Highly dispersed ultrafine Pt and PtRu nanoparticles on graphene: formation mechanism and electrocatalytic activity†
C.
Nethravathi
a,
E. A.
Anumol
a,
M.
Rajamathi
b and
N.
Ravishankar
*a
aMaterials Research Centre, Indian Institute of Science, Bangalore, 560012, India. E-mail: nravi@mrc.iisc.ernet.in; Tel: +91-80-22922566; Fax: +91-80-22922566
bDepartment of Chemistry, St. Joseph's College, 36 Lalbagh Road, Bangalore, 560 027, India
First published on 11th November 2010
Abstract
We demonstrate a robust strategy for obtaining a high dispersion of ultrafine Pt and PtRu nanoparticles on graphene by exploiting the nucleation of a metal precursor phase on graphite oxide surfaces. Our method opens up new possibilities to engineer graphene-based hybrids for applications in multifunctional nanoscale devices.
Nanoscale heterostructures with engineered interfaces offer significant advantages in a variety of applications in nanoelectronics,1,2 solar cells3 and catalysis.4–6Graphene, owing to its interesting electronic properties, has been considered a promising material in field-effect devices,7 sensors8 and transparent electrodes.9 Hybridisation of graphene with metal or semiconductor nanoparticles has been demonstrated to be an effective strategy for photovoltaic devices,10lithium batteries,11catalysts12 and biosensors.13Graphene/Pt (G/Pt) and graphene/PtRu (G/PtRu) composites have been investigated as potential systems for anodic oxidation of methanol in PEM fuel cell applications. However, in most of these cases, the dispersed nanoparticles exhibit a wide size range, a non-uniform spatial distribution and the synthesis method does not provide significant control over the metal loading.
For the preparation of catalysts for fuel cell applications, graphite oxide is typically the starting material where the oxygen-bearing functional groups on the surface induce solubilisation of oxidised graphene sheets in solvents and also allow for the intercalation of molecules in the interlamellar space14 and the preparation of graphene-based hybrids.4,10,15–24 Although it has been suggested that these functional groups, whose number density on the surface can be varied, could potentially be used as anchors/nucleating centres for the controlled growth of nanoparticles,25 their exact role in controlling the nucleation and growth of the particles has not been clarified. Here, we demonstrate a robust strategy for obtaining a high dispersion of ultrafine Pt and PtRu nanoparticles on graphene by exploiting the nucleation of a metal precursor phase on graphite oxide (GO) surfaces. Detailed mechanistic investigations of the formation of such hybrids show that the oxygen-bearing functional groups on graphite oxide play a key role in the anchoring of the metal ions to the surface. A subsequent reduction step allows for the reduction of GO to graphene and the formation of a highly uniform dispersion of nanoparticles on the reduced GO surface. We show here that such composites of G/Pt and G/PtRu exhibit exceptional activity for methanol oxidation. Our method opens up new possibilities to engineer graphene-based hybrids for applications in multifunctional nanoscale devices.
The synthesis of the nanocomposites of graphene with Pt or PtRu was carried out in a toluene medium using the reaction scheme indicated in Scheme 1. While the powder XRD pattern from the precursor GO is characteristic of turbostratically disordered GO (Supporting Information, Fig. S1a†), the XRD pattern of the G/Pt nanocomposite (Supporting Information Fig. S1b†) shows broad peaks only due to Pt metal nanoparticles (JCPDS 87-0646) and a very broad, low intensity peak due to graphite indicating nearly complete exfoliation of graphite oxide sheets and their reduction in the composite. Similar features are observed in the case of the G/PtRu nanoparticle composite (Supporting Information Fig. S1c†). Bright-field TEM images of the G/Pt and G/PtRu composites in Fig. 1 show highly dispersed metal nanoparticles of 2–3 nm uniformly distributed on the graphene sheets in both cases. This observation is in contrast to control experiments on the formation of Pt nanoparticles on a graphite surface where particles of varied sizes largely exist as aggregates and are distributed randomly (Supporting Information Fig. S2†).
