Preparation of functional magnetic nanocomposites and hybrid materials: recent progress and future directions
Silke
Behrens
*
Karlsruhe Institute of Technology, Institute for Technical Chemistry, Karlsruhe, Germany. E-mail: silke.behrens@kit.edu; Fax: +49 7247 822244; Tel: +49 7247 826512
First published on 17th December 2010
Abstract
The aim of this article is to provide an overview of current research activities on functional, magnetic nanocomposite materials. After a brief introduction to general strategies for the synthesis of superparamagnetic nanoparticles (NPs), different concepts and state-of-the-art solution chemical methods for their integration into various types of functional, magnetic nanocomposite materials will be reviewed. The focus is on functional materials which are based on discrete magnetic NPs, including multicomponent nanostructures, colloidal nanocrystals, matrix-dispersed composite materials and mesoscaled particles. The review further outlines the magnetic, structural, and surface properties of the materials with regard to application.
 Silke Behrens | Silke Behrens studied chemistry at Karlsruhe University, the Ecole Européenne de Chimie, Polymères, Matériaux (ECPM) de Strasbourg, and Emory University, Atlanta. She obtained her PhD on synthesis and crystal structures of cadmium selenide and telluride clusters in 1997. In 1998 she joined the Institute of Technical Chemistry of the Karlsruhe Institute of Technology where she is currently leading the nanochemistry group. Her research interests include the synthesis of inorganic protein hybrid materials, and the preparation, properties, and application of nanomaterials and nanocomposites. |
1 Introduction
Nearly all natural and synthetic materials are heterogeneous, i.e. they are microscopically built by different components or phases. In nanocomposite materials, one of the solid constituents traditionally exhibits a nanoscale structure, i.e. one-, two-, or three-dimensions of less than 100 nm. Nacre (mother of pearl), for example, is a natural nanocomposite with a layered structure, just composed of calcium carbonate layers and a small amount of organic biopolymers (proteins and chitin).1 It exhibits superior structural robustness, despite the brittle nature of its predominant ceramic constituent (CaCO3). Multifold-toughening mechanisms based on the interaction of the different components and their nanoscopic structure work together in nacre to achieve a thousand-fold increase in toughness. Although this high degree of structuring and complexity of natural materials has not yet been reached for synthetic systems, many research groups around the world have been working on the fabrication of nanocomposite materials where nanoscale inclusions are embedded in a matrix material in order to improve the material properties. The modification and improvement of materials, e.g., by incorporating NPs present already a field of application of great industrial importance.2 Applications range from hardening, ultraviolet stabilization to self-cleaning, antimicrobial, and fire-resistant surfaces.NPs, with large surface areas and quantum confinement effects, possess distinct size-dependent optical,3 electronic,4catalytic,5–7 and magnetic properties.8 In particular magnetic NPs offer innovative prospects in current nanotechnology. Magnetic NPs have been widely studied for various fields of application in biology and medicine, e.g., as novel imaging and therapeutic agents. Magnetic NPs may guide drugs to a site of interest in the body by an external magnetic field, thus acting as site-specific drug delivery vehicles.9 They have further been used as contrast agents for magnetic resonance imaging (MRI).10 Moreover, magnetic NPs can be made to heat up by an AC magnetic field, thus delivering cytotoxic amounts of thermal energy to malignant cancer cells in hyperthermic cancer treatment. They can also be very useful as magnetic carriers not only for the separation and purification of biochemical products (e.g., nucleic acids, proteins) but also for the magnetically driven recovery of catalysts.11 Magnetic NPs offer other exciting opportunities for many technical applications as well, e.g., for data storage and permanent magnetic nanocomposites. Magnetic media, e.g., made from self-organised magnetic NPs promise to have a data storage density of several terabits per square centimetre.12
In view of many applications, efforts have been made to disperse the magnetic NPs in a matrix, e.g., to prevent superparamagnetic NPs from aggregating into large ferromagnetic species. Matrix-dispersed NPs can be created in a variety of different states, e.g. dispersed in a continuous amorphous matrix, grafted on larger, mesocale particles, or well defined, three-dimensional superstructures of NPs. Nanocomposite materials not only provide the material with an improved stability of the nanoparticulate building blocks, but may further introduce new physical and biological properties and multifunctional behaviour. Recently, efforts have been made to combine different types of NPs, e.g., luminescent and magnetic NPs into bimodal nanocomposite materials, allowing the manipulation by an external magnetic field and real time optical visualization at the same time.13Fig. 1 displays various morphologies of magnetic hybrid nanocomposite materials. The field of magnetic nanocomposite materials, however, is still developing fast, and a clear classification of these materials remains difficult.
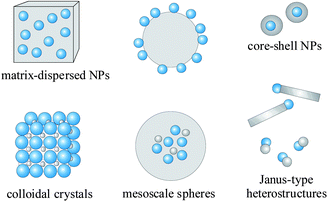 | ||
| Fig. 1 Typical morphologies of magnetic nanocomposite materials. Blue spheres represent magnetic NPs. Non-magnetic entities and matrix materials are displayed in grey colour. The nonmagnetic entity may provide the composite material with further functionalities and properties, providing multifunctional hybrid systems. | ||
The present review tries to give an overview on concepts and state-of-the-art solution chemical methods for various types of functional, magnetic nanocomposite materials. The focus is on functional materials which are based on discrete magnetic NPs, including multicomponent hybrid nanostructures and colloidal nanocrystals as well as matrix-dispersed composite materials and mesoscaled particles. Hybrid systems of magnetic nanoparticles (magnetic metal and iron oxide NPs) combined with organic and inorganic constituents (e.g., organic polymer and silica matrices, semiconductor and metal NPs) will be highlighted.
2 Synthesis of superparamagnetic NPs
Syntheses of magnetic NPs of various elemental compositions and phases have been reported in the literature, e.g., Fe3O4 and γ-Fe2O3,14,15 metallic Co, Fe, Ni,16,17 spinel-type ferrite MgFe2O4, MnFe2O4, CoFe2O4,18 and alloyed FePt, FeCo, CoPt3,19–21 or FeCoPt22 NPs.Recent advances in the synthesis of magnetic NPs using colloidal chemical approaches have been reviewed by several authors. A short overview will be given in the following section by some representative examples. For a more detailed description the reader is referred to current review articles on this subject.23–28 Besides the physical techniques, solution-phase syntheses offer a unique way for producing monodisperse NPs and include (1) coprecipitation methods, (2) thermal29 or sonochemical30decomposition of metal precursors (e.g., metal carbonyls, transition metal oleate complexes), (2) reduction of metal salts (e.g., by electrochemical reduction or by using triorganoborohydrides, NaBH4, H2, or alcohols as reducing agents), (3) solvothermal syntheses, and (4) microemulsion techniques. Different precursors, solvents, capping agents have been systematically investigated for producing monodisperse superparamagnetic NPs and various wet-chemical procedures have been developed that permit a good control over size, shape, composition, and internal structure of the resulting NPs.
A commonly used and simple procedure for magnetic NPs has been the coprecipitation of M2+ and Fe3+ ions by a base, usually NaOH or NH3OH in aqueous solution, yielding γ-Fe2O3 NPs or spinel-type ferrites (Fe3O4, MgFe2O4, MnFe2O4, CoFe2O4).31 Although this precipitation method is suitable for large-scale production of magnetic NPs, it requires the careful adjustment of the reaction parameters and often results in a rather broad size distribution of the NPs.
An alternative approach yielding highly monodisperse magnetic NPs has been the thermal decomposition of metal precursors, including metal carbonyls (Co2(CO)8, Fe(CO)5, Ni(CO)4) and metal oleates. Typically the metal precursors are injected into a hot organic solvent containing stabilizing ligands, e.g., oleic acid, oleylamine, or phosphines or are decomposed in the presence of aluminium trialkyls. Various magnetic metal NPs have been obtained by this method, e.g., using metal carbonyl precursors such as Co2(CO)816,29,32–35 and Fe(CO)5.17,36,37 Neutral, low-valent alkene or polyene metal complexes can be decomposed with H2 at room temperature, yielding magnetic metal NPs, e.g., Co38–40 or Ni.41 Magnetic metal nanocrystals (NCs) (e.g., Co, Fe, and Ni NCs) have superior magnetic moments than iron oxide particles. However, these NPs tend to be easily oxidized even under ambient conditions, which makes them often unsuitable for a direct biomedical or technical application. Thus, stabilization of the particles is an important issue. Magnetic metal particles have been stabilized by surface passivation with low doses of oxygen42,43 or by coating with an inert protective (e.g., carbon) shell.44 Presently, the thermolysis of metal–organic complexes (e.g., metal oleate complexes) seems to be the best procedure for the synthesis of iron oxide particles in terms of particle size control and narrow size distribution, yielding highly monodisperse Fe3O4 and γ-Fe2O3 NPs.14,15
Solution-phase reduction of metal salts in the presence of stabilizing agents has also been employed to prepare magnetic colloids. Co NCs, e.g., have been obtained by reduction of the metal salts using various reducing agents such as polyols (e.g., 1,2-dodecane diol,45ethylene gylcol,46etc.), and triorganoborohydrides (e.g., superhydride)47 or electrochemically using a sacrificial anode as metal source.48
Miroemulsion-based syntheses are typically performed by accomplishing the aforementioned chemical transformation reactions (e.g., co-precipitation or reduction) in microemulsions in order to control particle sizes, that is by subsequent mixing and coalescence of two separate micellar systems, each of them containing one reactant, or by thermally induced reaction inside a single micelle containing both reactants. Various magnetic metals and spinel ferrites have been synthesized in microemulsions.49,50
Magnetic alloys have many advantages, such as high magnetic anisotropy, large coercivities, and enhanced magnetic susceptibilities.51 Nanoalloyed particles such as FePt,52CoPt3,53FeCo,54,55 and SmCo556 are currently of great scientific interest in material science and chemistry. In general, nanoalloyed systems may be synthesized by the co-transformation and co-aggregation of different metal precursors, that is by simultaneous co-decomposition of the metal precursors or by simultaneously performing two different chemical transformation reactions. Alloyed FePt NPs, for example, have been prepared by combining the thermal decomposition of Fe(CO)5 with the polyol reduction of platinum acetylacetonate.57 For nanoalloy formation the reaction kinetics of the processes has to be well-controlled. Individual reaction rates of the present species must be similar; otherwise the formation of single cluster domains or core–shell morphology may occur.58 Often solution-phase syntheses yield the chemically disordered NPs and additionally thermal annealing is required for phase transformation into the desired compositionally ordered phases or superstructures.
Besides their size, structure and composition, the shape of the particles is an important parameter with impact on magnetic properties (e.g., coercitivities, magnetic anisotropy). During the last few years there has been a significant progress in the development of protocols for shape-controlled NC synthesis.59 Most of the available strategies are built upon manipulation of the growth process rather than nucleation by precise control of the reaction parameters, e.g., precursor concentration, reduction/decomposition rates, or use of specific capping agents. The synthesis of magnetic nanostructures exhibiting various shapes has been described, e.g., Co nanorods60 or nanoskeletons,61 and alloyed FePt,62 or FePd63nanostructures. However, a detailed understanding of the reaction mechanisms is still a challenge to be faced in the coming years.
