Twist-boat conformation in graphene oxides†
Duminda K.
Samarakoon
and
Xiao-Qian
Wang
*
Department of Physics & Center for Functional Nanoscale Materials, Clark Atlanta University, Atlanta, Georgia 30314, USA. E-mail: xwang@cau.edu
First published on 3rd November 2010
Abstract
We have investigated the structural, electronic, and vibrational properties of graphene oxide based on first-principles density-functional calculations. A twist-boat conformation is identified as the energetically most favorable nonmetallic configuration for fully oxidized graphene. The calculated Raman G-band blue shift is in very good agreement with experimental observations. Our results provide important insight into structural and electronic characteristics that are useful for further development of graphene-based nanodevices.
Graphene, a single-layer planar sheet of carbon atoms with chicken wire structure, is being actively explored as a viable candidate for next-generation nanoscale electronics and photonics.1,2 As a chemically derived configuration, graphene oxide (GO) has recently attracted a great deal of attention due to its solubility in a diversity of solvents and promising potential for large-scale production of graphene-based devices.2–5 GO can be regarded as graphene with hydroxyl and epoxide functional groups decorating the basal plane and edges.6 The sp3 hybridized carbon-oxygen bonds in GO disrupt the extended sp2 conjugated network of graphene and induce the formation of an energy gap. The reduction of oxygen can convert the material to a semiconductor and eventually to a graphene-like semi-metal.2–5 A detailed understanding of the associated energy gap tuning6 and the shift in G-band Raman feature9 is thus of great importance for practical applications in electronic and photonic devices.
Functional groups of epoxide or hydroxyl on graphene can induce pronounced roughness in the oxide layers. Several first-principles studies have been performed for structural and electronic properties of GO.6–11 Despite the progress made, some questions remain regarding the characteristic structure of the oxidized phases,6,10 as well as identifying the mechanisms that promote the blue shifts observed in the experimental Raman G-band.7–9,12,13 This is, to a large extent, attributed to the composition variations and intrinsic inhomogeneities in the oxidization. Here we present results from first-principles density-functional calculations focusing on the structural, electronic, and vibrational properties of fully-oxidized GO layer. Specifically, we demonstrate that a twist-boat model is capable of accounting for the electronic structure and Raman blue shift characteristics of GO.
Our investigation of the optimal GO structure was built on the current body of knowledge from previous first-principles calculations6 encompassing the aggregation of functional groups in GO and various low-energy conformations of graphane—fully hydrogenated graphene.14,15 The aggregation is attributed to a dwindling of vertical structural distortion when functional groups are chemically bonded on both sides of the sheet,6 while the graphane can arrange in a randomly distributed regions consisting of a few low-energy membranes referred to as chair, boat, and twist-boat conformations.15,22 Specifically, oxygen atoms prefer “epoxy” bonded positions and form epoxide above the middle of a carbon-carbon bond. Hydroxyl groups, on the other hand, reside in the symmetric on-site position above the carbon. A dilute ensemble of epoxide groups on graphene tends to form spatially correlated patterns, which originates from the electron-mediated interaction among epoxides.16
We have employed first-principles density-functional approach as in our previous study of graphane.15 The structure and electronic properties of all conformations were investigated using first-principles density-functional calculations. Perdew–Burke–Ernzerhof (PBE) parametrization17 of the generalized gradient approximation (GGA) was used in majority of the calculations.18 A kinetic energy cutoff of 340 eV in the plane-wave basis and appropriate Monkhorst-Pack k-points (6 × 6 × 1 for graphene oxide and 10 × 10 × 1 for graphene) were sufficient to converge the grid integration of the charge density. Although the first-principles approach systematically underestimates the band gaps,19 we are interested primarily in the relative stability of the conformations and the validity of the structural models. Furthermore, we rectified the GGA results by employing Becke 3-parameter and Lee–Yang–Parr (B3LYP) hybrid functional.20,21 The initial search for stable structures was carried out through semi-empirical molecular dynamics by means of density-functional tight-binding (DFTB) method.26,27 The obtained local energy-minimum structures were further optimized through first-principles calculations with forces less than 0.01 eV/Å. For GGA structural calculations, Vanderbilt ultrasoft pseudopotentials were employed.19 The optimization of atomic positions proceeds until the change in energy is less than 1 × 10−6 eV per cell.