 | ||
| Scheme 1 Reaction scheme for the formation of G/Pt and G/PtRu nanocomposites. | ||
 | ||
| Fig. 1 Bright-field TEM images of (left) G/Pt and (right) G/PtRu nanocomposites. The chemical interaction between the Pt ions and the oxygen-containing functional groups provides anchoring sites where the nucleation of particles takes place. This interaction is also responsible for minimizing surface mobility leading to highly dispersed ultrafine particles on the reduced GO surface. | ||
To investigate the mechanistic details of the formation of the composite, we carried out detailed XPS investigations at various stages of the synthesis. Fig. 2a is the core-level Pt spectrum from the GO sample refluxed with the Pt salt in toluene medium. The core-level Pt 4f XPS spectrum (Fig. 2a and Table 1) indicates the presence of Pt4+ with a lower binding energy (74.57 and 77.89 eV) compared to Pt4+ in the starting H2PtCl6 (75.5 and 78.85 eV) indicating interaction of Pt with the oxygen groups on the graphene oxide surface. The significant shift in the binding energy (∼1 eV) indicates that there is a chemical interaction between the Pt ions and the oxygen-containing functional groups on the GO surface. This is further corroborated by the C 1s and the O 1s spectra (Supporting Information Fig. S3†) that also indicate shifts in the –C–OH, –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and –COOH sub-bands to higher binding energies compared to pristine GO and reduced GO indicating the formation of Pt–O linkages. We propose that the uniform dispersion of the Pt nanoparticles on the surface is due to the chemical interaction and the formation of these Pt–O linkages on the GO surface that provide heterogeneous nucleation sites on which the Pt or PtRu nanoparticles form by reduction. Control experiments on the formation of Pt nanoparticles on graphite surface (Supporting Information Fig. S2†) show a highly non-uniform distribution of larger Pt particles that show significant aggregation in many regions further confirming that the functional groups do play a significant role in the case of GO surfaces.
O and –COOH sub-bands to higher binding energies compared to pristine GO and reduced GO indicating the formation of Pt–O linkages. We propose that the uniform dispersion of the Pt nanoparticles on the surface is due to the chemical interaction and the formation of these Pt–O linkages on the GO surface that provide heterogeneous nucleation sites on which the Pt or PtRu nanoparticles form by reduction. Control experiments on the formation of Pt nanoparticles on graphite surface (Supporting Information Fig. S2†) show a highly non-uniform distribution of larger Pt particles that show significant aggregation in many regions further confirming that the functional groups do play a significant role in the case of GO surfaces.
| Sample | Binding Energies/eV | ||
|---|---|---|---|
| C 1s | O 1s | Pt 4f | |
| GO | 284.51 [–C–C] sp2 | 529.75 | |
| 286.07 [–C–OH] | 532.42 | ||
| 287.46 [–CO] |
534.18 | ||
| 289.14 [–COOH] | |||
| GO–Pt intermediate | 284.51 [–C–C] sp2 | 529.76 | 74.57 & 77.89 (Pt4+) |
| 285.76 [–C–C] sp3 | 532.79 | ||
| 286.83 [–C–OH/–C–N] | 534.45 | ||
| 288.29 [–CO] |
|||
| 289.18 [–COOH] | |||
| G/Pt | 284.51 [–C–C] sp2 | 530.03 | 70.91 & 74.31 (Pt0) |
| 285.99 [–C–OH] | 531.91 | 72.23 & 75.66 (Pt2+) | |
| 287.31 [–CO] |
533.14 | 73.62 & 76.92 (Pt2+) | |
| 288.81 [–COOH] | 535.04 | ||
 | ||
| Fig. 2 Core-level Pt 4f XPS spectra (a) before reduction and (b) after reduction with NaBH4. The formation of Pt4+ that strongly interacts with the GO surface via Pt–O linkages is evident here. For comparison, the Pt4+ position for H2PtCl6 is shown as a dotted line in (a). After reduction, two types of Pt2+ corresponding to Pt–O linkages to the reduced GO surface and free surface Pt–O can be seen as illustrated schematically in (c) and (d). | ||
On reduction using NaBH4, the –C–C– (sp2) component corresponding to graphene is restored largely (Fig. S3†) and the binding energy of –C–OH, –CO and –COOH and the corresponding O 1s components shift to lower values compared to GO. The Pt 4f core-level XPS spectrum (Fig. 2b) shows doublets corresponding to Pt0. In addition, two different types of Pt2+ that correspond to oxygen linkages of the Pt nanoparticles to the reduced GO surface and the formation of oxide layers on the surface of Pt are seen as indicated in the schematic in Fig. 2. The extremely small size of the nanoparticles can be explained on the basis of these linkages. While metals (including Pt) show significant mobility on graphene surfaces and hence tend to agglomerate, the anchoring of the metal on the defect sites significantly reduces the mobility leading to the retention of fine particles on the surface.26 This is also clear from the larger particle size and aggregation of the particles formed on a graphite surface (Supporting Information Fig. S2†).