3 Multicomponent magnetic NPs: core–shell type NPs and heterooligomers
The combination of two nanoscaled entities into a single hybrid particle has recently attracted much attention due to the numerous possibilities of application.64 Hybrid NPs may provide a platform with dual imaging capabilities for medical diagnosis (e.g., simultaneous magnetic and optical imaging), dual action combining magnetic imaging and therapy, and multiplexing in sensors. By this approach, the respective properties of the components may be combined and optimized independently. In addition, cooperatively enhanced performances due to collective interactions between the constituents have been achieved. Otherwise, however, the direct combination of the different entities may lead to undesired effects such as luminescence quenching by direct contact of magnetic NPs and quantum dots (QDs).To date, several morphologies of multicomponent, magnetic hybrid NPs have been reported, including core–shell and heterodimeric NPs.65 The general strategy for multicomponent nanostructures is to first prepare NPs of one material, and then use them as nucleation seeds to deposit the other material. This strategy has been well established for the synthesis of semiconductor QDs with epitaxial shells, while the controlled synthesis of uniform NPs that combine materials with different crystallographic structures, lattice dimensions, chemical stabilities and reactivities still faces many challenges. To date, a number of heterostructures has been synthesized by applying a seed-mediated approach.
Coating has been routinely applied for magnetic core stabilization and surface functionalization in view of biomedical and technical applications. One of the simplest methods for preparing core–shell type NPs has been the partial oxidation of magnetic metal NPs to form a shell of the native oxide on the particle surface. Polycrystalline Fe3O4 shells, e.g., which were generated by chemical oxidation on Fe particles, were shown to successfully protect and stabilize Fe NPs against full oxidation.66 For Co@CoO NPs, additionally to their stabilization,42,43 an exchange bias effect (a shift of the hysteresis loop along the field axis) was observed as a result of a strong interaction between the nanometre scale antiferromagnetic CoO layer and the ferromagnetic Co core. Bimagnetic core–shell systems such as FePt@Fe3O4 or FePt@CoFe2O4, where both core and shell are strongly magnetic (ferro- or ferrimagnetic), show effective exchange coupling phenomena and facilitate the fabrication of magnetic materials with tunable properties.67 The magnetic properties, e.g., magnetization and coercivity, can be readily controlled by tuning the chemical composition and the geometrical parameters of the core and the shell (Fig. 2).
 | ||
| Fig. 2 FePt@Fe3O4 NP assembly: (a) TEM image, (b) magnetization curve measured at 10 K (Fe3O4 shell thickness 1 nm), and (c) normalized coercivity hc as a function of the Fe3O4 volume fraction (ref. 67). | ||
Core–shell Co@MFe2O4 (M: Fe, Mn) NPs, which were prepared by decomposing Fe(acac)3 and accordingly Fe(acac)3/Mn(acac)2 in the presence of 10 nm sized Co seeds, revealed a greatly enhanced stability in aqueous solution.68 Core–shell type NPs of two metals with a different electrochemical potential have been synthesized by galvanic displacement at the surface of seed NPs. Co@Pt NPs, e.g., were synthesized by combining a redox transmetallation reaction of Pt hexafluoroacetylacetonate with presynthesized Co NPs.69 As the Pt grows around the Co core, Co metal is replaced by Pt. A variety of core–shell structures (e.g., Co@Au, Co@Pd, Co@Au) have been available accordingly.70
An impressive example for multicomponent hybrids is Au–Fe3O4 structure which is of interest for problems in molecular biology and medicine. The combination of magnetic and high-contrast X-ray materials is promising, e.g., for the design of dual agents allowing simultaneous MRI and X-ray detection. This system nicely shows how the morphology of hybrids can be modulated by tuning the parameters of synthesis. Shevchenko et al. have reported the synthesis of Au NPs encapsulated in a hollow, polycrystalline iron oxide shell by applying a three-step protocol (Fig. 3a): (1) synthesis of Au seeds, (2) deposition of a Fe shell around the Au core via the thermolysis of Fe(CO)5, and (3) air-oxidation of Fe to form hollow iron oxide shells by means of the Kirkendall effect.71 The growth of these structures was based on the random nucleation of multiple Fe seeds on the Au NPs. Encapsulation of multiple Au cores in a single iron oxide shell was achieved by adjusting Au particle–particle interactions with subsequent organic molecules. In other work, the morphology of Au–Fe3O4 hybrid particles was modulated by adjusting the solvent polarity: unpolar solvents (e.g., octadecene) yielded epitaxially grown, dumbbell-like heterostructures, whereas more polar solvents (e.g., diphenyl ether) yielded core–shell type structures (Fig. 3c).72 In the case of a close match of the lattice parameters, the growth of the second material at the surface of the seeds may be crystallographically controlled. The epitaxial growth of a Fe3O4 shell on Au seeds, e.g., was achieved by decomposing Fe(acac)3 on the surface of preformed Au NPs in high-boiling-point solvents in the presence of oleic acid and oleylamine (Fig. 3b).73 Depending on the choice of solvent, different types of morphologies were obtained: spherical core–shell NPs, spherical core–cubic shell NPs, peanut-like NPs, and dumbbell-like particles with two Fe3O4 NPs connected by a Au NP. Both components were single crystalline and the interfaces were coherent. The lattice spacings of the {111} lattice planes were 2.36 and 4.86 Å for Au and Fe3O4, respectively. As a result of the small lattice mismatch (∼3%) between 2d111(Au) and d111(Fe3O4), the Fe3O4 grows epitaxially on the Au NPs. Ternary heterostructures were manufactured when peanut-like Au–Fe3O4 hybrid particles were used as seeds for Fe3O4–Au–PbS or Fe3O4–Au–PbSe particles (Fig. 3d). The other way around, Au has been deposited on pre-synthesized Fe3O4 seeds to form Fe3O4@Au core–shell type nanocomposites.74
 | ||
| Fig. 3 Au@hollow, polycrystalline Fe3O4 NPs (a), epitaxial grown Au@Fe3O4 NPs (b), heterodimeric Au–Fe3O4 ensembles (c), and ternary Fe3O4–Au–PbS hybrids (d) ((a) ref. 71, (c) ref. 72, and (b and d) ref. 73). | ||
The interaction between Au and Fe3O4 was manifested both in the optical as well as in the magnetic properties of the NPs (Fig. 4). By coating the metal particles with a dielectric layer, the peak position of the surface plasmon resonance shifted from 520 nm (10 nm sized Au NPs) to 546 nm (partial Fe3O4 shell), 559 nm (2 nm shell), and 573 nm (3 nm shell). A red-shift of the surface plasmon resonance band from 520 nm (Au NPs) to 538 nm (Au–Fe3O4 heterodimers) was also observed for heterodimeric Au–Fe3O4 dumbbells.72 The authors suggested that the interface communication between Au and Fe3O4 resulted in a deficient electron population on Au, and, thus, in a shift of the plasmon absorption to longer wavelengths. In contrast to pure Fe3O4 NPs, the core–shell structure of the Au@Fe3O4 NPs induced a magnetic anisotropy, which was manifested by an increase of the saturation field and the coercivity as well as by a decrease of the remanence ratio. Dumbbell Au–Fe3O4 particles also showed a loop of slow increase in moment with the field up to 5 T due to spin canting in the Fe3O4 NPs at the interface.
 | ||
| Fig. 4 (a) Absorption spectra, (c) magnetization curve (10 K) of core–shell Au@Fe3O4 NPs (ref. 73); (b) absorption spectra, (d) magnetization curve (room temperature) of dumbbell Au–Fe3O4 NPs (ref. 72). | ||
Sun and coworkers just recently demonstrated the great potential of heterostructures for highly sensitive diagnostic and highly efficient therapeutic applications.75 They further heterofunctionalized the dumbell-like Au–Fe3O4 NPs with cisplatin at the Au NP site and with the Her2-specific monoclonal antibody Herceptin at the Fe3O4 NP site. These multifunctional nanocarriers were shown to deliver platin into Her2-positive breast cancer cells with a strong therapeutic effect.
When adsorbed at a liquid–liquid interface, colloidal particles may act in many ways like surfactant molecules, forming Pickering emulsions or “colloidosomes”.76,77 Xu and coworkers have used such o/w colloidosomes of Fe3O4, hollow iron oxide or FePt NPs to form heterodimeric structures by reacting the water-exposed surface of the NPs with Ag+ (Fig. 5).78,79
 | ||
| Fig. 5 Synthesis of heterodimeric Ag–hollow iron oxide nanostructures by using colloidosomes (ref. 79). | ||
In other work, lattice incompatibilties between FePt and CdS have been exploited to form CdS–FePt heterodumbbells. Initially an amorphous CdS layer was deposited on FePt NPs, followed by crystallization of CdS upon heating. The crystallization was accompanied by a dewetting process resulting in heterodimers of FePt and CdS NPs (Fig. 6).80
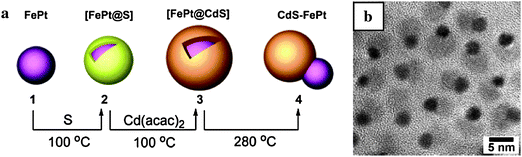 | ||
| Fig. 6 Synthesis of CdS–FePt dumbbells (ref. 80). | ||
Moreover, hybrid materials combining nanorods with NPs have been obtained by using a seed-growth approach. Wetz et al. have reported the synthesis of Au-tipped ferromagnetic Co nanorods. The selective choice of both, Au precusors and ligands, allowed to control the growth process, viz. heterogeneous seed-mediated growthversus galvanic displacement and location of the Au NPs (tip or whole body). Magnetic metals could also be grown anisotropically on the tip of semiconductor nanorods (Fig. 7a).81 Such semiconducting-magnetic CdSe–Co heterostructures remained fluorescent, however, their emission quantum yield was considerably lower than for CdSe nanorods. It was assumed that the quenching of photoluminescence was due to a new nonradiative pathway arising from the interaction of the CdSe with Co. The magnetic properties were similar to the pure Co NPs, with a large magnetization (163 A m2 kgCo−1 at 2 K) and effective magnetic anisotropy (5 × 105 J m−3). The topologically controlled growth of superparamagnetic NPs (such as Fe3O4 or Co) on preformed semiconductor oxide nanorods (viz.TiO2 nanorods) has been reported by Cozzoli and coworkers (Fig. 7b).82,83

Hybrid NPs composed of luminescent CdS or CdTe QDs and superparamagnetic iron oxide NPs were synthesized based on a ligand exchange mechanism.84 Initially, the QDs were functionalised and stabilized in aqueous solution by dual-functional ligands (2-mercaptopropionic acid, mercaptoacetic acid), bearing a thiol group binding to the QD surface together with a free carboxylate. Lauric acid-stabilized iron oxide NPs were then extracted from chloroform to aqueous phase by adsorption of the carboxylate-functionalised QDs. The resulting hybrid displayed the characteristics of its components: they revealed ferrofluidic behaviour (saturation magnetisation Ms 7.3–7.5 A m2 kg−1) and luminescence (quantum yield 17–59%, depending on the iron oxide to QD ratio). Fluorescent and magnetic nanocomposites (saturation magnetisation Ms 2.3 A m2 kg−1) were prepared from thiol-functionalised silica coated Fe3O4@SiO2–SH NPs and CdTe QDs.85 The antibody-functionalised Fe3O4/CdTe nanocomposites were successfully used to label and image HeLa cells.