Given the preferred on-site and epoxy site adsorption for hydroxyl and epoxide functional groups, respectively, the fully-oxidized phases can be modeled through a graphene sheet randomly decorated with hydroxyl and epoxy groups.23–25 The geometry optimization of the decorated graphene sheet was performed using DFTB method with self-consistent charges.26
The fully-oxidized GO with randomly decorated hydroxyl and epoxy groups closely resembles the fully hydrogenated graphene in that there exist broad distribution of corrugated membranes that can be classified in accordance to the chair, boat, and twist-boat conformations of graphane (see Fig. 1).15 However, there exist crucial differences from graphane due to the existence of predominantly randomly-oriented epoxides along zigzag or armchair directions and a paucity of on-site hydroxyl groups forming a chair membrane. The boat conformation (Fig. 1a), recognized as the fully oxidized structure in previous calculations,6 is free from angle strain. In the boat conformation parallel bonds along the zigzag direction lift out alternatively from the graphene plane with two of six bonds being eclipsed, which have severe steric crowding on the “bow” and “stern” of the boat conformation.
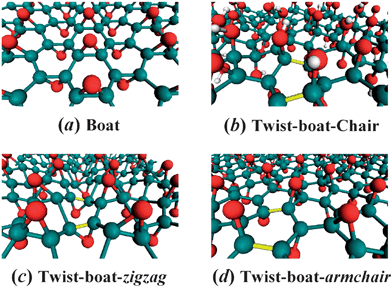 | ||
| Fig. 1 Prospective views of GO atomic structures for selected phases. (a) Fully-oxidized epoxide-only boat phase C2O with oxygen rows on both sides of the plane along the zigzag direction; (b) C8O2(OH)4 structure with hydroxyl-epoxide strips separated by twist-boat epoxide conformations; (c) and (d) Fully-oxidized epoxide-only twist-boat C2O phase along zigzag and armchair directions, respectively. The twisted bonds are indicated with yellow. Carbon, oxygen, and hydrogen atoms are colored with green, red, and white, respectively. | ||
In the presence of both hydroxyl and epoxide functional groups, the local epoxide configuration typically assumes a twist-boat form adjacent to chair membranes of hydroxyl groups (see Fig. 1b for a prototype twist-boat-chair structure).15 By twisting the boat structure, the steric hindrance can be partially relieved with completely eclipsed bonds. A twist-boat conformation of fully-oxidized GO (C/O = 2) can be constructed along either zigzag or armchair directions. The twist-boat-zigzag conformation (Fig. 1c) is locally stable but converts to boat under cell optimization. Careful examination of various structures indicates that the twist-boat along the armchair direction (Fig. 1d) is the lowest-energy conformation for fully-oxidized GO.
Summarized in Table 1 are the calculated binding energy, energy gap, and unit cell dimensions for boat and twist-boat conformations, along with those for low-energy epoxy-pair (EP) structures. EP chain conformations (Figure S1), although higher in energy than the twist-boat-armchair, are relevant to the oxidative breakup process.11 Closer scrutiny of the energy contributions from various conformations reveals that EP prefers to aggregate together such that the EP chains can break up into carbonyl pairs through “twisting boat” process, in conformity with the conclusion obtained from previous studies.11
| Structure | E PBE B (eV) | E B3LYP B (eV) | E g (eV) | a × b (Å2) |
|---|---|---|---|---|
| Boat | −23.39 | 23.29 | 2.95 | 2.52 × 4.49 |
| Twist-boat | −23.65 | 23.33 | 4.40 | 5.10 × 4.28 |
| EP-zigzag | −23.06 | 22.79 | 0.40 | 2.52 × 4.50 |
| EP-alt | −23.54 | 23.27 | 1.85 | 5.47 × 4.60 |
An important ramification of our simulation results is that the epoxide twist-boat membranes represent as a realistic model for describing the structure of GO. As can be seen from Table I (and Table S1), among various fully-oxidized structures,6 the effective periodicity of 2.55 Å × 4.28 Å for twist-boat conformation has the best agreement with experimentally observed local periodicity of 2.73 Å × 4.06 Å.28,29 In view of the experimentally observed fully-oxidized GO does not have well-ordered structures,23–25 we investigated the randomly distributed carbonyl and hydroxyl groups using DFTB approach (see Fig. 2). As is readily observable from Fig. 2, there exists a paucity of regular-ordered patterns, while local membranes have prominent corrugations with twisted bonds. This suggests that the corrugations built intrinsically in the twist-boat model capture the essential structural features of the randomly distributed membranes.22
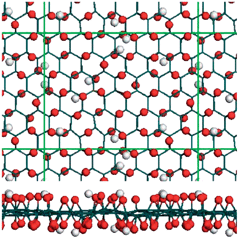 | ||
| Fig. 2 Top and side views of randomly distributed, fully oxidized GO conformation functionalized by carbonyl and hydroxyl using DFTB method. | ||
We illustrate in Fig. 3 the calculated band structures for boat and twist-boat conformations, along with the extracted charge density distribution of valence and conduction states, respectively. The extracted gap for optimized twist-boat structure is 4.4 eV while the boat counterpart has a gap of 3.0 eV. By contrast, all EP structures have much smaller gap, which is attributed to the coexistence of sp2 and sp3 configurations. As seen from insets of Fig. 3, the charge density follows the epoxide chains, which is more apparent for the conduction state of the twist-boat-armchair.