The electrochemical surface area (ECSA) of the G/Pt and G/PtRu composites was estimated from the hydrogen adsorption/desorption peaks using cyclic voltammetry (Supporting Information Fig. S4†). The observed ECSA is enhanced significantly compared to the values reported in the literature owing to the highly dispersed nature of the ultrafine particles on the reduced GO surface.10,19–24 The electrocatalytic oxidation of methanol using G/Pt and G/PtRu was investigated using cyclic voltammetry and chronoamperometry. Fig. 3a and b show the cyclic voltammograms in 1M H2SO4 containing 2M CH3OH recorded at a sweep rate of 40 mV s−1. The oxidation potential, current and the tolerance (IF/IB) are listed in Table 2, which shows that the performance of these composites is much better than those reported thus far. In comparison to the G/Pt composite, the G/PtRu nanoparticle composite exhibits enhanced electrochemical activity in terms of lower oxidation potential, higher current and tolerance to CO poisoning.
| Electrode | Metal (%) | ECSA (m2 g−1 of Pt) | Forward sweep | Reverse sweep | I F/IB | ||
|---|---|---|---|---|---|---|---|
| I F/A g−1 | E/V | I B/A g−1 | E/V | ||||
| a The oxidation current is normalised to per gram of the G/Pt or G/PtRu nanocomposite. | |||||||
| G/Pt (after 5 cycles) | 40 | 66.5 | 17.4 | 0.7 | 7.25 | 0.5 | 2.4 |
| G/Pt (after 50 cycles) | 16.2 | 0.7 | 5.17 | 0.5 | 3.1 | ||
| G/PtRu (after 5 cycles) | 43 | 158 | 33.5 | 0.7 | 7.32 | 0.4 | 4.7 |
| G/PtRu (after 50 cycles) | 28.0 | 0.6 | 7.01 | 0.4 | 4.0 | ||
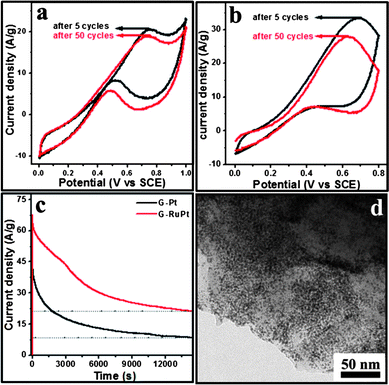 | ||
| Fig. 3 Cyclic voltammograms of the oxidation of methanol on (a) G/Pt and (b) G/PtRu catalysts showing excellent activity. (c) Chronoamperometry scans showing the long term stability of the catalysts. (d) Bright field TEM image of the G/PtRu catalyst after 4 h of catalyst use (steady state experiment) showing that there is no significant difference in the morphology and distribution. | ||
Long term oxidation of methanol was conducted at 0.74 V (G/Pt) and 0.65 V (G/PtRu) and the variation of current with time was recorded (Fig. 3c). The oxidation currents of 9 A g−1 (G/Pt) and 21.5 A g−1 (G/PtRu) at the end of 4 h of experiment time suggests a durable catalytic activity of the G/Pt and G/PtRu nanoparticle composites for the electro-oxidation of methanol. Fig. 3d is a bright field TEM image of the G/PtRu composite subjected to a steady state experiment for 4 h which shows that there is no significant change in the morphology after the electrochemical experiments.
The enhanced electrochemical performance observed in these two cases can be attributed to the presence of ultrafine Pt and PtRu nanoparticles with high ECSA and the uniform spatial distribution of metal particles on the high surface area graphene sheets. The enhanced anchoring of nanoparticles to the graphene sheets helps in maintaining the high active surface areas after several hours of catalyst use.