4 Colloidal crystals
The assembly of small building blocks (e.g., atoms, molecules, and NPs) into ordered macroscopic superstructures has been an important issue in various areas of chemistry, biology, and material science. Self-assembly of NPs into two-dimensional and three-dimensional superlattices with a high degree of translational order has attracted a lot of attention since the early observation of iron oxide super crystals by Bentzon et al.86 More recently, Bergström and coworkers self-assembled super crystals of iron oxide nanocubes by a drying-mediated process, applying a magnetic field at the initial stage of the process. These super crystals did not only reveal a translational order but further an orientational order with a crystallographic alignment of the nanocubes.87The assembly of NPs of different materials into defined colloidal crystals or quasi crystals provides a general path to a large variety of composite materials (metamaterials) with new collective properties arising from the interaction of the different NCs in the assembly.88 O'Brien and co-workers have reported the formation of three-dimensionally ordered binary superlattices with a large structural diversity, by combining two sets of NCs, e.g., magnetic NCs with semiconductor QDs or metal particles.89 In a model system, PbSe semiconductor QDs and superparamagnetic γ-Fe2O3 NCs with independently tuneable optical and magnetic properties were co-assembled by slow solvent evaporation.90 The PbSe NCs displayed a size-dependent, near-infrared (NIR) absorption and emission, whereas the as-synthesized, superparamagnetic γ-Fe2O3 NCs revealed a weak absorption in the NIR at 1400 nm. It was shown that electrical charges on sterically stabilized NCs determine the stoichiometry of the superlattices together with entropic, van der Waals, steric and dipolar forces. The charge state of the NCs could be tuned by adding small amounts of ligands, e.g., carboxylic acids, TOPO, or dodecylamine. The addition of carboxylic acid to solutions of PbSe–Fe2O3 NC mixtures resulted in the growth of AB or AB2 superlattices, whereas the addition of TOPO to the same mixtures favoured growth of AB13 or AB5 structures (Fig. 8). The single domain regions of the AB2 and AB13 superlattices ranged from 0.16 to 2 µm2.
 | ||
| Fig. 8 TEM micrographs and sketches of AB13 superlattices of 11 nm γ-Fe2O3 and 6 nm PbSe NCs. (a) Cubic subunit of the AB13 unit cell. (b) AB13 unit cell built up of eight cubic subunits. (c) Projection of a {100}SL plane at high magnification. (d) As (c) but at a low magnification (inset: small-angle electron diffraction pattern). (e) Depiction of a {100} plane. (f) Projection of a {110}SL plane. (g) As (f) but at a high magnification. (h) Depiction of the projection of the {110} plane. (i) Small-angle electron diffraction pattern. (j) Wide-angle electron diffraction pattern of an AB13-superlattice (ref. 90). | ||
As there are a growing number of monodisperse NC systems available, the use of NCs with independently tuneable properties will enable the synthesis of divers materials with material responses which can be fine-tuned to magnetic, electrical, optical, and mechanical stimuli.
5 Matrix-dispersed magnetic NPs
Superparamagnetic NPs have further been incorporated or grafted on the surface of a non-magnetic matrix material, typically an organic polymer or SiO2. The choice between organic or inorganic polymers for magnetic composite materials is essentially determined by the intended application and the expertise of the experimentalists. The matrix material protects the superparamagnetic cores against aggregation into large ferromagnetic species and, to a certain degree, against oxidation. Other types of probes (e.g., fluorophores, catalysts, drugs) may be additionally incorporated into the matrix or grafted on the surface of mesoscale composite particles, thus providing multifunctional objects. In many cases the shell not only protects the magnetic core, but also prevents the direct contact of the magnetic particles with additional agents (e.g., fluorophores, QDs), and thus the problem of quenching may be avoided.91 Superparamagnetic NPs may further couple their physical properties to the matrix material, resulting in new functionalities, e.g. movement, change in shape or thermoresponsive behaviour.Moreover, many applications require magnetic particles with a large magnetization to achieve a quick response to an external magnetic field or a superior sensitivity for magnetic detection. Thus efforts have been made to produce mesoscale spheres of superparamagnetic NPs encapsulated in a non-magnetic matrix. Functional, mesoscale, magnetic particles have gained recently significant interest for diverse applications, including the magnetofiltration of catalysts,92 the purification of proteins, nucleic acids and cells and the adsorption of harmful environmental pollutants (e.g., heavy metal ions) from aqueous effluents.93,94 In view of these applications great experimental effort has been devoted to specifically tailor the morphological, magnetic, and textural properties of such mesoscale nanocomposites. The most important requirements for advanced application include a high and uniform magnetic loading of the beads, a narrow size distribution, homogeneity in shape, a high surface area, and a high sorption capacity. The preparation of mesoscale nanocomposite materials is often complex and involves multi-step syntheses. There are two general strategies for preparing composite particles with superparamagnetic properties: (1) direct encapsulation of the magnetic NPs within the non-magnetic matrix or (2) deposition of the magnetic NPs at the surface of previously synthesized nonmagnetic beads. The NPs can be either adsorbed from colloidal solution or generated in situ using appropriate metal precursors.
In the following, functional, magnetic nanocomposite materials, which are based on the most common, non-magnetic matrix materials (i.e., organic polymers and SiO2) will be highlighted.
5.1 Silica-based magnetic composites
SiO2 has been employed as a very common and promising material for coating and encapsulating magnetic NPs. There are several reasons for choosing SiO2 as coating or matrix material in magnetic composite materials. First of all, it reveals silanol surface groups which may easily be derivatized with a variety of functional groups, providing a convenient platform for further functionalization. Due to its optical transparency, fluorescing dyes or QDs may additionally be introduced. In this case, SiO2 may provide a barrier between fluorophore and magnetic core and prevents luminescence quenching. SiO2-based magnetic composites have been studied in view of various biomedical and environmental applications due to their biocompatibility, stability against degradation, and a hydrophilic character. In addition, there are procedures to manufacture mesoporous SiO2 materials with well-defined pores and large surface areas which have a great impact on catalytic and separation purposes. Magnetic SiO2 composite materials have been prepared by several procedures, including aerosol routes (e.g., aerosol pyrolysis, spray-drying), microemulsion polymerization, and sol–gel processes (e.g., the Stöber method).The Stöber method has been an effective technique for incorporating magnetic NPs into SiO2, by hydrolysis and condensation of silicon alkoxide precursors in alcohol/water mixtures.95 By this procedure, additional functionalities (e.g., QDs, fluorescing agents, catalysts) may directly be incorporated during SiO2 synthesis. Using a sol–gel approach, Philipse and coworkers were able to individually coat Fe3O4 NPs with SiO2. As a crucial step in their procedure, they had to lower the isoelectric point of the starting Fe3O4 ferrofluid from pH 7 to pH 3 by initially generating a thin SiO2 coating with Na2SiO3 in order to avoid flocculation of the ferrofluid during subsequent SiO2 growthvia Stöber process.96 In general, there are various sol–gel based synthetic procedures for SiO2 composite materials based on iron oxide NPs,97 however, the reports on materials based on magnetic metal NPs (e.g., Co, Fe, or Ni NPs) are rather scarce even though much better magnetic properties are expected. Table 1 shows the magnetic properties of some selected Co-based SiO2 spheres.
| Size/nm | d Co /nm | Co (wt%) | M s/A m2 kg−1 | |
|---|---|---|---|---|
| a Ferromagnetic Co particles. b the Ms increases to 180 A m2 kgCo-1 after annealing the sample at 500 °C. | ||||
| Co@SiO242 | 840 | 8.2 | 64.3 | 103, 160.9 (per kgCo) |
| Co@SiO298 | 520 | 9 | 24.4 | 21, 87.5 (per kgCo) |
| SiO2@Co 102 | 250 | 10 | 4.92 | 3.5,a 70 (per kgCo) |
| Tubular Co@SiO299 | 100–220 × 30 | 10–70 | 8.9 | 2.0, 22.3 (per kgCo) |
| Co@SiO2101 | 40–100 | 20–75 | — | 20–180 (per kgCo)b |
| Co@SiO2100 | 5 | 0.003 | — | <15 |
Co NPs, which were prepared by NaBH4 reduction of CoCl2 in the presence of citrate, were individually coated with SiO2 by hydrolysis/condensation of 3-aminopropytriethoxysilane and TEOS.101 In contrast to Fe3O4 NPs,96 a surface priming with NaSiO3 was not needed. Both the core size as well as the thickness of the SiO2 shell could be controlled through the parameters of synthesis (e.g., Co2+/citrate ratio, subsequent addition of ammonia and TEOS). The as-synthesized NPs were amorphous with a rather low saturation magnetization, which only increased after annealing and crystallization at high temperatures. Many applications require particles with a large magnetization and a high magnetic loading. Gorschinski et al. have prepared mesoscale Co@SiO2 particles with a saturation magnetization of up to 103 A m2 kg−1.42 The Co2(CO)8 precursor was initially thermolyzed in the presence of a bifunctional siloxane (e.g., 3-aminopropyltriethoxysilane), which served both as a ligand during NP synthesis as well as a coupling agent between the particle surface and the SiO2 coating. Eventually, TEOS was hydrolysed to form mesoscale Co@SiO2 particles (Fig. 9).
 | ||
| Fig. 9 Mesoscale magnetic SiO2 spheres (ref. 42). | ||
Alternatively, magnetic NPs have been adsorbed or synthesized in situ on mesoscale SiO2 particles, by using the SiO2 spheres as a template for NP deposition. For example, Co NPs were generated in situ on SiO2 microspheres (diameter 225–250 nm) via sonochemical decomposition of a [Co(CO)3NO] precursor.102Water-based FePt NPs could be deposited on SiO2 spheres through layer-by-layer assembly, by exploiting electrostatic interactions of polyelectrolytes.103 This approach allows good control over the shell composition and thickness, since it involves a sequential adsorption of preformed magnetic NPs and oppositely charged polymers on a SiO2 template. At first, a three-layer polyelectrolyte film was deposited on the negatively charged SiO2 surface providing a smooth, uniform, and positively charged surface. Finally, the negatively charged FePt NPs were attracted to the positively charged surface. The pH value was important: at pH > 11 the FePt NPs were surrounded by ammonium ions and no interaction occurred, whereas for pH < 10.5 aggregate formation was observed. This approach was further extended to Co NPs which were adsorbed on the modified SiO2 surface after their in situ generation with NaBH4 and citrate.104 In addition, positively charged SiO2 particles and negatively charged magnetic particles were combined and heterocoagulated through electrostatic interaction.105 After modification of the Fe3O4 NPs with a cationic silane coupling agent (N-trimethoxysilylpropyl-N,N,N-trimethylammonium chloride), the positive region of the magnetic NPs was broadened, and their isoelectric point shifted from pH 6 to 11. Almost 30% of the 8 nm sized magnetic NPs could be adsorbed onto negatively charged, 234 nm sized SiO2 particles. The magnetic content of the resulting composites ranged from 2.7 to 10% and the saturation magnetisation from 1.8 to 6.9 A m2 kg−1, respectively. Repetitive heterocoagulation leads to an increase in magnetic loading (41.3 wt% for quintuple heterocoagulation) and magnetic saturation (27.7 A m2 kg−1).106 In order to prepare fluorescent magnetic particles, the authors further introduced a thin layer of silica and an additional fluorescent polymer layer on the magnetic composites.