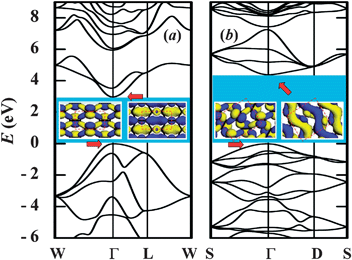 | ||
| Fig. 3 Calculated band structure for fully-oxidized epoxide-only C2O: (a) boat and (b) twist-boat along armchair direction, respectively. Insets: extracted charge density (isovalue of 0.05 au) at the band center (Γ point) for conduction band minimum (CBM) and valence band maximum (VBM), respectively. Dashed rectangles indicate unit cells (Table S2). | ||
It is worth mentioning that energy order for various GO conformations remains intact based on calculations using hybrid B3LYP functional20,21 (see Table 1). The energy gap for the twist-boat conformation is 6.42 eV using B3LYP as compared to 4.40 eV using GGA. Notwithstanding the corrections to the band gaps, the overall band structures are qualitatively similar. On the other hand, there exists specially-patterned GO EP-structures that are metallic and low in energy (see Table S1).30 Those structures are separated from the twist-boat conformation with a much larger barrier than from the simple EP-models as shown in Table 1. The low-energy metallic phase may be relevant to the experimentally observed flash reduction and patterning of GO.31
The stretching of the carbon-carbon bond in graphene gives rise to the G-band Raman feature at ∼1582 cm−1.7,9,32 Our calculation yields a dominant peak at 1589 cm−1 associated with the E2g mode, in very good agreement with experimental and other first-principles calculation results.7,9,12,13 For Raman spectrum calculations, the norm-conserving potentials and GGA were used. The calculation was based on the linear-response scheme.33,34
When the bond lengths and angles of graphene are modified by the oxidization, the hexagonal symmetry of graphene is broken. As a result, the G-band is highly sensitive to strain effects and can be used to probe the degree of oxidization.9 In the experimental work on the transformation from graphene to GO, there is a blue shift of the graphene G-band in GO, while the converse is observed after partial reduction of GO.9,12,13
Shown in Fig. 4 are the calculated Raman spectrum of the twist-boat-armchair conformation along with that for graphene. We also summarize in Table 2 the corresponding vibrational frequency at the band center (Γ point). The effects of epoxides give rise to one dominant peak in the G-band spectrum around 1650 cm−1, which is attributed to two vibrational modes at 1652 and 1662 cm−1 with Ag and B2g symmetry, respectively. As is readily observable in the insets of Fig. 4, contrary to in-plane motions of the Raman G-band for graphene, both Ag and B2g modes in the Raman G-band for GO engage out-of-plane motions as well. The in-phase and out-of-phase stretching among the epoxide bonds correlates with the differences between these Raman-active modes. The calculated G-band blue shift in GO (∼50–70 cm−1) is in very good agreement with experimental observations,9,12,13 supporting the validity of the twist-boat model for GO.
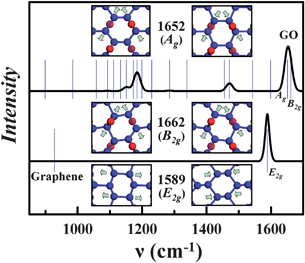 | ||
| Fig. 4 Calculated Raman spectrum for the twist-boat GO and graphene, respectively. The vibrational frequencies are displayed as blue bars (Table 2). Insets: vibrational motions of GO at 1652 and 1662 cm−1, and the vibrational motion for graphene at 1589 cm−1, respectively. | ||
In summary, we have demonstrated that the twist-boat model accounts for the structural and vibrational characteristics of GO, in good agreement with experimental observations. Our findings are useful for developing effective graphene manipulation means and for understanding the GO structure. We remark, before closing, that it is straightforward to employ the first-principles approach to partial reduction in GO, as the structural relaxation plays a vital role in “debundling” of oxygen from the twist-boat conformation. The investigation of the relevant electronic structures will provide an important tool for developing future graphene-based nanodevices.
Acknowledgements
This work was supported by the National Science Foundation (Grant DMR-0934142), Army Research Office (Grant W911NF-06-1-0442), and Air Force Office of Scientific Research (Grant FA9550-10-1-0254).References
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183 CrossRef CAS.
- T. Ohta, A. Bostwick, T. Seyller, K. Horn and E. Rotenberg, Science, 2006, 313, 951 CrossRef CAS.
- M. Y. Han, B. Ozyilmaz, Y. B. Zhang and P. Kim, Phys. Rev. Lett., 2007, 98, 206805 CrossRef.
- X. Li, X. Wang, L. Zhang, S. Lee and H. J. Dai, Science, 2008, 319, 1229 CrossRef CAS.
