- T. G. Schaaff, G. Knight, M. N. Shafigullin, R. F. Borkman and R. L. Whetten, J. Phys. Chem. B, 1998, 102, 10643–10646 CrossRef CAS.
- T. G. Schaaff and R. L. Whetten, J. Phys. Chem. B, 2000, 104, 2630–2641 CrossRef CAS.
- N. Nishida, H. Yao, T. Ueda, A. Sasaki and K. Kimura, Chem. Mater., 2007, 19, 2831–2841 CrossRef CAS.
- H. Yao, K. Miki, N. Nishida, A. Sasaki and K. Kimura, J. Am. Chem. Soc., 2005, 127, 15536–15543 CrossRef CAS.
- C. Gautier and T. Bürgi, J. Am. Chem. Soc., 2006, 128, 11079–11087 CrossRef CAS.
- O. Lopez-Acevedo, H. Tsunoyama, T. Tsukuda, H. H. äkkinen and C. M. Aikens, J. Am. Chem. Soc., 2010, 132, 8210–8218 CrossRef CAS.
- C. Gautier and T. Bürgi, J. Am. Chem. Soc., 2008, 130, 7077–7084 CrossRef CAS.
- Z. Wu, C. Gayathri, R. R. Gil and R. Jin, J. Am. Chem. Soc., 2009, 131, 6535–6542 CrossRef CAS.
- M. R. Provorse and C. M. Aikens, J. Am. Chem. Soc., 2010, 132, 1302–1310 CrossRef CAS.
- A. Sánchez-Castillo, C. Noguez and I. L. Garzón, J. Am. Chem. Soc., 2010, 132, 1504–1505 CrossRef CAS.
- H. Yao, T. Fukui and K. Kimura, J. Phys. Chem. C, 2007, 111, 14968–14976 CrossRef CAS.
- H. Yao, T. Fukui and K. Kimura, J. Phys. Chem. C, 2008, 112, 16281–16285 CrossRef CAS.
- S. Si, C. Gautier, J. Boudon, R. Taras, S. Gladiali and T. Bürgi, J. Phys. Chem. C, 2009, 113, 12966–12969 CrossRef CAS.
- N. Nishida, H. Yao and K. Kimura, Langmuir, 2008, 24, 2759–2766 CrossRef CAS.
- N. Cathcart, P. Mistry, C. Makra, B. Pietrobon, N. Coombs, M. Jelokhani-Niaraki and V. Kitaev, Langmuir, 2009, 25, 5840–5846 CrossRef CAS.
- H. Qi, J. O'Neil and T. Hegmann, J. Mater. Chem., 2008, 18, 374–380 RSC.
- J. M. Ha, A. Solovyov and A. Katz, Langmuir, 2009, 25, 153–158 CrossRef CAS.
- H. Qi and T. Hegmann, J. Am. Chem. Soc., 2008, 130, 14201–14206 CrossRef CAS.
- C. E. Román-Velázquez, C. Noguez and I. L. Garzón, J. Phys. Chem. B, 2003, 107, 12035–12038 CrossRef CAS.
- M. R. Goldsmith, C. B. George, G. Zuber, R. Naaman, D. H. Waldeck, P. Wipfac and D. N. Beratan, Phys. Chem. Chem. Phys., 2006, 8, 63–67 RSC.
- I. L. Garzón1, M. R. Beltrán, G. González, I. Gutíerrez-González1, K. Michaelian1, J. A. Reyes-Nava1 and J. I. Rodríguez-Hernández, Eur. Phys. J. D, 2003, 24, 105–109 CrossRef CAS.
- H. Yao, Curr. Nanosci., 2008, 4, 92–97 Search PubMed.
- C. Gautier and T. Bürgi, ChemPhysChem, 2009, 10, 483–492 CrossRef CAS.
- C. Gautier and T. Bürgi, Chiral Nanoparticles. In Chirality at the Nanoscale: Nanoparticles, Surfaces, Materials and more, Wiley-VCH: Weinheim, 2009, Vol. 1, pp 67-111 Search PubMed.
