Nanoporous bimetallic Pt–Au alloy nanocomposites with superior catalytic activity towards electro-oxidation of methanol and formic acid†
Zhonghua
Zhang
*a,
Yan
Wang
*b and
Xiaoguang
Wang
a
aKey Laboratory for Liquid-Solid Structural Evolution and Processing of Materials (Ministry of Education), School of Materials Science and Engineering, Shandong University, Jingshi Road 17923, Jinan, 250061, P.R. China. E-mail: zh_zhang@sdu.edu.cn
bSchool of Materials Science and Engineering, University of Jinan, Jiwei Road 106, Jinan, 250022, P.R. China. E-mail: mse_wangy@ujn.edu.cn
First published on 11th February 2011
Abstract
We present a facile route to fabricate novel nanoporous bimetallic Pt–Au alloy nanocomposites by dealloying a rapidly solidified Al75Pt15Au10 precursor under free corrosion conditions. The microstructure of the precursor and the as-dealloyed sample was characterized using X-ray diffraction, scanning electron microscopy, transmission electron microscopy, high-resolution transmission electron microscopy, and energy dispersive X-ray (EDX) analysis. The Al75Pt15Au10 precursor is composed of a single-phase Al2(Au,Pt) intermetallic compound, and can be fully dealloyed in a 20 wt.% NaOH or 5 wt.% HCl aqueous solution. The dealloying leads to the formation of the nanoporous Pt60Au40 nanocomposites (np-Pt60Au40 NCs) with an fcc structure. The morphology, size and crystal orientation of grains in the precursor can be conserved in the resultant nanoporous alloy. The np-Pt60Au40 NCs consist of two zones with distinct ligament/channel sizes and compositions. The formation mechanism of these np-Pt60Au40 NCs can be rationalized based upon surface diffusion of more noble elements and spinodal decomposition during dealloying. Electrochemical measurements demonstrate that the np-Pt60Au40 NCs show superior catalytic activity towards the electro-oxidation of methanol and formic acid in the acid media compared to the commercial JM-Pt/C catalyst. This material can find potential applications in catalysis related areas, such as direct methanol or formic acid fuel cells. Our findings demonstrate that dealloying is an effective and simple strategy to realize the alloying of immiscible systems under mild conditions, and to fabricate novel nanostructures with superior performance.
Introduction
Pt is one of the most commonly used catalysts and electrocatalysts in a wide range of applications such as methanol oxidation, oxygen reduction reactions, and so on. However, monometallic Pt is susceptible to deactivation or poisoning during catalytic and electrocatalytic processes.1 For example, Pt catalysts can be deactivated due to carbon deposition or coking in many heterogeneous catalytic reactions, and can also be poisoned by CO adsorption due to the strong bonding of CO on the Pt surface. It is well known that adding a second metallic component can enhance the activity, selectivity, and stability of pure metal catalysts. It has been reported that the formation of bimetallic nanostructures with other metals, such as Ru, Ag and Au, can reduce CO poisoning. Thus, bimetallic nanomaterials constituting various combinations of noble metals (e.g. Ag–Au,2,3 Au–Ni,4 Au–Pd,5,6 Au–Pt,7–15 Pt–Ag,16 Pd–Ag,16) have attracted great attention owing to their unique catalytic, magnetic, optical and electronic properties that are distinct not only from the bulk metals but also from the corresponding monometallic nanoparticles. Among them, Au–Pt is of special interest for their potential applications in catalysis, specially for electrocatalysis in fuel cell reactions, and bimetallic Au–Pt nanostructures with various compositions, sizes and morphologies have recently been synthesized.7–15 Shen et al.7 have reported the synthesis of free standing Pt–Au bimetallic membranes with a leaf-like nanostructure from agarose-mediated electrodeposition. Monte Carlo simulations were carried out to systematically investigate the effects of composition, size, and temperature on the surface segregation and structural features of Au–Pt nanoparticles.8 A new, highly sensitive and selective sensor for the electrochemical assay of Hg(II) has been developed, whereby a glassy carbon electrode is modified with bimetallic Au–Pt nanoparticles/organic nanofibers.9 Peng and Yang14 have fabricated Pt–Au bimetallic heteronanostructures through post-synthesis modification of Pt-on-Au nanoparticles, and these Pt–Au heteronanostructures show much more activity than Pt in catalyzing the oxidation of formic acid.The properties of bimetallic nanostructures can vary dramatically not only with size, as happens in monometallic nanoclusters, but also with chemical composition. Controlling their structure and chemical ordering can be the starting point to prepare building blocks for specifically tailored cluster-assembled materials.17 The chemical synthesis of bimetallic nanostructures can be roughly classified into two approaches.16 One employs a galvanic replacement reaction. Disadvantages are that the two metals should have a large difference in electrochemical redox potential, which restricts the possible combinations of two metals. In addition, the tunability of alloy composition is limited. The other more popular approach is based on co-reduction of two metal salts by a strong reductant (such as NaBH4). The inconveniences include complex reaction microenvironments and/or tedious cleaning procedures after synthesis. There exist several possible bimetallic architectures including alloys, core-shell nanoparticles and contact aggregates (or heteroaggregates).12 The stabilities of bimetallic architectures are usually dictated by the thermodynamic miscibility of the two metals. According to the binary phase diagram, Au and Pt are immiscible over a wide composition and temperature range. As such, core–shell and contact aggregate structures are thermodynamically more stable whereas Au–Pt alloy nanostructures are metastable. In addition, theoretical calculations have also suggested that Pt and Au prefer core–shell or core–shell-like structures due to their thermodynamic immiscibility.18 Above all, bimetallic PtAu alloy nanoparticles display unique physicochemical properties that are different from those of monometallic and nonalloyed solids. Thus, the performance of alloyed PtAu samples as electrocatalysts for the oxygen reduction reaction is superior to that of the PtAu-segregated samples.19 However, the main remaining challenge is that the preparation of alloyed Au–Pt nanostructures either requires severe thermal treatments, generally yielding aggregated particles, or follows milder but complicated routes to obtain nanostructured materials.15
Recently, dealloying has attracted wide attention as an ideal method to produce nanoporous (np) metals,20,21 which refers to selective dissolution of one or more components out of an alloy.22 Nanoporous metals fabricated by dealloying generally assume a three-dimensional bicontinuous interpenetrating ligament-channel structure at the nanometre scale, and have been widely investigated in such applications as catalysis and fuel cells, sensing, actuation, microfluidic controlling, and so forth.20,21,23 The dealloying process (also known as depletion gilding) has an ancient history, and has been used to colorate artifacts by Indians of pre-Columbian Central America and medieval artisans in the Near East and Europe.24 This process has also been developed to prepare high surface area metal catalysts such as RANEY® nickel since the days of RANEY®.25 Similar to the great success of RANEY® metals and nanoporous pure metals (like nanoporous gold, NPG20,21,23), dealloying has recently been recognized as an effective method to prepare useful alloy nanocatalysts.26–33 For example, the dealloying method has been employed to fabricate nanoporous Au–Pt and Pt–Ru alloys with pre-determined compositions through selectively etching Cu and Al from CuAuPt and PtRuAl precursors, respectively.27,28 Moreover, these alloy nanostructures show much enhanced specific activity towards the electro-oxidation of methanol and formic acid. Liu et al.32,33 have reported the synthesis of nanoporous Pt–Co and Pt–Ni alloy nanowires with an enhanced electrocatalytic activity by the combination of template (AAO) method and dealloying strategy.