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282 CrossRef CAS.
- J.-A. Yan, L. Xian and M. Y. Chou, Phys. Rev. Lett., 2009, 103, 086802 CrossRef.
- J.-A. Yan, W. Y. Ruan and M. Y. Chou, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 125401 CrossRef.
- J. L. Li, K. N. Kudin, M. J. McAllister, R. K. Prudhomme, I. A. Aksay and R. Car, Phys. Rev. Lett., 2006, 96, 176101 CrossRef.
- K. N. Kudin, B. Ozbas, H. C. Schniepp, R. K. Prud'homme, I. A. Aksay and R. Car, Nano Lett., 2008, 8, 36 CrossRef CAS.
- D. W. Boukhvalov and M. I. Katsnelson, J. Am. Chem. Soc., 2008, 130, 10697 CrossRef CAS.
- Z. Y. Li, W. H. Zhang, Y. Luo, J. L. Yang and J. G. Hou, J. Am. Chem. Soc., 2009, 131, 6320 CrossRef CAS.
- L. Liu, S. Ryu, M. R. Tomasik, E. Stolyarova, N. Jung, M. S. Hybertsen, M. L. Steigerwald, L. E. Brus and G. W. Flynn, Nano Lett., 2008, 8, 1965 CrossRef CAS.
- D. C. Kim, D.-Y. Jeon, H.-J. Chung, Y. Woo, J. K. Shin and S. Seo, Nanotechnology, 2009, 20, 375703 CrossRef.
- D. C. Elias, R. R. Nair, T. M. G. Mohiuddin, S. V. Morozov, P. Blake, M. P. Halsall, A. C. Ferrari, D. W. Boukhvalov, M. I. Katsnelson, A. K. Geim and K. S. Novoselov, Science, 2009, 323, 610 CrossRef CAS.
- D. K. Samarakoon and Wang, ACS Nano, 2009, 3, 4017 CrossRef CAS.
- R. J. W. E. Lahaye, H. K. Jeong, C. Y. Park and Y. H. Lee, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 125435 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr., 2005, 220, 567 CrossRef CAS.
- M. C. Payne, M. P. Teter, D. C. Allan, T. A. Arias and J. D. Joannopoulos, Rev. Mod. Phys., 1992, 64, 1045 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785 CrossRef CAS.
- S. B. Legoas, P. A. S. Autreto, M. Z. S. Flores and D. S. Galvão, Nanotechnology, 2009, 20, 465704 CrossRef.
- N. R. Wilson, P. A. Pandey, R. Beanland, R. J. Young, I. A. Kinloch, L. Gong, Z. Liu, K. Suenaga, J. P. Rourke, S. J. York and J. Sloan, ACS Nano, 2009, 3, 2547 CrossRef CAS.
- D. W. Lee, L. De Los Santos V., J. W. Seo, L. Leon Felix, A. Bustamante D., J. M. Cole and C. H. W. Barnes, J. Phys. Chem. B, 2010, 114, 5723 CrossRef CAS.
- L. B. Casabianca, M. A. Shaibat, W. W. Cai, S. Park, R. Piner, R. S. Ruoff and Y. Ishii, J. Am. Chem. Soc., 2010, 132, 5672 CrossRef CAS.
- M. Elstner, D. Porezag, G. Jungnickel, J. Elsner, M. Haugk, T. Frauenheim, S. Suhai and G. Seifert, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 7260 CrossRef CAS.
- C. Z. Wang, K. M. Ho and C. T. Chan, Phys. Rev. Lett., 1993, 70, 611 CrossRef CAS.
- C. Gomez-Navarro, J. C. Meyer, R. S. Sundaram, A. Chuvilin, S. Kurasch, M. Burghard, K. Kern and U. Kaise, Nano Lett., 2010, 10, 1144 CrossRef CAS.
- D. Pandeya, R. Reifenberger and R. Piner, Surf. Sci., 2008, 602, 1607 CrossRef.
- H. J. Xiang, S.-H. Wei and X. G. Gong, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 035416 CrossRef.
- L. J. Cote, R. Cruz-Silvaand and J.-X. Huang, J. Am. Chem. Soc., 2009, 131, 11027 CrossRef CAS.
- M. S. Dresselhaus, A. Jorio, M. Hofmann, G. Dresselhaus and R. Saito, Nano Lett., 2010, 10, 751 CrossRef CAS.
- S. Baroni, S. de Gironcoli, A. dal Corso and P. Giannozzi, Rev. Mod. Phys., 2001, 73, 515 CrossRef CAS.
- D. Porezag and M. R. Pederson, Phys. Rev. B: Condens. Matter, 1996, 54, 7830 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The optimized atomic coordinates, binding energies, and vibrational modes. See DOI: 10.1039/c0nr00710b |
| This journal is © The Royal Society of Chemistry 2011 |