References
- W. Lu and C. M. Lieber, Nat. Mater., 2007, 6, 841–850 CrossRef CAS.
- P. Kundu, A. Halder, B. Viswanath, D. Kundu, G. Ramanath and N. Ravishankar, J. Am. Chem. Soc., 2009, 132, 20–21.
- P. V. Kamat, J. Phys. Chem. C, 2007, 111, 2834–2860 CrossRef CAS.
- A. K. Nicholas, D. Imre and H. F. Janos, Adv. Mater., 1996, 8, 637–641 CrossRef CAS.
- M. Valden, X. Lai and D. W. Goodman, Science, 1998, 281, 1647–1650 CrossRef CAS.
- A. Halder, S. Sharma, M. S. Hegde and N. Ravishankar, J. Phys. Chem. C, 2009, 113, 1466–1473 CrossRef CAS.
- I. Jung, D. A. Dikin, R. D. Piner and R. S. Ruoff, Nano Lett., 2008, 8, 4283–4287 CrossRef CAS.
- F. Schedin, A. K. Geim, S. V. Morozov, E. W. Hill, P. Blake, M. I. Katsnelson and K. S. Novoselov, Nat. Mater., 2007, 6, 652–655 CrossRef CAS.
- X. Wang, L. Zhi and K. Mullen, Nano Lett., 2007, 8, 323–327.
- S. Guo, S. Dong and E. Wang, ACS Nano, 2010, 4, 547–555 CrossRef CAS.
- Z.-S. Wu, W. Ren, L. Wen, L. Gao, J. Zhao, Z. Chen, G. Zhou, F. Li and H.-M. Cheng, ACS Nano, 2010, 4, 3187–3194 CrossRef CAS.
- G. M. Scheuermann, L. Rumi, P. Steurer, W. Bannwarth and R. Mulhaupt, J. Am. Chem. Soc., 2009, 131, 8262–8270 CrossRef CAS.
- S. Mao, G. Lu, K. Yu, Z. Bo and J. Chen, Adv. Mater., 2010, 22, 3521–3526 CrossRef CAS.
- R. C. Croft, Quartz. Rev., 1960, 14.1, 11–13 Search PubMed.
- T. Cassagneau, J. H. Fendler, S. A. Johnson and T. E. Mallouk, Adv. Mater., 2000, 12, 1363–1366 CrossRef CAS.
- K. Jasuja and V. Berry, ACS Nano, 2009, 3, 2358–2366 CrossRef CAS.
- C. Nethravathi, T. Nisha, N. Ravishankar, C. Shivakumara and M. Rajamathi, Carbon, 2009, 47, 2054–2059 CrossRef CAS.
- D. Wang, D. Choi, J. Li, Z. Yang, Z. Nie, R. Kou, D. Hu, C. Wang, L. V. Saraf, J. Zhang, I. A. Aksay and J. Liu, ACS Nano, 2009, 3, 907–914 CrossRef CAS.
- Y. Si and E. T. Samulski, Chem. Mater., 2008, 20, 6792–6797 CrossRef CAS.
- E. Yoo, T. Okata, T. Akita, M. Kohyama, J. Nakamura and I. Honma, Nano Lett., 2009, 9, 2255–2259 CrossRef CAS.
- B. Seger and P. V. Kamat, J. Phys. Chem. C, 2009, 113, 7990–7995 CrossRef CAS.
- Y. Li, L. Tang and J. Li, Electrochem. Commun., 2009, 11, 846–849 CrossRef CAS.
- L. Dong, R. R. S. Gari, Z. Li, M. M. Craig and S. Hou, Carbon, 2010, 48, 781–787 CrossRef CAS.
- Y. Li, W. Gao, L. Ci, C. Wang and P. M. Ajayan, Carbon, 2010, 48, 1124–1130 CrossRef CAS.
- H. Wang, J. T. Robinson, G. Diankov and H. Dai, J. Am. Chem. Soc., 2010, 132, 3270–3271 CrossRef CAS.
- J. A. Rodriguez-Manzo, O. Cretu and F. Banhart, ACS Nano, 2010, 4, 3422–3428 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, MD simulation details, XRD and size distribution. See DOI: 10.1039/c0nr00664e |
| This journal is © The Royal Society of Chemistry 2011 |