Since their discovery in 1992, ordered mesoporous materials have gained much attention.107,108 Recently, considerable efforts have been devoted to magnetic mesoporous composite materials in order to combine the extremely high surface area and magnetic properties, most of them focused on adsorption, drug delivery or catalytic applications. In principle, there are three basic approaches for integrating magnetic NPs into mesoporous materials: (1) growth of the mesoporous oxide on pre-synthesized, magnetic NPs (typically coated by a non-porous SiO2 layer), (2) the space-confined formation of the NPs inside the pores of the SiO2 matrix, and (3) the adsorption/binding of pre-synthesized magnetic NPs on/to the mesoporous oxide material. In (2) and (3) a blocking of the pores by the NPs has to be avoided.
Periodic mesoporous SiO2 matrices with well-defined pores, e.g., SBA or MCM-type matrices, have been exploited as hard templates for the in situ synthesis of magnetic NPs. In this case, the pores act as “dispersing agents”, increasing the average spacing between the NP nucleation sites and limiting NP growth during calcination. The two-solvent technique has been an effective approach to load the iron precursor inside the pores and to avoid the deposition of NP outside the SiO2 grains.109 In this approach, the dehydrated, mesoporous SiO2 is suspended in a hydrophobic solvent (e.g., hexane). The iron precursor is added subsequently in aqueous solution, with a quantity equal to the pore volume of the SiO2 matrix. Magnetic composites based on iron oxide and SBA-15 with large surface areas were prepared by this technique. An alternative procedure is based on incipient wetness impregnation. For example, mesoporous FePt–SiO2 composite materials were prepared by incipient wetness impregnation of SBA-15 with stoichiometric mixtures of Fe(acac)3 and Pt(acac)2 and H2 redution.110
In recent years, the synthesis of functional mesoporous magnetic microspheres with a defined size and narrow size distribution has attracted increased attention as promising materials for various applications. Fig. 11 displays a typical four-step procedure for the synthesis of mesoporous superparamagnetic microspheres consisting of: (1) synthesis of superparamagnetic NPs, (2) development of a dense, nonporous SiO2 layer, (3) templated growth of the porous SiO2 shell, and (3) template removal by calcination or solvent extraction. Etching of the magnetic cores in harsh media is typically prevented by introduction of an intermediate, nonporous SiO2 layer in step (2). Particles (500 nm) with magnetic core and an ordered, mesoporous SiO2 shell with perpendicular oriented accessible pores were obtained by such a four-step procedure using cetyltrimethylammonium bromide (CTAB) as mesopore template.111 The template was finally removed by extraction with acetone. The authors suggested that the perpendicular arrangement of the pores resulted from a preferred alignment of the rod-shaped siliceous/CTAB micelles on the carved surface of the Fe3O4@SiO2 microspheres, by decreasing the interface energy of the system. The obtained microspheres displayed a magnetization saturation of 53.3 A m2 kg−1, accessible 2.3 nm sized mesochannels, and a surface area of 365 m2 g−1. They could be further applied for an effective removal of toxic bacterial heptapeptides (microcystins) from aqueous solution. In a similar work, the authors introduced additional Au catalysts. Au NPs were bound to the initial, amino-functionalized Fe3O4@SiO2 particles before deposition of the mesoporous SiO2 (Fig. 10a).112 Due to its open mesochannels and good accessibility, the system was active for both the catalytic reduction of 4-nitrophenol and the epoxidation of styrene. The rate constant was dependent on the thickness of the mesoporous layer. Due to shorter diffusion distances and better mass diffusion, the rate constant was increased from 0.20 to 0.35 min−1 when decreasing the thickness of the mesoporous layer from 90 to 20 nm. In the production of bulk mesoporous materials such as MCM-41, the template is typically removed by calcination at high temperatures. For nano- and mesoparticulate systems, however, calcination at high temperatures may lead to the formation of large aggregates. Morales et al. reported the calcination of similar particles for template removal.113 Although aggregate formation was observed even at relatively low calcination temperatures (300 °C), the calcinated material revealed a very high specific surface area of 1023 m2 g−1. A similar procedure was used by Kim et al.: (1) hydrothermal synthesis of α-Fe2O3 particles, (2) coating with a dense SiO2 layer, (3) subsequent formation of a mesoporous SiO2 shell by the sol–gel reaction of tetraethoxysilane (TEOS) and n-octadecyltrimethoxysilane, (4) removal of the organic groups of n-octadecyltrimethoxysilane by calcination at 500 °C.114 The resulting mesoporous nanocomposite was loaded with Ni2+ ions and treated at 500 °C under H2/N2, thereby reducing hematite to magnetite, as well as producing Ni NPs on the surface. After oxidation in air, the NiO NPs were applied to magnetically separate His-tagged proteins from mixed-protein solution and Escherichia coli lysate.
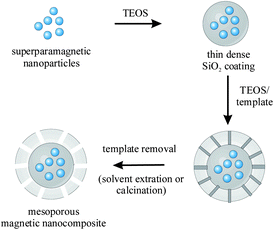 | ||
| Fig. 10 Schematic illustration of a typical four-step procedure for the synthesis of superparamagnetic mesoporous SiO2 spheres. | ||
Monodisperse mesoporous Fe3O4–SiO2 microspheres were also prepared by inducing urea-formaldehyde (UF) polymerization in Fe3O4 NPs/SiO2 sols.115 The UF polymer was removed by calcination at 300 °C, yielding 1.72 µm sized, superparamagnetic γ-Fe2O3–SiO2 composite particles with open-pore structure (pore size 6.62 nm, BET surface 240 m2 g−1). An additional thin, amorphous SiO2 layer was formed on the embedded γ-Fe2O3 NPs in order to avoid Fe leaching. The particles were used to extract genomic DNA from pea and green pepper samples. Julián-López et al. have demonstrated a one-pot synthesis of mesoporous γ-Fe2O3-organosilica microspheres via spray-dyring, using γ-Fe2O3 NPs, TEOS, a template (CTAB or Pluronic P123), and a volatile catalyst.116 These aerosol-generated particles were spherical with a size ranging over 100 to 500 nm and a large surface area (935 m2 g−1 for CTAB and 400 m2 g−1 for Pluronic P123) (Fig. 11d); the saturation magnetisation was 40 A m2 kg−1 of γ-Fe2O3 and a γ-Fe2O3 content of 5 wt%.
![TEM images displaying different types of magnetic mesoporous SiO2 particles: (a) Fe3O4@SiO2–Au@mSiO2 particles, (b) mesoporous SiO2 particles decorated with multiple Fe3O4 NPs, (c) uniform Fe3O4@mSiO2 particles with a single Fe3O4 core and (d) Fe3O4–mSiO2 prepared viaspray-drying [reprinted from (a) ref. 112, (b) ref. 118, (c) ref. 117, and (d) ref. 116].](/image/article/2011/NR/c0nr00634c/c0nr00634c-f11.gif) | ||
| Fig. 11 TEM images displaying different types of magnetic mesoporous SiO2 particles: (a) Fe3O4@SiO2–Au@mSiO2 particles, (b) mesoporous SiO2 particles decorated with multiple Fe3O4 NPs, (c) uniform Fe3O4@mSiO2 particles with a single Fe3O4 core and (d) Fe3O4–mSiO2 prepared viaspray-drying [reprinted from (a) ref. 112, (b) ref. 118, (c) ref. 117, and (d) ref. 116]. | ||
Non-aggregated, mesoporous SiO2 particles that possess a small enough size to allow long blood circulation (smaller than 100 nm) are of great interest for biomedical applications. Kim et al. reported the direct encapsulation of individual Fe3O4 NPs into mesoporous Fe3O4@mSiO2 particles with diameters of 45 to 105 nm (Fig. 11c).117 In their protocol, CTAB was not only used as a stabilizer for transfer of hydrophobic Fe3O4 NPs into the aqueous phase but also as a template for the formation of mesopores via sol–gel reaction. The template was removed by extraction in acidic ethanol solution at pH 1.4. The pH value during extraction was important since the Fe3O4 cores were fully removed when the pH decreased below pH 1, resulting in hollow mesoporous SiO2 spheres. The Fe3O4@mSiO2 particles were further modified with PEG to render them biocompatible by preventing non-specific adsorption of proteins. The Fe3O4@mSiO2 particles could be further loaded with fluorescing dyes (fluorescein isothiocyanate (FITC) and rhodamine B isothiocyanate (RITC)) and doxorubicin (DOX) and were tested for MR and fluorescence imaging.