In our former work, we have fabricated an ultrafine nanoporous Ag–Pd alloy by dealloying a ternary Mg–Ag–Pd precursor, and have found that the addition of the third element Pd into Mg–Ag can realize the design and functionalization of a nanoporous bimetallic alloy structure which exhibits a superior catalytic activity towards electro-oxidation of ethanol.34 In the present work, we mainly focus upon the chemical dealloying of a ternary Al–Pt–Au precursor in acid and alkaline solutions. Nanoporous Pt–Au alloy nanocomposites (NCs) can thus be synthesized and their formation mechanism has been analyzed based upon surface diffusion of more noble elements and spinodal decomposition. In addition, these nanoporous Pt–Au nanocomposites exhibit a superior catalytic activity towards electro-oxidation of small organic molecules such as methanol and formic acid.
Compared to the CuAuPt precursor,27 it should be noted that the Al–Pt–Au precursor has some advantages for the fabrication of nanoporous Pt–Au alloys. Firstly, there exists a greater difference in standard electrode potentials between Al and Pt/Au than that between Cu and Pt/Au. Thus, the dealloying can be easier performed under potential control and even under free corrosion conditions, and the dealloying duration is much shorter (typically less than 1 h) than that (3–10 h27) of the CuAuPt precursor. Secondly, due to the amphoteric nature of Al, the dealloying of Al–Pt–Au can be carried out in either acidic (such as HCl) or alkaline (such as NaOH) solutions. Furthermore, the nanoporous structure of the as-obtained Pt–Au alloy can be tuned by simply changing the dealloying solution. Thirdly, using the pitting tendency of Al in Cl−-containing solutions, the Al–Pt–Au precursor can even be dealloyed in neutral NaCl aqueous solutions under potential control, which is important for the ‘green’ fabrication of nanoporous Pt–Au alloys. And the investigation on the electrochemical dealloying of Al–Pt–Au in the neutral NaCl solution is in progress.
Experimental section
The Al75Pt15Au10 (at.%) precursor alloy was prepared from elemental Al (purity, 99.95 wt.%), Pt (purity, 99.9 wt.%) and Au (purity, 99.9 wt.%) in a quartz crucible using a high-frequency induction furnace. Using a single roller melt spinning apparatus, the pre-alloyed ingots were remelted by high-frequency induction heating in a quartz tube and then melt-spun onto a copper roller with a diameter of 0.35 m at a speed of 1000 revolutions per minute (rpm) in a controlled argon atmosphere. The rapidly solidified ribbons obtained were typically 20–50 μm in thickness, 2–5 mm in width and several centimetres in length. The dealloying of the rapidly solidified Al75Pt15Au10 alloy was carried out in a 20 wt.% NaOH aqueous solution firstly at room temperature until no obvious bubbles emerged, and then at 90 ± 5 °C to further leach out the residual Al in the samples. The total dealloying time is less than 1.0 h. In addition, the dealloying was also performed in a 5 wt.% HCl aqueous solution in order to investigate the influence of chloride ion (Cl−) on the dealloying process; the total dealloying duration is less than 0.5 h. The as-dealloyed samples were rinsed using distilled water and dehydrated alcohol.The phases present in the rapidly solidified Al75Pt15Au10 precursor and as-dealloyed samples were identified using an X-ray diffractometer (XRD, Hitachi Rigaku D/max-RB)) with Cu Kα radiation. The microstructure of the Al75Pt15Au10 precursor was observed using a scanning electron microscope (SEM, LEO 1530VP) in a back scattered mode. The microstructure of the as-dealloyed samples was characterized using SEM (LEO 1530VP in an Inlens mode), transmission electron microscopy (TEM, Philips CM 20), and high-resolution TEM (HRTEM, FEI Tecnai G2). Selected-area electron diffraction (SAED) and fast Fourier transform (FFT) were also used to document the crystalline nature of the as-dealloyed samples. The chemical compositions of the rapidly solidified Al75Pt15Au10 precursor and as-dealloyed samples were determined by an energy-dispersive X-ray (EDX) analyzer which was attached to SEM. In addition, scanning transmission electron microscopy (STEM) images and nanobeam-EDX (NB-EDX) spectra were also obtained by the FEI Tecnai G2 microscope under high-angle annular dark filed (HAADF) mode. For one composition, at least 3–5 EDX experiments were carried out and an average value was given in the results.
The electrochemical measurements were performed in a standard three-electrode cell using a LK 2005A Potentiostat. The catalyst suspensions were prepared as follows: 2 mg fine ground as-dealloyed samples, 3 mg Vulcan XC-72 carbon powders, 300 μL isopropanol, and 100 μL Nafion solution (0.5 wt.%) were ultrasonically mixed. Then, 5 μL of the homogeneously mixed catalyst ink was placed on a freshly polished glassy carbon (GC) electrode with a diameter of 4 mm. These modified GC electrodes were used as the working electrode (Here, the Vulcan XC-72 carbon powders were used to better disperse and support the ground catalyst samples. And the catalyst can better adhere onto the GC electrode surface.27,28,34). The counter electrode was a bright Pt plate, and a saturated calomel electrode (SCE) was used as the reference electrode. Voltammetric behavior was characterized in a 0.5 M H2SO4 solution deaerated with N2. The electrocatalytic activity measurements were carried out in solutions of 0.5 M H2SO4 + 0.5 M CH3OH and 0.5 M H2SO4 + 0.5 M HCOOH. For comparison, the commercial JM-Pt/C (40 wt.% Pt) catalyst was also measured under the identical experimental conditions. All electrochemical experiments were performed at ambient temperature (∼ 25 °C).
Results and discussion
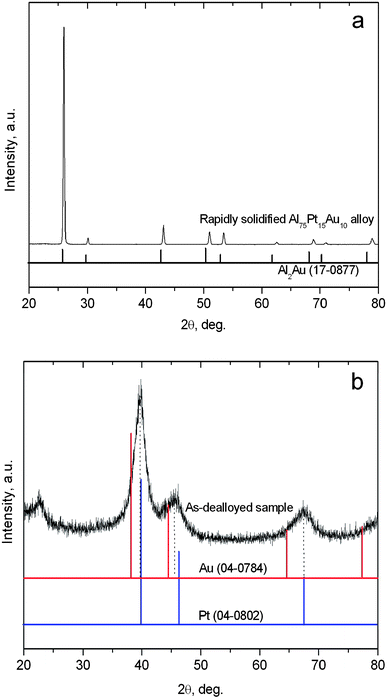
Fig. 1a shows the XRD pattern of the rapidly solidified Al75Pt15Au10 precursor. By comparison with the reference profile of Al2Au (PDF No. 17-0877), it is interesting to note that the Al75Pt15Au10 precursor is composed of a single-phase intermetallic compound with an Al2Au-type structure. According to the binary phase diagram,35 the Al-rich Al–Pt alloy (such as Al75Pt25) normally consists of α-Al and Al21Pt8/Al21Pt5. In the present case, no AlxPty-type intermetallic phase can be identified in the rapidly solidified Al75Pt15Au10 precursor. Although the atomic ratio of Pt![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Au is 3:2, the Al2Au-type intermetallic phase is present in the rapidly solidified precursor. Therefore, it is reasonable to assume that Pt exists in a solid solution form partially substituting for Au in the Al2Au lattice. This phase in the Al75Pt15Au10 precursor can be denoted as Al2(Au,Pt). In addition, all diffraction peaks shift to higher angles compared to the reference profile of Al2Au, especially for the high-index reflections (Fig. 1a). This demonstrates that the introduction of Pt causes the lattice contraction of Al2Au. Besides, it should be noted that the intensity of the diffraction peak at ∼25.7° is much higher than that of the other peaks in the XRD pattern, indicating the preferred orientation (111) of the rapidly solidified alloy. It is obvious that rapid solidification plays a significant role in the formation and orientation of Al2(Au,Pt) in the Al75Pt15Au10 precursor. The single-phase microstructure of the rapidly solidified Al75Pt15Au10 precursor can be further verified by SEM observation. Fig. 2 shows the microstructure of the rapidly solidified Al75Pt15Au10 precursor. It is clear that equiaxed and columnar dendrites can be observed throughout the whole section of the precursor ribbons (Fig. 2a). The grains are several microns in size as can be seen at a higher magnification in Fig. 2b. Based upon the XRD analysis, these dendrites have the structure of Al2(Au,Pt). The EDX results show that the real composition of the rapidly solidified Al75Pt15Au10 precursor is 73.5 at.% Al, 15.8 at.% Pt and 10.7 at.% Au, which is highly consistent with the nominal composition of the alloy. A typical EDX spectrum is given in Fig. 2c.