- C. Gautier and T. Bürgi, J. Phys. Chem. C, 2010, 114, 15897–15902 CrossRef CAS.
- C. Noguez and I. L. Garzón, Chem. Soc. Rev., 2009, 38, 757–771 RSC.
- M. P. Moloney, Y. K. Gun'ko and J. M. Kellya, Chem. Commun., 2007, 3900–3902 RSC.
- S. D. Elliott, M. P. Moloney and Y. K. Gun'ko, Nano Lett., 2008, 8, 2452–2457 CrossRef CAS.
- Y. Zhou, M. Yang, K. Sun, Z. Tang and N. A. Kotov, J. Am. Chem. Soc., 2010, 132, 6006–6013 CrossRef CAS.
- T. Nakashima, Y. Kobayashi and T. Kawai, J. Am. Chem. Soc., 2009, 131, 10342–10343 CrossRef CAS.
- A. J. Mastroianni, S. A. Claridge and A. P. Alivisatos, J. Am. Chem. Soc., 2009, 131, 8455–8459 CrossRef CAS.
- W. Chen, A. Bian, A. Agarwal, L. Liu, H. Shen, L. Wang, C. Xu and N. A. Kotov, Nano Lett., 2009, 9, 2153–2159 CrossRef CAS.
- A. O. Govorov, Z. Fan, P. Hernandez, J. M. Slocik and R. R. Naik, Nano Lett., 2010, 10, 1374–1382 CrossRef CAS.
- G. Shemer, O. Krichevski, G. Markovich, T. Molotsky, I. Lubitz and A. B. Kotlyar, J. Am. Chem. Soc., 2006, 128, 11006–11007 CrossRef CAS.
- J. M. Ha, A. Solovyov and A. Katz, Langmuir, 2009, 25, 153–158 CrossRef CAS.
- I. Lieberman, G. Shemer, T. Fried, E. M. Kosower and G. Markovich, Angew. Chem., Int. Ed., 2008, 47, 4855–4857 CrossRef CAS.
- S. M. Nie and S. R. Emory, Science, 1997, 275, 1102–1106 CrossRef CAS.
- B. Knoll and F. Keilmann, Nature, 1999, 399, 134–137 CrossRef CAS.
- J. R. Lakowicz, Anal. Biochem., 2005, 337, 171–194 CrossRef CAS.
- M. Naito, K. Iwahori, A. Miura, M. Yamane and I. Yamashita, Angew. Chem., Int. Ed., 2010, 49, 7006–7009 CrossRef CAS.
- Z. Fan and A. O. Govorov, Nano Lett., 2010, 10, 2580–2587 CrossRef CAS.
- J. George and K. G. Thomas, J. Am. Chem. Soc., 2010, 132, 2502–2503 CrossRef CAS.
- C. Han and H. Li, Small, 2008, 4, 1344–1350 CrossRef CAS.
- I. S. Lim, D. Mott, M. H. Engelhard, Y. Pan, S. Kamodia, J. Luo, P. N. Njoki, S. Zhou, L. Wang and C. J. Zhong, Anal. Chem., 2009, 81, 689–698 CrossRef CAS.
- C. Carrillo-Carrión, S. Cárdenas, B. M. Simonet and M. Valcárcel, Anal. Chem., 2009, 81, 4730–4733 CrossRef CAS.
- J. E. Govan, E. Jan, A. Querejeta, N. A. Kotov and Y. K. Gun'ko, Chem. Commun., 2010, 46, 6072–6074 RSC.
- P. D. Jadzinsky, G. Calero, C. J. Ackerson, D. A. Bushnell and R. D. Kornberg, Science, 2007, 318, 430–433 CrossRef CAS.
- V. Kitaev, J. Mater. Chem., 2008, 18, 4745–4749 RSC.
| This journal is © The Royal Society of Chemistry 2011 |