Alternatively, 2-bromo-2-methylpropionic acid-modified Fe3O4 NPs were reacted with amine-functionalized, dye-doped mesoporous SiO2 spheres (Fig. 11b).118 The pores of the nanocomposite could be further loaded with the anti-cancer drug doxorubicin and thus served as a multimodal platform for optical imaging, MR contrast enhancement, and drug delivery. When other types of functional NPs (Au, CdSe/ZnS, or Pd) were coupled to the residual surface amino groups, multifunctional composite NP assemblies were obtained.119 In Table 2 the properties of some selected magnetic, mesoporous nanocomposite materials are summarized.
| Size/nm | M s/A m2 kg−1 | Magnetic loading (wt%) | Surface area/m2 g−1 | Properties | |
|---|---|---|---|---|---|
| a mSiO2: mesoporous silica. b Sample shows weak ferrimagnetic behaviour and low coercivity, size of magnetic core 500 nm. c M s may be increased to 42 A m2 kg−1 by changing the Fe3O4/SiO2 ratio during synthesis. d Prepared viaspray-drying. | |||||
| Fe3O4@SiO2@mSiO2111 | 500 | 53.3 | — | 365 | Sorption of microcystins |
| γ-Fe2O3@SiO2@mSiO2114 | 760 | 82.1b | Highb | 61 | NiO NPs for separation of His-tagged proteins |
| mSiO2@Fe3O4118 | 70 | — | 78 | 250 | Fluorescence, drug-delivery, optical/MR imaging |
| Fe3O4@SiO2–Au@mSiO2112 | ∼365 to 535 | 18.6 | 29 | 236 | Catalyst for reduction of 4-nitrophenol and epoxidation of styrene |
| mSiO2–Fe3O4@SiO2115 | 1720 | 17.22c | 24 | 240 | Separation of genomic DNA |
| mSiO2–Fe3O4d116 | 100–500 | ∼2 | 5 | 935 (CTAB) 400 (P123) | MRI, hyperthermia |
5.2 Functional magnetic polymers
Polymer coatings have been formed on magnetic NPs to simply change the surface properties of superparamagnetic NPs. The polymer then acts as a stabilizer or improves the biocompatibility of the NPs. However, magnetic NPs are also able to couple their physical properties with those of the polymer matrix. For example, magnetic NPs can be used to transfer forces applied by an external magnetic field to a surrounding polymer matrix, resulting in a change of shape or movement. This can be utilized for a variety of applications, such as actuators, switches, or magnetic separation. Moreover, magnetic NPs have been combined with polymer matrices which are sensitive to temperature changes induced by an AC magnetic field. Inductive heating of thermoresponsive polymers has been exploited for temperature-responsive flocculation of NPs, drug delivery, and shape transition applications. In addition, magnetic NPs have been aligned inside polymer matrices by use of a magnetic field, which influences strength and rigidity of the polymer. Moreover, well-defined, superparamagnetic particle agglomerates can be used to generate photonic crystals in a photocurable resin, with optical properties that are tuneable by an external magnetic field. Up to date, a plethora of functional composite materials has been reported in the literature, ranging from polymer-coated, magnetic NPs, mesoscaled magnetic spheres to polymer-dispersed magnetic NPs (e.g., ferrogels).120 A variety of superparamagnetic polymer microspheres is also currently available by various commercial suppliers. The uniform incorporation of large amounts of magnetic NPs into the polymer matrix is again one of the most crucial aspects to obtain a strong and uniform response of the composite material. In the following an outline of some emerging developments is given.Polymer-coated NPs can be produced directly by in situ precipitating or thermolysing a metal precursor in the presence of the polymer which then acts as a stabilizer. Polymeric surfactants can be designed with tunable composition viacopolymerization or post-functionalization techniques. Such polymeric surfactants must include a few general features, such as functional groups to anchor the particle surface (e.g., carboxylic acid, phosphine oxide, amine, or amide groups) located either along the polymeric backbone and/or at the end. A wide range of polymers has been used to date to synthesize and colloidally stabilize magnetic NPs via polymeric coatings.121–123 For example, polymeric surfactants with benzylamine and dioctylphosphine oxide end groups have been used to synthesize poly(styrene)-coated Co NPs by thermolysis of Co2(CO)8.124 These end-groups were used to mimic the small molecule ligand system developed by Alivisatos and coworkers.125 Moreover, the amphiphilic character of coblock polymers and their ability to form micelles have been exploited to template NP formation. Stable reverse micelles of a surfactant/polymer complex (poly(sodium 4-styrenesulfonate)/CTAB) have been used to generate amorphous Co NPs.126 The crystallinity of the NPs was subsequently enhanced through hydrothermal treatment at 165 °C. Thermo-responsive γ-Fe2O3@Au NPs have been prepared by using amphiphilic organic diblock copolymer chains (Fig. 12).127 The diblock copolymer chains included a thermally responsive poly(N-isopropylacrylamide) (pNIPAAm) block and an amine-containing poly(N,N-dimethylaminoethylacrylamide) (DMAEAm) block. An additional –C12H25hydrocarbon tail drived the formation of micelles. The micelles were loaded with Fe(CO)5, followed by subsequent thermolysis. The amine of the pDMAEAm block further served as electron donor for reducing AuCl4− to form a Au shell. Thermal aggregation of the particles above their lower critical solution temperature leads to dielectric coupling and to changes in the surface plasmon spectra. Further work, however, will be required for obtaining more homogeneous populations of particles.
 | ||
| Fig. 12 Schematic illustration of the synthesis of magnetic-core, Au-shell NPs with amphiphilic diblock copolymers (ref. 127). | ||
Alternatively, the surface functionalization of superparamagnetic NPs with polymeric surfactants may be achieved by replacing the ligands used in the initial NP synthesis. The NPs are synthesized separately and mixed with the polymer to enable ligand exchange by either physical or chemical adsorption of the polymer onto the magnetic material.
Surface initiated polymerization allows the controllable polymerization of end-tethered polymer coronas directly from the NP surface. A key requirement of this approach is the introduction of appropriate, surface attached initiators, while suppressing undesirable flocculation of the colloid. Thermoresponsive magnetic core–shell NPs were prepared by Schmidt et al.via surface initiated atom transfer radical polymerization (ATRP) of 2-methoxyethyl methacrylate (MEMA) from a colloidal Fe3O4 initiator.128ATRP is a transition metal catalyzed, controlled (“living”) radical polymerization mechanism, offering the advantage of good control on the molecular weight and polydispersity. In a high-frequency AC magnetic field the Fe3O4 particles heat up due to relaxation processes and magnetic induction. A thermosensitive dispersibility of the iron oxide polymer brush particles was observed in methanol with an upper critical solution temperature at 50 °C: above this temperature a stable colloidal solution was formed, whereas below this temperature the particles formed aggregates and precipitated. Via surface-initiated ATRP, iron oxide NPs were also coated by a copolymer shell of oligo(ethylene glycol) methylether methacrylate (OEGMA) and MEMA with adjustable thermoresponsive dispersion behaviour in water (Fig. 13).129 The phase transition temperature was tuneable by the copolymer composition and adjustable in a temperature range that is of interest for biomedical application. A poly(ε-caprolactone) shell was obtained via surface initiated ring-opening polymerization from a colloidal Co initiator.130
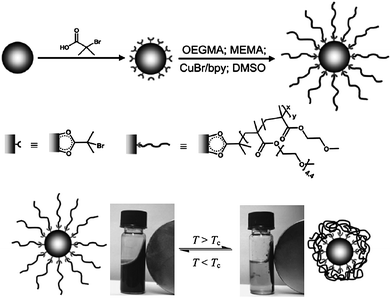 | ||
| Fig. 13 Synthesis of thermo-responsive magnetic iron oxide polymer brush particles via surface-initiated ATRP. Thermo-flocculation of the particles in water (ref. 129). | ||
For water-based thermo-responsive systems, poly(N-isopropylacrylamide) (PNIPAM) is often chosen due to its lower critical solution temperature of 32 °C, which is near to the body temperature and nearly insensitive to environmental conditions such as ionic strength and pH value. Brush-type PNIPAM-coated iron oxide NPs were prepared by Sun et al.131Methacryloxypropyltrimethoxysilane was covalently bound to the magnetic NPs in order to modify their surface with methacrylate double bonds, followed by a graft polymerization of NIPAM from the NP surface.
Magnetic NPs can also be efficiently encapsulated into small magnetic polymer particles via miniemulsion processes. The process of miniemulsification allows the generation of small, homogeneous, and stable droplets of 30–100 nm by high shear devices (e.g., ultrasound or high-pressure homogenizers). These droplets can be loaded with monomers and magnetic NPs and subsequently transferred by polymer reactions to the final latexes.132 Landfester and co-workers have applied a three-step preparation route including two miniemulsion processes to generate functionalized fluorescent and magnetic dual reporter poly(styrene-co-acrylic acid) particles.133 First, a dispersion of oleic acid-coated Fe3O4 NPs in octane was prepared. In a second step, NP aggregates were produced in a miniemulsion process by using sodium dodecylsulfate as surfactant. Finally, the miniemulsion of the NP aggregates was mixed with a styrene miniemulsion containing a fluorescent dye and polymerized. Hydrophilic acrylic acid was added and the polymerization continued. The particles could be further functionalized via surface carboxyl groups with transfection agents such as poly-L-lysine. Recently, Hu and Gao reported the synthesis of Janus type nanocomposite particles via ultrasonic emulsification.134Oleic acid-coated iron oxide NPs and an amphiphilic coblock polymer (poly(styrene-b-allyl alcohol)) in an organic solvent and polyvinyl alcohol in the aqueous phase were emulsified by sonication. A fluorescing dye (i.e., pyrene) was additionally added to label the polymer phase for optical detection. The organic phase was subsequently evaporated, which leads to the formation of uniform Janus type spheres, with diameter 120–220 nm depending on the experimental conditions. TEM images (Fig. 14) revealed the biphasic structure of the composite particles with the magnetic NPs assembled on one side and polymers on the other side, resulting from evaporation-induced phase separation of the two building blocks.
 | ||
| Fig. 14 Schematic illustration of the synthesis and TEM image of magneto-optical polymer composite particles with spatially separated functionalities (ref. 134). | ||
Matrix-dispersed composite materials of rather rigid polymer matrices filled with magnetic particles, viz. magnetic elastomers or magnetoelasts, have been known for many years. These materials are used as permanent magnets, magnetic cores, connecting and fixing elements in many areas. They display a low flexibility and do not change their size, shape, and elastic properties in the presence of an external magnetic field. More recently, a new generation of magnetic elastomers, consisting of mainly nanosized, superparamagnetic particles dispersed in a highly elastic polymer matrix, has attracted increasing interest in basic research as well as in certain applications.135 “Smart” ferrogels show unique magneto-elastic properties, i.e., they undergo a quickly controllable change in shape upon exposure to a magnetic field. These peculiar magnetoelastic properties may be used to create a wide range of motion and allow a smooth change in shape and movement.136 Hence, ferrogels are a promising class of materials for many applications, including actuators, switches, artificial muscles, and drug delivery systems. Ferrogels usually consist of a crosslinked polymer forming the gel matrix, and magnetic NPs dispersed in the matrix. Owing to interactions between the NPs and the polymer chains, the incorporated magnetic NPs connect the shape and physical properties of the gel to an external magnetic field. A ferrogel composed of crosslinked poly(N-tert-butylacrylamide-co-acrylamide) and Fe3O4 NPs, e.g., has been prepared by a two-step procedure.137 First, the hydrogel was synthesized by free-radical crosslinking copolymerization of the corresponding monomers, followed by subsequent co-precipitation of Fe2+ and Fe3+ in alkaline medium. A cylinder of the ferrogel was placed in a nonuniform magnetic field switching on and off, where the average magnetic field gradient was perpendicular to the axis of the ferrogel. Fig. 15 shows the reversible bending process of this ferrofluid cylinder due to the magnetic field.