:Au is 3:2, the Al2Au-type intermetallic phase is present in the rapidly solidified precursor. Therefore, it is reasonable to assume that Pt exists in a solid solution form partially substituting for Au in the Al2Au lattice. This phase in the Al75Pt15Au10 precursor can be denoted as Al2(Au,Pt). In addition, all diffraction peaks shift to higher angles compared to the reference profile of Al2Au, especially for the high-index reflections (Fig. 1a). This demonstrates that the introduction of Pt causes the lattice contraction of Al2Au. Besides, it should be noted that the intensity of the diffraction peak at ∼25.7° is much higher than that of the other peaks in the XRD pattern, indicating the preferred orientation (111) of the rapidly solidified alloy. It is obvious that rapid solidification plays a significant role in the formation and orientation of Al2(Au,Pt) in the Al75Pt15Au10 precursor. The single-phase microstructure of the rapidly solidified Al75Pt15Au10 precursor can be further verified by SEM observation. Fig. 2 shows the microstructure of the rapidly solidified Al75Pt15Au10 precursor. It is clear that equiaxed and columnar dendrites can be observed throughout the whole section of the precursor ribbons (Fig. 2a). The grains are several microns in size as can be seen at a higher magnification in Fig. 2b. Based upon the XRD analysis, these dendrites have the structure of Al2(Au,Pt). The EDX results show that the real composition of the rapidly solidified Al75Pt15Au10 precursor is 73.5 at.% Al, 15.8 at.% Pt and 10.7 at.% Au, which is highly consistent with the nominal composition of the alloy. A typical EDX spectrum is given in Fig. 2c.
 | ||
| Fig. 1 XRD patterns of the (a) rapidly solidified Al75Pt15Au10 alloy and (b) as-dealloyed sample in the 20 wt.% NaOH solution. | ||
 | ||
| Fig. 2 (a,b) Back-scattered SEM images showing the section-view microstructure of the rapidly solidified Al75Pt15Au10 alloy, and (c) a typical EDX spectrum. | ||
Fig. 1b shows the XRD pattern of the as-dealloyed sample in the 20 wt.% NaOH solution. Obviously, only one set of broad diffraction peaks can be observed, which can be assigned to the (111), (200) and (220) reflections of an fcc structure. All diffraction peaks appear between the reference peaks of Au (PDF No. 04-0784) and Pt (PDF No. 04-0802), but are closer to those of Pt. The present results indicate the formation of Pt(Au) alloy after dealloying. It is well known that Pt and Au are not miscible within a wide range of concentrations. Also, the equiatomic AuPt alloy is only thermodynamically stable above 1200 °C.35 In fact, Xu et al.27 have reported that the dealloying of CuPtAu alloys results in the formation of Au/Pt alloy structures. The present dealloying temperature is only 90 ± 5 °C, which is much lower than the thermodynamically stable temperature of the equiatomic AuPt alloy. It is well recognized that the dealloying process involves the dissolution of the less noble element (here, Al) and the surface diffusion/rearrangement of the more noble element (here, Pt and Au) at the nanoscale.22,24 Moreover, the physical and chemical properties of nanoscale materials are different from the bulk crystalline state even at low temperatures.36 Hence, the nanoscale alloying along the alloy/solution interface contributes to the formation of the Pt(Au) alloy during dealloying. The alloying is the main challenge for the preparation of Au–Pt nanostructures under mild reaction conditions by other methods such as galvanic replacement reaction, co-reduction of two metal salts, etc.15 The present results demonstrate that dealloying is an effective and facile route to realize the alloying of immiscible systems like Pt–Au even at low temperatures (less than 100 °C).
Fig. 3 shows the section-view SEM microstructure of the as-dealloyed sample in the 20 wt.% NaOH solution. At low magnification (Fig. 3a), the morphology of the as-dealloyed sample resembles that of the rapidly solidified Al75Pt15Au10 precursor (Fig. 2b), suggesting that the grain morphology and size of the precursor can be inherited in the resultant nanoporous sample during dealloying. An ultrafine nanoporous microstructure can be observed at a higher magnification (Fig. 3b), which exhibits a three-dimensional bicontinuous interpenetrating ligament-channel structure. The EDX analysis shows that the as-dealloyed sample is composed of 60.1 at.% Pt and 39.9 at.% Au, which is very consistent with the Pt/Au atomic ratio in the precursor. For simplicity, the as-dealloyed nanoporous sample is designated as np-Pt60Au40. A typical EDX spectrum is shown in Fig. 3c. In addition, a small amount of Al (several atom percent) can be detected in the as-dealloyed sample.
 | ||
| Fig. 3 (a,b) SEM images showing the section-view microstructure of the np-Pt60Au40 NCs fabricated by dealloying the rapidly solidified Al75Pt15Au10 precursor in the 20 wt.% NaOH solution, and (c) a typical EDX spectrum. | ||
For clarity, the microstructure of the np-Pt60Au40 alloy was further examined by TEM and HRTEM. Fig. 4 shows the TEM image and corresponding SAED patterns of the np-Pt60Au40 alloy fabricated by dealloying in the 20 wt.% NaOH solution. It is interesting to note that the microstructure of the np-Pt60Au40 alloy is inhomogeneous at the nanoscale and composed of two nanoporous zones with different ligament-channel sizes, as highlighted by A and B in Fig. 4a. The finer zones (A) have a ligament-channel size of 3.0 ± 0.5 nm, while the ligament size is 4.0 ± 1.0 nm and the channel size is 8.0 ± 2.0 nm in the coarser zones (B). As shown in Fig. 4b and c, the corresponding SAED patterns (close to [110] zone axis of the fcc Pt(Au) structure) show that both the finer and coarser zones have a single crystalline orientation in the selected areas (∼ 200 nm in diameter). The coarser and finer zones are interpenetrating and have a length scale of several hundred nm (Fig. 4a and Fig. S1a in ESI†). Despite different nanoporous structures, these two zones have almost the same crystalline orientation in a given area, suggesting that they belong to an identical grain (Fig. 4 and Fig. S1 in ESI†). It should be noted that the diffraction spots are diffused (not sharp) together with spot pairs/arcs (Fig. 4b) and weak diffraction rings (Fig. 4c). Although the overall orientation of the selected area is single crystalline, small nanocrystals with slightly different orientations as well as dislocations, stacking faults, and lattice distortions can be observed in the microstructure (see Fig. 5), which may contribute to the non-sharp appearance of the diffraction spots. It is well established that the crystal orientation is retained during dealloying with the conservation of the grain size of the master alloy.37 For example, NPG made by dealloying Ag/Au alloys often adopts a single-crystalline porous grain structure.38 In our previous work, we have found that the morphology, size, and orientation of the grains in the RS Al66.6Au33.4 alloy (composed of a single-phase Al2Au) can be conserved into the resulting NPG ribbons during dealloying.39 It is obvious that the dealloying of the Al2(Au,Pt) phase is quite similar to that of Al2Au in this respect.
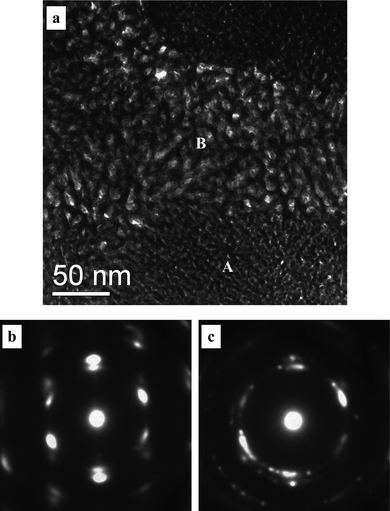 | ||
| Fig. 4 (a) TEM image showing the microstructure of the np-Pt60Au40 NCs fabricated by dealloying the rapidly solidified Al75Pt15Au10 precursor in the 20 wt.% NaOH solution, and (b,c) SAED patterns corresponding to zone A and B, respectively. | ||
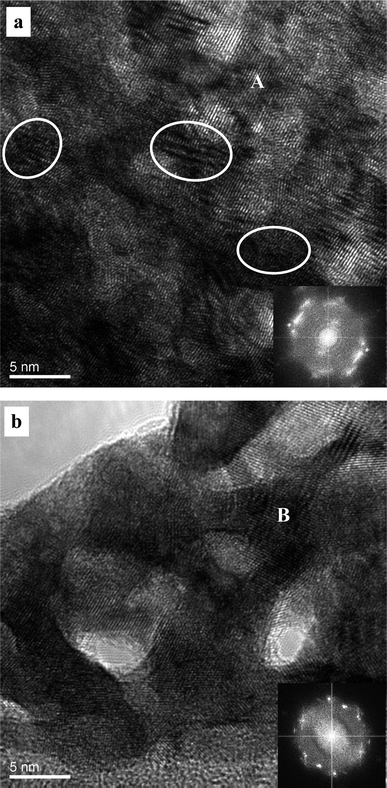 | ||
| Fig. 5 HRTEM images showing the microstructure of the np-Pt60Au40 NCs fabricated by dealloying the rapidly solidified Al75Pt15Au10 precursor in the 20 wt.% NaOH solution. Insets: corresponding FFT patterns. Some stacking faults, dislocations and lattice distortions are highlighted by ellipses in (a). | ||
Fig. 5 shows the HRTEM images and corresponding FFT patterns of the finer and coarser zones in the np-Pt60Au40 alloy fabricated by dealloying in the 20 wt.% NaOH solution. The ligament-channel size and structure can be clearly observed in both zones. It can be seen that lattice fringes extend throughout all the ligaments in the whole frame of observation, further confirming the single crystalline nature of these two zones. The corresponding FFT patterns also verify the single crystalline characteristic of the nanoporous structure (insets of Fig. 5). In addition, dislocations, stacking faults, and lattice distortions can also be observed in the HRTEM image, as highlighted by ellipses in Fig. 5a. To accurately determine the chemical compositions of these finer and coarser zones, STEM observation was performed under the HAADF mode and one typical image is shown in Fig. 6a. Both the coarser and finer nanoporous zones can be clearly observed in the z-contrast STEM image. The positions for the NB-EDX measurements were marked in the STEM image (see Fig. S2 in ESI†). Typical NB-EDX spectra are shown in Fig. 6b and c for the finer (A) and coarser (B) zones, respectively. It is clear that the chemical composition of the finer zones is different from that of the coarser zones. The chemical composition of the finer zones is 70.4 at.% Pt and 29.6 at.% Au (can be denoted as Pt70Au30), while that of the coarser zones is 52.6 at.% Pt and 47.4 at.% Au (can be denoted as Pt50Au50). However, this difference in composition between these two zones cannot be distinguished by XRD (Fig. 1b). Different structures and compositions of these two zones demonstrate that the present np-Pt60Au40 alloy eventually has a nanocomposite structure and can be designated as np-Pt60Au40 NCs.
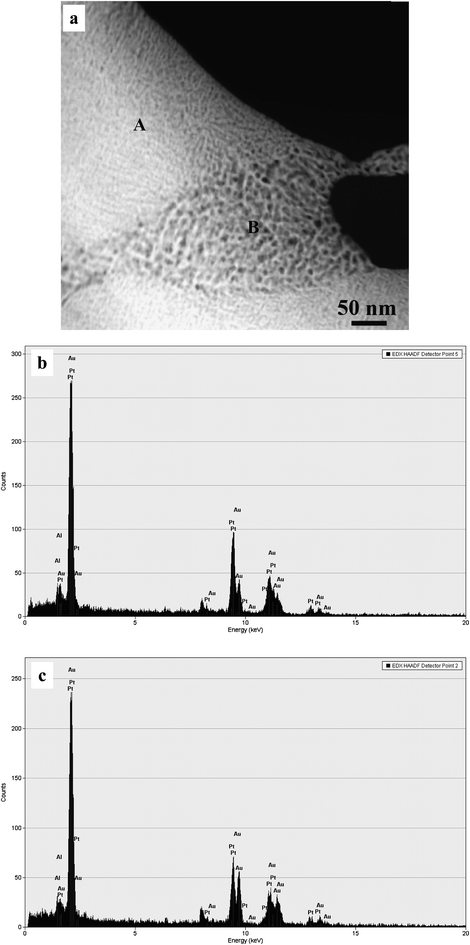 | ||
| Fig. 6 (a) HAADF-STEM image showing the microstructure of the np-Pt60Au40 NCs fabricated by dealloying the rapidly solidified Al75Pt15Au10 precursor in the 20 wt.% NaOH solution, and (b,c) NB-EDX spectra corresponding to zone A and B, respectively. | ||
The ligament size in the np-Pt60Au40 NCs is less than 5 nm, which is comparable to that (3–6 nm) of nanoporous Pt prepared by dealloying an Al–Pt alloy, but much smaller than that (10–20 nm) of NPG by dealloying the Al2Au alloy under similar corrosion conditions.39 It is known that surface diffusion of more noble elements along alloy/solution interfaces during dealloying plays a key role in the formation of nanoporous metals and has a significant influence on the length scale of ligaments/channels.22,24 Extrapolation to room temperature (298 K) from the literature data yields surface mass transfer diffusivities of Pt and Au of 3.6 × 10−22 and 2.2 × 10−19 cm2s−1 in vacuum.40 The surface diffusion of Pt is about three orders of magnitude slower than that of Au in the electrolyte. Snyder et al.41 have argued that as Pt possesses much slower surface diffusion rates than Au, Pt embedded in exposed terraces should segregate to the edges of the growing vacancy island step, stabilizing them, ultimately reducing the scale of porosity as well as leading to a Pt-rich shell. Similarly, the diffusion of Pd has a serious pinning effect on that of Ag during dealloying of Mg–Ag–Pd, which thus affects the porosity evolution/coarsening resulting in the formation of ultrafine ligaments/channels of as small as ∼5 nm in the nanoporous Ag80Pd20 alloy.34 It is obvious that the interaction between Pt and Au during dealloying of Al2(Au,Pt) leads to the formation of the ultrafine nanoporosity of the present np-Pt60Au40 alloy.