 | ||
| Fig. 15 Bending process of a ferrogel cylinder due to a magnetic field (ref. 137). | ||
Chemical crosslinking reactions typically result in the formation of covalent polymer networks and, thus, in irreversible ferrogels, i.e. once the gel has been formed, the shape and mechanical properties are fixed. In contrary, ferrogels, which are obtained by physical crosslinking, are non-permanently crosslinked and represent reversible gel systems. Thermo-reversible organoferrogels, e.g., have been synthesized by Lattermann et al.138,139Triblock copolymers (i.e., poly(styrene-b-(ethylene-co-butylene)-b-styrene)) of different molecular weights were used to produce gels from ferrofluids based on Fe3O4 NPs in paraffin oil. The paraffin oil acted as solvent for the poly(ethylene butylene) middle block and precipitated the polystyrene end-blocks, which aggregated into micellar domains. The sol–gel transition was induced by crosslinking through microphase separation. Investigations by TEM indicated that the Fe3O4 NPs were preferably located as clusters in the free paraffin oil areas between the crosslinked micellar domains of the gelator. At high concentrations, chain-like aggregates of magnetic NPs were formed and a NP network was built up in addition to the gelator network, resulting in an increase of the gel–sol transition temperature at high Fe3O4 loadings. Hydroferrogels based on Pluronic F127 copolymer micelles have just recently been reported by Qin et al.140Pluronic is a biocompatible, ABA-type triblock copolymer consisting of one poly(propylene oxide) (PPO) block and two poly(ethylene oxide) (PEO) blocks. Each Pluronic micelle comprised a hydrophobic PPO core, in which the hydrophobic drug (i.e., indomethacin) or the oleic acid-covered iron oxide NPs were solubilised and a hydrophilic PEO corona, acting as the scaffold of the ferrogel (Fig. 16). Without magnetic field, the release profile of indomethacin was a quasi linear curve (t1/2 3195 min), whereas in the presence of a magnetic field the release of the drug was accelerated (t1/2 1500 min). The hydrophobic drug can be loaded at the sol state at low temperatures. When injected in the body, the sol rapidly gelates at the body temperature, then acting as a reservoir for the controlled release of hydrophobic drugs.
 | ||
| Fig. 16 Ordered microstructure of the Pluronic ferrogel (a) before applying the magnetic field and (b) when the magnetic field is on. (c) Release profiles of indomethacin (pH 7.4, 37 °C) with magnetic field (a) on and (b) off (ref. 140). | ||
Hydroferrogels were also obtained recently by using γ-Fe3O4 NP/triblock terpolymer hybrid micelles with a thermo-responsive corona which were reversibly able to build up a network structure by an open association at elevated temperatures.141
Magnetic particles have been further exploited to generate ordered colloidal crystals, viz. photonic crystals by assembly in an external magnetic field. In combination with photocurable resins, these colloidal crystals may be fixed and used to manufacture photonic crystal thin films with versatile optical properties (Fig. 17).142
 | ||
| Fig. 17 High-resolution multiple structural colour patterns based on magnetic colloids in a photocurable resin: (a) reflection micrographs with increasing magnetic field (ii–viii 130 G to 700 G) and corresponding transmission micrographs (scale bars 100 µm). (b) Multicoloured photonic crystal pattern and (c) photonic crystal film (ref. 142). | ||
In a photonic crystal a specific wavelength of light is blocked and therefore the corresponding colour displayed. Such physical structures display unique colours which are iridescent and metallic, and cannot be mimicked by chemical dyes or pigments. Because the band gap of a photonic crystal depends on the size of the colloid used, different sizes of colloids are needed to produce multicoloured structures. In principle, colloidal particles of magnetic materials may be directly used to construct colloidal photonic crystals. However, as magnetic particles are grown into larger domains a transition from superparamagnetic to ferromagnetic behaviour occurs which limits their dispersability due to strong magnetic interaction. Highly water dispersable colloidal nanocrystal clusters with uniform size (30 to 180 nm) were synthesized by using a high-temperature hydrolysis reaction of FeCl3 in diethyleneglycol with poly(acrylic acid) as surfactant.143 The as-synthesized colloidal nanocrystal clusters were composed of many 10 nm sized magnetite crystallites, thus retaining superparamagnetic properties at room temperature. However, a dramatic increase of the magnetic moment was observed with increasing size of the crystallite clusters, indicating that a single colloidal nanocrystal cluster has a much stronger response to an external magnetic field than a single nanodot. The strong interaction of the clusters with an external magnetic field which were balanced by repulsive electrostatic forces of the polyacrylate-coated surface were exploited for constructing colloidal photonic crystals with highly tuneable stop bands that could be moved across the entire spectral region. The reflection spectra of such colloidal clusters in aqueous solution could be tuned either by varying the external magnetic field or by cluster size.144 In this case, the optical response to the changes in external magnetic field was fully reversible. The magnetic clusters were then further combined with a solvation liquid (ethanol) and a photocurable resin. This three-phase material (called M-Ink) could be used to generate photonic crystals which were fixed by solidifying the photocurable resin through ultraviolet exposure. By this approach, multicoloured high resolution patterns could be obtained with a single material by sequential fine tuning and fixing of the structural colour.
Overall, by combining superparamagnetic NPs with polymers, a variety of functional composite materials has recently been obtained with properties that may be modulated by an external magnetic filed and a widespread potential for application.
6 Conclusion and outlook
In this review, the recent progress of designing novel types of functional magnetic nanocomposite materials has been highlighted. The relevant chemistry deals with the synthesis of the mainly nanosized, superparamagnetic nanoparticles and their introduction into a non-magnetic matrix. At present, a variety of protocols exist for the synthesis of superparamagnetic nanoparticles with a good control over size and, to a certain degree, over shape. Once synthesised, the nanoparticles may be incorporated into or grafted onto a nonmagnetic matrix (e.g., an organic polymer or SiO2) to generate composite materials with novel properties. The integrated magnetic particles are able to produce physical changes in the surrounding matrix material: for example, forces applied by an external magnetic field may be transferred to the surrounding matrix, resulting in a change of shape or movement. This is interesting for divers technical and biomedical applications (e.g., actuators, switches, drug delivery, and magnetic separation). By inductive heating with an AC magnetic field, temperature changes may be induced in the surrounding matrix, thus generating a thermo-responsive behaviour of the matrix material. Magnetic nanocomposites with thermo-responsive qualities promise new applications, including temperature-responsive flocculation of NPs, thermally induced, controlled release of entrapped drugs, and shape transition applications. Many applications require a large magnetization in order to achieve a quick response to the magnetic field and a large surface area. Therefore, mesoscaled composite particles which incorporate several superparamagnetic nanoparticles have gained recently significant interest with new perspectives in catalysis, in the separation of biomacromolecules and cells, and waste water treatment. In respect thereof, great experimental effort has been devoted to specifically tailor the morphological, magnetic, and textural properties of such particles. However, the controlled synthesis of magnetic composite particles with a high and uniform magnetic loading, a narrow size distribution, homogeneity in shape, and a maximum surface area and sorption capacity still remains challenging. Moreover, the design of multifunctional platforms, by combining several functional components into a single hybrid system, has been an emerging direction in the biomedical area. There has been a great demand for dual action probes, e.g.probes with dual imaging (magnetic/optical) or selective diagnostic/therapeutic properties. Multifunctional polymer or SiO2 composite beads, which include superparamagnetic nanoparticles for manipulation, optical probes (e.g., fluorescing dyes, quantum dots) for tracking, functional groups for selective targeting and bioconjugation, and entrapped drugs for therapy, provide new prospects for a simultaneous highly sensitive disease assessment and highly efficient therapeutic treatment. Recently, systems have been reported, where two or more functional components (e.g., Fe3O4 and Au) are directly integrated into a single hybrid nanostructure. Such heterodimer or core/shell type nanomaterials may not only combine the properties of their single consituents but also lead to cooperatively enhanced material properties. Heterodimeric nanostructures have been applied as selective nanocarriers, by further functionalization with drugs and antibodies.Overall, the possibilities offered by these types of materials will be substantially larger in the future. Future developments will open up exciting opportunities in various areas, such as medicine, catalysis and others. However, there always remains a high demand to refine the current existing protocols as well as for new synthetic approaches.
References
- X. Li, JOM, 2007, 59, 71 CrossRef CAS.
- H. Goesmann and C. Feldmann, Angew. Chem., Int. Ed., 2010, 49, 1362 CAS.
- A. Smith and S. Nie, Acc. Chem. Res., 2010, 43, 190 CrossRef CAS.
- U. Simon, Adv. Mater., 1998, 10, 1487 CrossRef CAS.
- N. Toshima, Y. Shiraishi, T. Teranishi, M. Miyake, T. Tominaga, H. Watanabe, W. Brijoux, H. Bönnemann and G. Schmid, Appl. Organomet. Chem., 2001, 15, 178 CrossRef CAS.
- I. I. Moiseev, T. A. Stromnova, M. N. Vargafik, S. T. Orlova, T. V. Chernysheva and I. P. Stolarov, Catal. Today, 1999, 51, 595 CrossRef CAS.
- S. Behrens and G. Spittel, Dalton Trans., 2005, 868 RSC.
- A. H. Lu, E. L. Salabas and F. Schüth, Angew. Chem., Int. Ed., 2007, 46, 1222 CrossRef CAS.
- Q. A. Pankhurst, J. Connolly, S. K. Jones and J. Dobson, J. Phys. D: Appl. Phys., 2003, 36, R167 CrossRef CAS.
- H. B. Na, I. C. Song and T. Hyeon, Adv. Mater., 2009, 21, 1.
- C. Wang, L. Yin, L. Zhang, L. Kang, X. Wang and R. Gao, J. Phys. Chem. C, 2009, 113, 4008 CrossRef CAS.
- G. Reiss and A. Hütten, Nat. Mater., 2005, 4, 725 CrossRef CAS.
- S. Corr, Y. Rakovitch and Y. Gun'ko, Nanoscale Res. Lett., 2008, 3, 87 CrossRef CAS.
- N. R. Jana, Y. Chen and X. Peng, Chem. Mater., 2004, 16, 3931 CrossRef CAS.
- J. Park, K. An, Y. Hwang, J. G. Park, H. J. Noh, J. Y. Kim, J. H. Park, N. M. Hwang and T. Hyeon, Nat. Mater., 2004, 3, 891 CrossRef CAS.
- V. F. Puntes, K. M. Krishnan and P. Alivisatos, Appl. Phys. Lett., 2001, 78, 2187 CrossRef CAS.
- S. J. Park, S. Kim, S. Lee, Z. Khim, K. Char and T. Hyeon, J. Am. Chem. Soc., 2000, 122, 8581 CrossRef CAS.
- S. Sun, H. Zeng, D. Robinson, S. Raoux, P. Rice, S. Wang and G. Li, J. Am. Chem. Soc., 2004, 126, 273 CrossRef CAS.