According to the classification for the dealloying of a biphasic alloy,42 the nanoporous composite structure can be obtained through dealloying the alloy with two proper phases. For example, nanoporous gold composites (NPGCs) can be fabricated through chemical dealloying of biphasic Al–Au alloys comprising Al2Au and AlAu intermetallic compounds. Both the Al2Au and AlAu phases can be fully dealloyed, and the dealloying of these two phases in the Al–Au alloys separately proceeds, which results in the formation of the NPGCs.43 Nanoporous Pd composites can be fabricated by dealloying an Al70Pd30 precursor alloy composed of Al3Pd and Al3Pd2, where the Al3Pd phase can be fully dealloyed forming the nanoporous palladium matrix with the undealloyed Al3Pd2 phase as embeddings.44 Here, it is astonishing that a nanocomposite structure forms during dealloying of the single-phase Al75Pt15Au10 precursor, accompanying the nanoporosity evolution. Moreover, the overall composition of the np-PtAu NCs is Pt60Au40, while the compositions of the finer and coarser zones are Pt70Au30 and Pt50Au50, respectively. Spinodal decomposition is a mechanism by which a solid solution of two or more components can separate into distinct regions (or phases) with distinctly different chemical compositions and physical properties.45,46 Erlebacher et al.22 has explained the nanoporosity evolution during dealloying of Ag–Au alloys based upon spinodal decomposition. They believed that regions of the surface with high supersaturation of gold adatoms sit within the spinodal. Within the spinodal, composition fluctuations of infinitesimal amplitude lead to a lower overall free energy for the system, and involve atomic diffusion against concentration gradients (uphill diffusion). But fluctuations of long length scale grow slowly due to the required diffusion times, and short length scale fluctuations create much energetically unfavourable incipient interface between the phases, inhibiting their growth. Hence, phase separation is manifested most rapidly at an intermediate length scale that roughly corresponds to the spacing between the observed gold-rich clusters. Because of the existence of the immiscibility gap in the Au–Pt phase diagram, the formation of the nanocomposite structure can be rationalized by the spinodal decomposition mechanism. During dealloying of the Al75Pt15Au10 precursor, the Pt60Au40 nanostructure is unstable and spinodally decomposes into thermodynamically metastable nanoporous zones with different compositions of Pt70Au30 and Pt50Au50. Conversely, the different compositions give rise to the formation of the finer and coarser zones with different ligament-channel sizes. The detailed formation mechanism of this unique nanocomposite structure will be further probed in the undergoing work. In addition, Erlebacher and Seshadri47 have argued that surface Ni is oxidized and not mobile during the synthesis of RANEY® Ni through the selective dissolution of AlNi alloys in highly basic conditions. RANEY® Ni is normally in a powder form and no visible nanoporous structure can be observed. In spite of similar alkaline solutions, the dealloying of the present Al75Pt15Au10 precursor is different from that of AlNi alloys. The more noble elements (Pt and Au) are not oxidized but aggregate to form the nanoporous structure through surface diffusion along the alloy/solution interface. Moreover, the as-dealloyed samples are monolithic and have a good mechanical integrity. Additionally, we have shown that the ligament/channel sizes of NPG can be adjusted through control over surface diffusion of Au adatoms during dealloying of Al2Au in the alkaline solution.48
In order to probe the influence of chloride ion on the nanoporosity evolution, dealloying was also performed in the 5 wt.% HCl solution. A nanoporous nanocomposite structure can also be obtained as shown in Fig. 7 and 8. Fig. 7 shows the SEM images of the np-Pt60Au40 NCs fabricated by dealloying the rapidly solidified Al75Pt15Au10 precursor in the 5 wt.% HCl solution. Both the low-magnification and high-magnification microstructures are similar to those of the np-Pt60Au40 alloy prepared in the NaOH solution (Fig. 3). The TEM observation verifies the nanoporous nanocomposite structure in the np-Pt60Au40 NCs, and two zones with different ligament-channel structures are marked by A′ and B′ in Fig. 8a. The corresponding SAED patterns confirm the single crystalline nature of these two zones (Fig. 8b and c). Similar to the patterns in Fig. 4b and c, the diffraction spots are also diffused together with spot pairs/arcs and weak diffraction rings. Moreover, the ligament-channel sizes of both zones are also close to those of the np-Pt60Au40 NCs prepared in the NaOH solution. The halide ions like Cl− are well known to enhance the surface diffusivity of the adatoms during dealloying, and have a significant coarsening effect on the nanoporous structure. Newman and Sieradzki49 have reported that the coarsening process of NPG is affected by the environment; in particular, adsorption of Cl− increases the diffusion rate. Non-doped Al2Au precursor can be dealloyed to form the nanoporous structure with the length scale of 10–20 nm in the 20 wt.% NaOH solution. However, the dealloying of Al2Au in the 5 wt.% HCl solution will lead to the formation of a coarse nanoporous structure with a length scale of 60–80 nm.39 The present results demonstrate that the chloride ion has no obvious influence on the nanoporous structure and size of the np-Pt60Au40 NCs. The high Pt content in the precursor and the low surface diffusivity of Pt may balance out the coarsening effect of chloride ion during dealloying. The exact reason will be pursued in future work.
 | ||
| Fig. 7 SEM images showing the microstructure of the np-Pt60Au40 NCs fabricated by dealloying the rapidly solidified Al75Pt15Au10 precursor in the 5 wt.% HCl solution. | ||
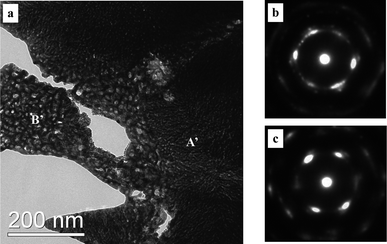 | ||
| Fig. 8 (a) TEM image showing the microstructure of the np-Pt60Au40 NCs fabricated by dealloying the rapidly solidified Al75Pt15Au10 precursor in the 5 wt.% HCl solution, and (b,c) SAED patterns corresponding to zone A′ and B′, respectively. | ||
The present nanoporous Au–Pt alloy NCs have a unique nanostructure with a high specific surface area, and the interconnected interstices and channels extending in all three dimensions allow unblocked transport of medium molecules and electrons, which is particularly desirable to catalysis.27 To evaluate their potential in important energy conversion applications, such as anode catalysts in fuel cells, it is necessary to investigate the electrocatalytic performance of the np-Pt60Au40 NCs. The sample prepared in the 20 wt.% NaOH solution was taken as an example. Fig. 9 shows the cyclic voltammograms (CVs) of the np-Pt60Au40 NCs and commercial JM-Pt/C catalyst in the 0.5 M H2SO4 solution. The voltammetric patterns observed for pure Au and Pt in sulfuric acid are well-established.50 In the forward scan, the peak (I) at the high potential is associated with the formation of Pt and Au oxides. In the back scan, the reduction of Au oxides (peak II) and Pt oxides (peak III) successively occur. Then, the adsorption of hydrogen and its evolution (peaks IV) in the negative potential region can be observed. Finally, hydrogen oxidation and desorption (peak V) take place at the low potential side in the positive scan. The peak positions for the reduction of Au and Pt oxides are comparable to those reported in the literature.15 In comparison, no reduction peak for Au oxides can be observed in the CV of the commercial JM-Pt/C catalyst. Moreover, the reduction peak for Pt oxides in the CV of the np-Pt60Au40 NCs shifts in the positive potential direction as compared to that of the commercial JM-Pt/C catalyst, as indicated by dotted lines in Fig. 9. This can be caused by the interaction (alloying effect14) between Pt and Au in the np-Pt60Au40 NCs. The electrochemical surface area (ECSA) can normally be calculated according to hydrogen adsorption/desorption (denoted as the under-potentially deposited hydrogen, Hupd).51 The ECSA values were evaluated to be 22.5 and 39.4 m2 g−1Pt for the np-Pt60Au40 NCs and JM-Pt/C, respectively. Compared with the commercial Pt/C catalysts, the lower ECSA of the Pt-based alloy nanostructures has also been reported in the literature.32,52 The smaller ECSA value of the np-Pt60Au40 NCs may be due to the alloy surface (that is, both Pt and Au atoms cover the surface of the ligaments, and also some Pt atoms are buried below the surface). In addition, it is known that Au does not show measurable hydrogen adsorption. Therefore, some researchers have argued that such electrochemical methods cannot be used to measure the true surface areas of PtAu alloys.14 Here, the current density in the following was normalized according to the unit mass of Pt rather than the ECSA. Moreover, the mass current density is also the preferred measure for practical applications.
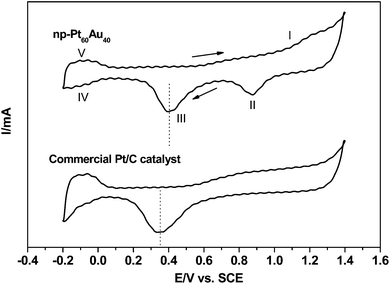 | ||
| Fig. 9 Cyclic voltammograms of the np-Pt60Au40 NCs (prepared in the 20 wt.% NaOH solution) and commercial JM-Pt/C catalyst in the 0.5 M H2SO4 solution. Scan rate: 50 mV s−1. | ||
Fig. 10 shows the electrocatalytic performance of the np-Pt60Au40 NCs and commercial JM-Pt/C catalyst for the methanol oxidation reaction (MOR) and formic acid oxidation (FAO) in the H2SO4 media. Fig. 10a presents the CV profiles for the MOR. As reported in previous literatures, the sharp rising current in the forward scan can be ascribed to the characteristic methanol oxidation on the electrode surface, forming adsorbed carbonaceous intermediates such as CO and HCO−, which will be oxidized at the higher potential due to the formation of Pt–OH and Pt–O species.27,53 In the back scan, the desorption of OHads or reduction of Pt oxides regenerates the active metallic Pt surface which allows further oxidation of methanol molecules at lower potentials. Therefore, the peak potential and current density for the forward anodic oxidation peak can be used to evaluate the catalytic activity of the electrocatalyst. As shown in Fig. 10a, the peak potential of the np-Pt60Au40 NCs for the forward anodic oxidation is 0.67 V (vs.SCE), which is comparable to that (0.66 V vs.SCE) of the commercial JM-Pt/C catalyst. Strikingly, for the np-Pt60Au40 NCs, the specific activity reaches 348.4 mA mg−1Pt, which is much higher than that (224.6 mA mg−1Pt) of the commercial JM-Pt/C catalyst.
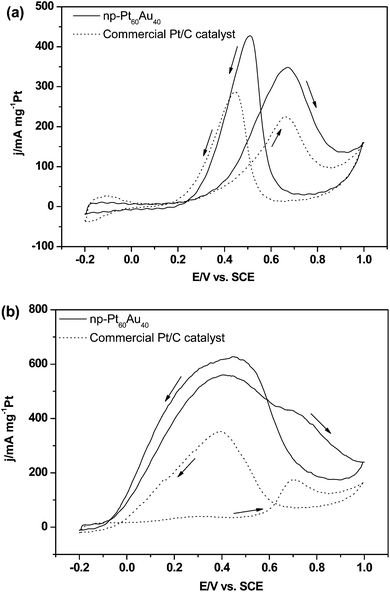 | ||
| Fig. 10 Cyclic voltammograms of the np-Pt60Au40 NCs (prepared in the 20 wt.% NaOH solution) and commercial JM-Pt/C catalyst in the solutions of (a) 0.5 M H2SO4 + 0.5 M CH3OH and (b) 0.5 M H2SO4 + 0.5 M HCOOH. Scan rate: 50 mV s−1. | ||
Fig. 10b shows the CV profiles for the FAO. For the commercial JM-Pt/C catalyst, the dehydrogenation occurs in the low potential range (0.1–0.45 V vs.SCE) in the forward scan, while the dehydration becomes dominant in the high potential range (0.6–0.9 V vs.SCE) resulting in a dramatic increase in the surface coverage by adsorbed CO intermediates. When the potential is above 0.7 V (vs.SCE), the adsorbed CO species begin to be oxidized. The observed high current density in the reverse scan is associated with direct formic acid oxidation after the removal of COads species, and can be used to measure the true activity of electrocatalyst because both COads and Pt oxides are stripped from the Pt surface.54,55 For the np-Pt60Au40 NCs, two peaks successively appear in the forward scan, but the dehydrogenation process is dominant. The current densities (560.8/428.8 mA mg−1Pt) for the dehydrogenation and dehydration processes are much higher than those (39.4/173.7 mA mg−1Pt) of the commercial JM-Pt/C catalyst. The ratio between these two peak current densities (I0.7 V/I0.4 V) in the positive scan decreases from 4.4 for Pt/C to 0.76 for Pt60Au40. This indicates that the dehydrogenation reaction is significantly enhanced for the np-Pt60Au40 NCs. In addition, the current density in the reverse scan is 628.0 mA mg−1Pt for Pt60Au40, which is almost double that (350.0 mA mg−1Pt) of Pt/C.