- A. Hütten, D. Sudfeld, I. Ennen, G. Reiss, K. Wojczykowski and P. Jutzi, J. Magn. Magn. Mater., 2005, 293, 93 CrossRef.
- S. Sun, C. B. Murray, D. Weller, L. Folks and A. Moser, Science, 2000, 87, 989.
- E. V. Shevchenko, D. V. Talapin, A. L. Rogach, A. Kornowski, M. Haase and H. Weller, J. Am. Chem. Soc., 2002, 124, 11480 CrossRef CAS.
- M. Chen and D. E. Nikles, Nano Lett., 2002, 2, 211 CrossRef CAS.
- B. Crushing, V. Kolesnichenko and C. O'Connor, Chem. Rev., 2004, 104, 3893 CrossRef CAS.
- T. Hyeon, Chem. Commun., 2003, 927 RSC.
- F. Dumestre, S. Martinez, D. Zitoun, M. C. Fromen, M. C. Casanove, P. Lecante, M. Respaud, A. Serres, R. E. Benfield, C. Amiens and B. Chaudret, Faraday Discuss., 2004, 125, 265 RSC.
- S. Sun, Adv. Mater., 2006, 18, 393 CrossRef CAS.
- U. Jeong, X. Teng, Y. Wang, H. Yang and Y. Xia, Adv. Mater., 2007, 19, 33 CrossRef CAS.
- C. B. Murray, C. R. Kagan and M. G. Bawendi, Annu. Rev. Mater. Sci., 2000, 30, 545 CrossRef CAS.
- H. Bönnemann, W. Brijoux, R. Brinkmann, N. Matoussevitch, N. Waldöfner, N. Palina and H. Modrow, Inorg. Chim. Acta, 2003, 350, 617 CrossRef CAS.
- D. De Caro, T. Ould Ely, A. Mari and B. Chaudret, Chem. Mater., 1996, 8, 1987 CrossRef CAS.
- R. Massart, IEEE Trans. Magn., 1981, 17, 1247 CrossRef.
- D. Dinega and M. G. Bawendi, Angew. Chem., Int. Ed., 1999, 38, 1788 CrossRef CAS.
- M. Giersig and M. Hilgendorff, J. Phys. D: Appl. Phys., 1999, 32, 111 CrossRef.
- U. Wiedwald, M. Spasova, E. L. Salabas, M. Ulmeanu and M. Farle, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 064424 CrossRef.
- Y. Ying, R. M. Rioux, C. K. Erdonmez, S. Hughes, G. Somorjai and P. Alivisatos, Science, 2004, 304, 711 CrossRef CAS.
- N. Burke, H. Stöver and F. Dawson, Chem. Mater., 2002, 14, 4752 CrossRef CAS.
- K. Butter, A. P. Philipse and G. J. Vroege, J. Magn. Magn. Mater., 2002, 252, 1 CrossRef CAS.
- J. Osuna, D. de Caro, C. Amiens and B. Chaudret, J. Phys. Chem., 1996, 100, 14571 CrossRef CAS.
- M. Respaud, J. M. Broto, H. Rakoto, A. R. Fert, L. Thomas, B. Barbara, M. Verelst, E. Snoeck, P. Lecante, A. Mosset, J. Osuna, T. Ould Ely, C. Amiens and B. Chaudret, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 2925 CrossRef CAS.
- F. Dumestre, S. Martinez, D. Zitoun, M. C. Fromen, M. C. Casanove, P. Lecante, M. Respaud, A. Serres, R. E. Benfield, C. Amiens and B. Chaudret, Faraday Discuss., 2004, 125, 265 RSC.
- T. Ould Ely, C. Amiens and B. Chaudret, Chem. Mater., 1999, 11, 526 CrossRef.
- A. Gorschinski, G. Khelashvili, D. Schild, W. Habicht, R. Brand, M. Ghafari, H. Bönnemann, E. Dinjus and S. Behrens, J. Mater. Chem., 2009, 19, 8829 RSC.
- S. Rudenkiy, M. Frerichs, F. Voigts, W. Maus-Friedrichs, V. Kempter, R. Brinkmann, N. Matoussevitch, W. Brijoux, H. Bönnemann, N. Palina and H. Modrow, Appl. Organomet. Chem., 2004, 18, 553 CrossRef CAS.
- R. N. Grass, E. Athanassiou and W. J. Stark, Angew. Chem., Int. Ed., 2007, 46, 4909 CrossRef CAS.
- C. B. Murray, S. Sun, W. Gaschler, H. Doyle, T. A. Betley and C. R. Kagan, IBM J. Res. Dev., 2001, 45, 47 CrossRef CAS.
- C. W. Kim, H. G. Cha, Y. H. Kim, A. P. Jadhav, E. S. Ji, D. I. Kang and Y. S. Kang, J. Phys. Chem. C, 2009, 113, 5081 CrossRef CAS.
- S. Sun and C. B. Murray, J. Appl. Phys., 1999, 85, 4325 CrossRef CAS.
- J. A. Becker, R. Schäfer, R. Festag, W. Ruland, J. H. Wendorff, J. Pebler, S. A. Quaiser, W. Helbig and M. T. Reetz, J. Chem. Phys., 1995, 103, 2520 CrossRef CAS.
- C. Petit, A. Taleb and M. P. Pileni, J. Phys. Chem. B, 1999, 103, 1805 CrossRef CAS.
- D. H. Chen and S. H. Wu, Chem. Mater., 2000, 12, 1354 CrossRef.
- D. Weller, A. Moser, L. Folks, M. Best, W. Lee, M. F. Toney, M. Schwickert, J. U. Thiele and M. F. Doerner, IEEE Trans. Magn., 2000, 36, 10 CrossRef CAS.
- S. Sun, Adv. Mater., 2006, 18, 393 CrossRef CAS.
- E. V. Shevchenko, D. V. Talapin, A. L. Rogach, A. Kornowski, M. Haase and H. Weller, J. Am. Chem. Soc., 2002, 124, 11480 CrossRef CAS.
- A. Hütten, D. Sudfeld, I. Ennen, G. Reiss, W. Hachmann, U. Heinzmann, K. Wojczykowski, P. Jutzi, W. Saikaly and G. Thomas, J. Biotechnol., 2004, 112, 47 CrossRef CAS.
- H. Bönnemann, R. A. Brand, W. Brijoux, H.-W. Hofstadt, M. Frerichs, V. Kempter, W. Maus-Friedrichs, N. Matoussevitch, K. S. Nagabhushana, F. Voigts and V. Caps, Appl. Organomet. Chem., 2005, 19, 790 CrossRef.
- Y. Hou, Z. Xu, S. Peng, C. Rong, J. P. Liu and S. Sun, Adv. Mater., 2007, 19, 3349 CrossRef CAS.
- S. Sun, C. B. Murray, D. Weller, L. Folks and A. Moser, Science, 2000, 287, 1989 CrossRef CAS.
- M. Zubris, R. B. King, H. Garmestani and R. Tannenbaum, J. Mater. Chem., 2005, 15, 1277 RSC.
- Y. Xia, Y. Xiong, B. Lim and S. Skrabalak, Angew. Chem., Int. Ed., 2008, 48, 60 CrossRef.
- F. Dumestre, B. Chaudret, C. Amiens, M. Respaud, P. Fejes, P. Renaud and P. Zurcher, Angew. Chem., Int. Ed., 2003, 42, 5213 CrossRef CAS.
- X. Wang, H. Fu, A. Peng, T. Zhai, Y. Ma, F. Yuan and J. Yao, Adv. Mater., 2009, 21, 1636 CrossRef CAS.
- C. Wang, Y. Hou, J. Kim and S. Sun, Angew. Chem., Int. Ed., 2007, 46, 6333 CrossRef CAS.
- D. Ung, L. Tung, G. Caruntu, D. Delaportas, I. Alexandrou, I. Prior and N. Thanh, CrystEngComm, 2009, 11, 1309 RSC.
- C. Wang, C. Xu, H. Zeng and S. Sun, Adv. Mater., 2009, 21, 3045 CrossRef CAS.
- L. Carbone and P. D. Cozzoli, Nano Today, 2010, 5, 449 CrossRef.
- S. Peng, C. Wang, J. Xie and S. Sun, J. Am. Chem. Soc., 2006, 128, 10677.
- H. Zeng, S. Sun, J. Li, Z. L. Wang and J. P. Liu, Appl. Phys. Lett., 2004, 85, 792 CrossRef CAS.
- S. Peng, J. Xie and S. Sun, J. Solid State Chem., 2008, 181, 1560 CrossRef CAS.
- J. Park and J. Cheon, J. Am. Chem. Soc., 2001, 123, 5743 CrossRef CAS.
- W. Lee, M. G. Kim, J. Choi, J. I. Park, S. J. Ko, S. J. Oh and J. Cheon, J. Am. Chem. Soc., 2005, 127, 16090 CrossRef CAS.
- E. V. Shevchenko, M. I. Bodnarchuk, M. V. Kovaleno, D. V. Talapin, R. K. Smith, S. Aloni, W. Heiss and A. P. Alivisatos, Adv. Mater., 2008, 20, 4323 CrossRef CAS.
- H. Yu, M. Chen, P. M. Rice, S. X. Wang, R. L. White and S. Sun, Nano Lett., 2005, 5, 379 CrossRef CAS.
- W. Shi, H. Zeng, Y. Sahoo, T. Y. Ohulchanskyy, Y. Ding, Z. L. Wang, M. Swihart and P. N. Prasad, Nano Lett., 2006, 6, 875 CrossRef CAS.
- L. Wang, H. Y. Park, S. I. Lim, M. Schadt, D. Mott, J. Luo, X. Wang and C. Zhong, J. Mater. Chem., 2008, 18, 2629 RSC.
- C. Xu, B. Wang and S. Sun, J. Am. Chem. Soc., 2009, 131, 4216 CrossRef CAS.
- B. P. Binks, Curr. Opin. Colloid Interface Sci., 2002, 7, 21 CrossRef CAS.
- Y. Lin, H. Skaff, T. Emrick, A. D. Dinsmore and T. P. Russell, Science, 2003, 299, 226 CrossRef CAS.
- H. Gu, Z. Yang, J. Gao, C. K. Chang and B. Xu, J. Am. Chem. Soc., 2005, 127, 34 CrossRef CAS.
- Y. Pan, J. Gao, B. Zhang and B. Xu, Langmuir, 2010, 26, 4184 CrossRef CAS.
- H. Gu, R. Zheng, X. Zhang and B. Xu, J. Am. Chem. Soc., 2004, 126, 5664 CrossRef CAS.
- J. Maynadié, A. Salant, A. Falqui, M. Respaud, E. Shaviv, U. Banin, K. Soulantica and B. Chaudret, Angew. Chem., Int. Ed., 2009, 48, 1814 CrossRef CAS.