It is obvious that the np-Pt60Au40 NCs exhibit better catalytic performance towards the electro-oxidation of methanol and formic acid than the commercial JM-Pt/C catalyst. It is known that the catalytic reaction is a complicated process, and many factors will affect this process. According to the Randles-Sevcik equation,56 the observed peak currents strongly depend on mass transport, diffusion of reactants, their concentration/activity in the electrolyte and also scan rate. Despite the similar conditions (diffusion of reactants, concentration of reactants and scan rate), the influence of mass transport on the observed catalytic characteristics cannot be excluded due to different microstructures of the np-Pt60Au40 NCs and commercial JM-Pt/C. However, the nanoporous structure of np-Pt60Au40 and the continuous channels extending in three dimensions allow unblocked transport of the involved methanol and formic acid molecules. Here, we do not plan to probe the detailed mechanism of the catalytic reaction, and mainly focus upon the role of the Pt–Au bimetallic effect. The reasons for the better electrocatalytic performance of np-Pt60Au40 NCs can be rationalized as follows. Firstly, bimetallic alloy formation modifies the surface electronic properties resulting from both the strain effect due to the lattice mismatch and the electronic effect from the formation of the hetero-atom bond.57 Density function theory (DFT) calculations have shown that the d-band center shifts from 2.25 eV for Pt to around 1.80 eV for either PtAu alloys or Pt overlayers on Au.58 Recently, DFT results have suggested that the binding energies of CO should be reduced on the Au/Pt bimetallic surfaces. Moreover, temperature-programmed desorption (TPD) results reveal that CO binds more weakly to Au/Pt bimetallic surfaces than to the Pt surface, suggesting that the formation of Au/Pt bimetallic surface can reduce the extent of CO poisoning.1 Secondly, the higher electronegativity of Au than Pt can cause an increase of the amount of charge transferred from Pt to Au, and an increase in the d-orbital vacancy in Au–Pt.59 Thirdly, it is generally recognized that the direct oxidation of formic acid on Pt surface (dehydrogenation) does not require the presence of continuous neighboring Pt sites while the dissociative adsorption of formic acid to form COads requires at least two ensemble binding sites, which is the so-called ensemble effect.60 This ensemble effect has recently been confirmed by DFT calculations. For the FAO reaction on a Pt-based electrode, three neighboring Pt atoms are required for the dehydration path to occur, while the dehydrogenation path proceeds preferentially on isolated Pt atoms.61 For the present np-Pt60Au40 NCs, the surface Pt atoms can be effectively isolated by the neighboring Au atoms.27 Finally, the unique nanoporous nanocomposite structure should also be responsible for the superior electrocatalytic activity of the np-Pt60Au40 NCs.
In addition, it should be noted that the peak potential in the reverse scan of the np-Pt60Au40 NCs shows a little positive shift compared to that of the commercial JM-Pt/C (Fig. 10). It is known that the observed current density in the reverse scan is associated with direct methanol/formic acid oxidation due to the reduction of Pt oxides which regenerates the active metallic Pt surface. As for the np-Pt60Au40 NCs, the reduction of Au oxides is inevitable and will occur at a higher potential in the reverse scan in comparison to the reduction of Pt oxides (Fig. 9). The reduction of Au oxides will also recover some Pt active sites, catalyzing the oxidation of methanol or formic acid. Therefore, the peak potential positively shifts in the reverse scan of the present np-Pt60Au40 NCs.
Summary and conclusions
Nanoporous bimetallic PtAu alloy nanocomposites can be facilely fabricated by dealloying the rapidly solidified Al75Pt15Au10 alloy in the NaOH or HCl aqueous solution. The rapidly solidified Al75Pt15Au10 precursor is composed of the single-phase intermetallic compound with an Al2Au-type structure (Al2(Au,Pt)). The Al75Pt15Au10 precursor can be fully dealloyed, resulting in the formation of the np-PtAu alloy NCs with an fcc structure and an overall composition of Pt60Au40. The np-Pt60Au40 NCs consist of two zones with distinct ligament/channel sizes and compositions. The formation mechanism of these np-Pt60Au40 NCs can be rationalized in the light of surface diffusion of more noble elements and spinodal decomposition. Moreover, the chloride ion has no obvious influence on the dealloying process and the formation of the np-Pt60Au40 NCs. In comparison to the commercial JM-Pt/C catalyst, the np-Pt60Au40 NCs show superior catalytic activity towards the electro-oxidation of methanol and formic acid in the acid media. These PtAu NCs can find potential applications in direct methanol or formic acid fuel cells. This dealloying strategy can be extended to other ternary or multicomponent alloy systems, realizing the alloying of immiscible systems under mild conditions. Furthermore, nanoporous alloys with tunable structures and compositions can be fabricated by dealloying, based upon alloy design and control over surface diffusion of more noble elements.Acknowledgements
The authors gratefully acknowledge financial support by the National Natural Science Foundation of China under grant 50971079 and 50801031, Independent Innovation Foundation of Shandong University (2010JQ015), 2nd special support from China Postdoctoral Science Foundation (200902555), and 43rd China Postdoctoral Science Foundation. Z. H. Zhang acknowledges support from the Alexander von Humboldt Foundation (Germany). The experimental assistance from Ruhr University at Bochum (Germany) is acknowledged.References
- H. Ren, M. P. Humbert, C. A. Menning, J. G. Chen, Y. Shu, U. G. Singh and W.-C. Cheng, Appl. Catal., A, 2010, 375, 303 CrossRef CAS.
- L. Gao, L. Fan and J. Zhang, Langmuir, 2009, 25, 11844 CrossRef CAS.
- J. F. Huang, S. Vongehr, S. C. Tang, H. M. Lu, J. C. Shen and X. K. Meng, Langmuir, 2009, 25, 11890 CrossRef CAS.
- H.-L. Jiang, T. Umegaki, T. Akita, X.-B. Zhang, M. Haruta and Q. Xu, Chem.–Eur. J., 2010, 16, 3132 CrossRef CAS.
- B. Lim, H. Kobayashi, T. Yu, J. Wang, M. J. Kim, Z. Y. Li, M. Rycenga and Y. N. Xia, J. Am. Chem. Soc., 2010, 132, 2506 CrossRef CAS.
- M. Suzuki, M. Abe, T. Ueno, S. Abe, T. Goto, Y. Toda, T. Akita, Y. Yamadae and Y. Watanabe, Chem. Commun., 2009, 45, 4871 Search PubMed.
- X. Shen, X. Chen, J.-H. Liu and X.-J. Huang, J. Mater. Chem., 2009, 19, 7687 RSC.
- L. Deng, W. Hu, H. Deng and S. Xiao, J. Phys. Chem. C, 2010, 114, 11026 CrossRef CAS.
- J. Gong, T. Zhou, D. Song, L. Zhang and X. Hu, Anal. Chem., 2010, 82, 567 CrossRef CAS.
- H. Ataee-Esfahani, L. Wang and Y. Yamauchi, Chem. Commun., 2010, 46, 3684 RSC.
- H.-X. Ren, X.-J. Huang, J.-H. Kim, Y.-K. Choi and N. Gu, Talanta, 2009, 78, 1371 CrossRef CAS.
- S. Zhou, G. S. Jackson and B. Eichhorn, Adv. Funct. Mater., 2007, 17, 3099 CrossRef CAS.
- K.-J. Kim and H.-G. Ahn, Appl. Catal., B, 2009, 91, 308 CrossRef CAS.
- Z. Peng and H. Yang, Nano Res., 2009, 2, 406 CrossRef CAS.
- M. Mirdamadi-Esfahani, M. Mostafavi, B. Keita, L. Nadjo, P. Kooyman and H. Remita, Gold Bull., 2010, 43, 49 CAS.
- W. He, X. Wu, J. Liu, X. Hu, K. Zhang, S. Hou, W. Zhou and S. Xie, Chem. Mater., 2010, 22, 2988 CrossRef CAS.
- N. Toshima and T. Yonezawa, New J. Chem., 1998, 22, 1179 RSC.
- M. M. Mariscal, S. A. Dassie and E. P. M. Leiva, J. Chem. Phys., 2005, 123, 184505 CrossRef.
- P. Hernández-Fernández, S. Rojas, P. Ocón, J. L. Gómez de la Fuente, J. San Fabián, J. Sanza, M. A. Peña, F. J. García-García, P. Terreros and J. L. G. Fierro, J. Phys. Chem. C, 2007, 111, 2913 CrossRef CAS.
- J. Biener, A. Wittstock, L. A. Zepeda-Ruiz, M. M. Biener, V. Zielasek, D. Kramer, R. N. Viswanath, J. Weissmüller, M. Bäumer and A. V. Hamza, Nat. Mater., 2009, 8, 47 CrossRef CAS.
- A. Wittstock, V. Zielasek, J. Biener, C. M. Friend and M. Bäumer, Science, 2010, 327, 319 CrossRef CAS.
- J. Erlebacher, M. J. Aziz, A. Karma, N. Dimitrov and K. Sieradzki, Nature, 2001, 410, 450 CrossRef CAS.
- C. Xu, J. Su, X. Xu, P. Liu, H. Zhao, F. Tian and Y. Ding, J. Am. Chem. Soc., 2007, 129, 42 CrossRef CAS.
- A. J. Forty, Nature, 1979, 282, 597 CrossRef CAS.
- M. Raney, U.S. Patent No. 1563587, 1925.
- S. Koh and P. Strasser, J. Am. Chem. Soc., 2007, 129, 12624 CrossRef CAS.
- C. X. Xu, R. Y. Wang, M. W. Chen, Y. Zhang and Y. Ding, Phys. Chem. Chem. Phys., 2010, 12, 239 RSC.
- C. X. Xu, L. Wang, X. L. Mu and Y. Ding, Langmuir, 2010, 26, 7437 CrossRef CAS.
- Y. Ding, M. W. Chen and J. Erlebacher, J. Am. Chem. Soc., 2004, 126, 6876 CrossRef CAS.
- J. T. Zhang, H. Y. Ma, D. J. Zhang, P. P. Liu, F. Tian and Y. Ding, Phys. Chem. Chem. Phys., 2008, 10, 3250 RSC.
- R. Zeis, A. Mathur, G. Fritz, J. Lee and Jonah Erlebacher, J. Power Sources, 2007, 165, 65 CrossRef CAS.
- L. F. Liu, E. Pippel, R. Scholz and U. Gösele, Nano Lett., 2009, 9, 4352 CrossRef CAS.
- L. F. Liu, R. Scholz, E. Pippel and U. Gösele, J. Mater. Chem., 2010, 20, 5621 RSC.
- H. Ji, J. Frenzel, Z. Qi, X. G. Wang, C. C. Zhao, Z. H. Zhang and G. Eggeler, CrystEngComm, 2010, 12, 4059 RSC.
- T. B. Massalski, H. Okamoto, P. R. Subramanian and L. Kacprzak (Ed.), Binary alloy phase diagrams, ASM International, 1996 Search PubMed.
- J. Luo, M. M. Maye, V. Petkov, N. N. Kariuki, L. Wang, P. Njoki, D. Mott, Y. Lin and C. J. Zhong, Chem. Mater., 2005, 17, 3086 CrossRef CAS.
- A. J. Forty and P. Durkin, Philos. Mag. A, 1980, 42, 295 CrossRef CAS.
- Y. Ding, Y. J. Kim and J. Erlebacher, Adv. Mater., 2004, 16, 1897 CrossRef CAS.
- Z. H. Zhang, Y. Wang, Z. Qi, W. H. Zhang, J. Y. Qin and Jan Frenzel, J. Phys. Chem. C, 2009, 113, 12629 CrossRef CAS.
- E. G. Seebauer and C. E. Allen, Prog. Surf. Sci., 1995, 49, 265 CrossRef CAS.
- J. Snyder, P. Asanithi, A. B. Dalton and J. Erlebacher, Adv. Mater., 2008, 20, 4883 CrossRef CAS.
- Q. Zhang and Z. H. Zhang, Phys. Chem. Chem. Phys., 2010, 12, 1453 RSC.
- Z. H. Zhang, Y. Wang, Z. Qi, C. Somsen, X. G. Wang and C. C. Zhao, J. Mater. Chem., 2009, 19, 6042 RSC.
- X. G. Wang, W. M. Wang, Z. Qi, C. C. Zhao, H. Ji and Z. H. Zhang, J. Alloys Compd., 2010, 508, 463 CrossRef CAS.
- J. W. Cahn and J. E. Hilliard, J. Chem. Phys., 1958, 31, 688.
- J. E. Hilliard, in Solidification, American Society for Metals, Metals Park, Ohio, 1971, 497–560 Search PubMed.
- J. Erlebacher and R. Seshadri, MRS Bull., 2009, 34, 561 CAS.
- Z. H. Zhang, Y. Wang, Y. Z. Wang, X. G. Wang, Z. Qi, H. Ji and C. C. Zhao, Scripta Mater., 2010, 62.
- R. C. Newman and K. Sieradzki, Science, 1994, 263, 1708 CrossRef CAS.
- M. N. Desic, M. M. Popovic, M. D. Obradovic, L. M. Vracar and B. N. Grgur, J. Serb. Chem. Soc., 2005, 70, 231 CrossRef CAS.
- J. Luo, L. Y. Wang, D. Mott, P. N. Njoki, Y. Lin, T. He, Z. C. Xu, B. N. Wanjana, I. S. Lim and C. J. Zhong, Adv. Mater., 2008, 20, 4342 CrossRef CAS.
- C. C. Qiu, J. T. Zhang and H. Y. Ma, Solid State Sci., 2010, 12, 822 CrossRef.
- T. J. Schmidt, H. A. Gasteiger and B. J. Behm, Electrochem. Commun., 1999, 1, 1 CrossRef CAS.
- Y. X. Chen, M. Heinen, Z. Jusys and R. B. Behm, Angew. Chem., Int. Ed., 2006, 45, 981 CrossRef CAS.
- G. Q. Lu, A. Crown and A. Wieckowski, J. Phys. Chem. B, 1999, 103, 9700 CrossRef CAS.
- P. Zanello, in Inorganic Electrochemistry: Theory, Practice and Application, The Royal Society of Chemistry, 2003 Search PubMed.
- J. R. Kitchin, J. K. Norskov, M. A. Barteau and J. G. Chen, Phys. Rev. Lett., 2004, 93, 156801 CrossRef CAS.
- J. Greeley, J. K. Norskov and M. Mavrikakis, Annu. Rev. Phys. Chem., 2002, 53, 319 CrossRef CAS.
- S. Senthil Kumar and K. L. N. Phani, J. Power Sources, 2009, 187, 19 CrossRef CAS.
- S. Park, Y. Xie and M. J. Weaver, Langmuir, 2002, 18, 5792 CrossRef CAS.
- M. Neurock, M. Janik and A. Wieckowski, Faraday Discuss., 2009, 140, 363 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S2. See DOI: 10.1039/c0nr00830c |
| This journal is © The Royal Society of Chemistry 2011 |