- M. Casavola, V. Grillo, E. Carlino, C. Giannini, F. Gozzo, E. F. Pinel, M. A. Garcia, L. Manna, R. Cingolani and P. D. Cozzoli, Nano Lett., 2007, 7, 1386 CrossRef CAS.
- R. Buonsanti, V. Grillo, E. Carlino, C. Giannini, F. Gozzo, M. Garcia-Hernandez, M. A. Garcia, R. Cingolani and P. D. Cozzoli, J. Am. Chem. Soc., 2010, 132, 2437 CrossRef CAS.
- R. Kas, E. Sevinc, U. Topal and H. Y. Acar, J. Phys. Chem. C, 2010, 114, 7758 CrossRef CAS.
- P. Sun, H. Zhang, C. Liu, J. Fang, M. Wang, J. Chen, J. Zhang, C. Mao and S. Xu, Langmuir, 2010, 26, 1278 CrossRef CAS.
- M. D. Bentzon, J. van Wonterghem, S. Mørup and A. Thölén, Philos. Mag. B, 1989, 60, 169 CAS.
- A. Ahniyaz, Y. Sakamoto and L. Bergström, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 17570 CrossRef CAS.
- D. V. Talapin, J. S. Lee, M. V. Kovalenko and E. Shevchenko, Chem. Rev., 2010, 110, 389 CrossRef CAS.
- E. V. Shevchenko, D. Talapin, N. A. Kotov, S. O'Brien and C. B. Murray, Nature, 2006, 439, 55 CrossRef.
- F. X. Redl, K. S. Cho, C. B. Murray and S. O. O'Brien, Nature, 2003, 423, 968 CrossRef CAS.
- S. A. Corr, Y. P. Rakovitch and Y. K. Gun'ko, Nanoscale Res. Lett., 2008, 3, 87 CrossRef CAS.
- S. Shylesh, V. Schünemann and W. Thiel, Angew. Chem., Int. Ed., 2010, 49, 3428 CAS.
- C. Wang, S. Tao, W. Wie, C. Meng, F. Liu and M. Han, J. Mater. Chem., 2010, 20, 4635 RSC.
- M. A. Morales, A. J. S. Mascarenhas, A. M. S. Gomes, C. A. P. Leite, H. M. C. Andrade, C. M. C. de Castilho and F. Galembeck, J. Colloid Interface Sci., 2010, 342, 269 CrossRef CAS.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62 CrossRef.
- A. P. Philipse, M. P. B. van Bruggen and C. Pathmamanoharan, Langmuir, 1994, 10, 92 CrossRef CAS.
- A. L. Morel, S. I. Nikitenko, K. Gionnet, A. Wattiaux, J. Lai-Kee-Him, C. Labrugere, B. Chevalier, G. Deleris, C. Petibois, A. Brisson and M. Simonoff, ACS Nano, 2008, 2, 847 CrossRef CAS.
- N. Matoussevitch, A. Gorschinski, W. Habicht, J. Bolle, E. Dinjus, H. Bönnemann and S. Behrens, J. Magn. Magn. Mater., 2007, 311, 92 CrossRef CAS.
- L. Ren, L. He, C. Chen, M. Wark, C. Li, P. Che and L. Guo, J. Magn. Magn. Mater., 2007, 312, 405 CrossRef CAS.
- T. Haeiwa, K. Segawa and K. Konishi, J. Magn. Magn. Mater., 2007, 310, e809 CrossRef CAS.
- Y. Kobayashi, M. Horie, M. Konno, B. Rodríguez-González and L. M. Liz-Marzán, J. Phys. Chem. B, 2003, 107, 7420 CrossRef CAS.
- S. Ramesh, Y. Cohen, R. Prozorov, K. Shafi, D. Aurbach and A. Gedanken, J. Phys. Chem. B, 1998, 102, 10234 CrossRef CAS.
- V. Salgueiriño-Maceira, M. Correa-Duarte and M. Farle, Small, 2005, 1, 1073 CrossRef CAS.
- V. Salgueiriño-Maceira, M. Spasova and M. Farle, Adv. Funct. Mater., 2005, 15, 1036 CrossRef CAS.
- D. Nagao, M. Yokoyama, N. Yamauchi, H. Matsumoto, Y. Kobayashi and M. Konno, Langmuir, 2008, 24, 9804 CrossRef CAS.
- H. Matsumoto, D. Nagao and M. Konno, Langmuir, 2010, 26, 4207 CrossRef CAS.
- C. T. Cresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710 CrossRef CAS.
- J. Y. Ying, C. P. Mehnert and M. S. Wong, Angew. Chem., Int. Ed., 1999, 38, 56 CrossRef CAS.
- E. Delahaye, V. Escax, N. El Hassan, A. Davidson, R. Aquino, V. Dupuis, R. Perzynski and Y. L. Raikher, J. Phys. Chem. B, 2006, 110, 26001 CrossRef CAS.
- E. Kockrick, P. Krawiec, W. Schnelle, D. Geiger, F. Schappacher, R. Pöttgen and S. Kaskel, Adv. Mater., 2007, 19, 3021 CrossRef CAS.
- Y. Deng, D. Qi, C. Deng, X. Zhang and D. Zhao, J. Am. Chem. Soc., 2008, 130, 28 CrossRef CAS.
- Y. Deng, Y. Cai, Z. Sun, J. Liu, C. Liu, J. Wei, W. Li, C. Liu, Y. Wang and D. Zhao, J. Am. Chem. Soc., 2010, 132, 8466 CrossRef CAS.
- M. Morales, A. Mascarenhas, A. Gomes, C. Leite, H. Andrade, C. de Castilho and F. Galembeck, J. Colloid Interface Sci., 2010, 342, 269 CrossRef CAS.
- J. Kim, Y. Piao, N. Lee, Y. Park, I. Lee, J. H. Lee, S. R. Paik and T. Hyeon, Adv. Mater., 2010, 22, 57 CrossRef CAS.
- F. Chen, R. Shi, Y. Xue, L. Chen and Q. H. Wan, J. Magn. Magn. Mater., 2010, 322, 2439 CrossRef CAS.
- B. Julián-López, C. Boissière, C. Chanéac, D. Grosso, S. Vasseur, S. Miraux, E. Duguet and C. Sanchez, J. Mater. Chem., 2007, 17, 1563 RSC.
- J. Kim, H. S. Kim, N. Lee, T. Kim, H. Kim, T. Yu, I. C. Song, W. K. Moon and T. Hyeon, Angew. Chem., Int. Ed., 2008, 47, 8438 CrossRef CAS.
- J. E. Lee, N. Lee, H. Kim, J. Kim, S. H. Choi, J. H. Kim, T. Kim, I. C. Song, S. P. Park, W. K. Moon and T. Hyeon, J. Am. Chem. Soc., 2010, 132, 552 CrossRef CAS.
- J. Kim, J. E. Lee, J. Lee, Y. Jang, S. W. Kim, K. An, J. H. Yu and T. Hyeon, Angew. Chem., Int. Ed., 2006, 45, 4789 CrossRef CAS.
- J. Pyun, Polym. Rev. (Philadelphia, PA, U. S.), 2007, 47, 231 Search PubMed.
- N. A. D. Burke, H. D. H. Stöver and F. P. Dawson, Chem. Mater., 2002, 14, 4752 CrossRef CAS.
- P. Y. Keng, I. Shim, B. D. Korth, J. F. Douglas and J. Pyun, ACS Nano, 2007, 1, 279 CrossRef CAS.
- J. Stevenson, M. Rutnakornpituk, M. Vadala, A. Esker, S. Charles, S. Wells, J. Dailey and J. Riffle, J. Magn. Magn. Mater., 2001, 225, 47 CrossRef CAS.
- B. D. Korth, P. Keng, I. Shim, S. E. Bowles, C. Tang, T. Kowalewski, K. W. Nebesny and J. Pyun, J. Am. Chem. Soc., 2006, 128, 6562 CrossRef CAS.
- V. F. Puntes, D. Zachnet, C. K. Erdonmez and A. P. Alivisatos, J. Am. Chem. Soc., 2002, 124, 12874 CrossRef CAS.
- X. H. Zhang, K. M. Ho, A. H. Wu, K. H. Wong and P. Li, Langmuir, 2010, 26, 6009 CrossRef CAS.
- M. A. Nash, J. J. Lai, A. S. Hoffman, P. Yager and P. S. Stayton, Nano Lett., 2010, 10, 85 CrossRef CAS.
- T. Gelbrich, M. Feyen and A. Schmidt, Macromolecules, 2006, 39, 3469 CrossRef CAS.
- T. Gelbrich, G. Marten and A. Schmidt, Polymer, 2010, 51, 2818 CrossRef CAS.
- C. Gürler, M. Feyen, S. Behrens, N. Matoussevitch and A. M. Schmidt, Polymer, 2008, 49, 2211 CrossRef.
- Y. Sun, X. Ding, Z. Zheng, X. Cheng, X. Hu and Y. Peng, Chem. Commun., 2006, 2765 RSC.
- K. Landfester, A. Musyanovych and V. Mailänder, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 493 CrossRef CAS.
- V. Holzapfel, M. Lorenz, C. K. Weiss, H. Schrezenmeier, K. Landfester and V. Mailänder, J. Phys.: Condens. Matter, 2006, 18, S2581 CrossRef CAS.
- S. H. Hu and X. Gao, J. Am. Chem. Soc., 2010, 132, 7234 CrossRef CAS.
- Z. Varga, J. Fehér, G. Filipcsei and M. Zrínyi, Macromol. Symp., 2003, 200, 93 CrossRef CAS.
- C. Gollwitzer, A. Turanov, M. Krekhova, G. Lattermann, I. Rehberg and R. Richter, J. Chem. Phys., 2008, 128, 164709 CrossRef.
- T. Caykara, D. Yörük and S. Demirci, J. Appl. Polym. Sci., 2009, 112, 800 CrossRef CAS.
- G. Lattermann and M. Krekhova, Macromol. Rapid Commun., 2006, 27, 1373 CrossRef CAS.
- M. Krekhova and G. Lattermann, J. Mater. Chem., 2008, 18, 2842 RSC.
- J. Qin, I. Asempah, S. Laurent, A. Fornara, R. N. Muller and M. Muhammed, Adv. Mater., 2009, 21, 1354 CrossRef CAS.
- S. Reinicke, S. Döhler, S. Tea, M. Krekhova, R. Messing, A. M. Schmidt and H. Schmalz, Soft Matter, 2010, 6, 2760 RSC.
- H. Kim, J. Ge, J. Kim, S. Choi, H. Lee, W. Park, Y. Yin and S. Kwon, Nat. Photonics, 2009, 3, 534 CrossRef CAS.
- J. Ge, Y. Hu, M. Biasini, W. Beyermann and Y. Ying, Angew. Chem., Int. Ed., 2007, 46, 4342 CrossRef CAS.
- J. Ge, Y. Hu and Y. Yin, Angew. Chem., Int. Ed., 2007, 46, 7428 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |