Two-layer membranes of calcium phosphate/collagen/PLGA nanofibres: in vitro biomineralisation and osteogenic differentiation of human mesenchymal stem cells†
Nora
Hild
a,
Oliver D.
Schneider
a,
Dirk
Mohn
a,
Norman A.
Luechinger
a,
Fabian M.
Koehler
a,
Sandra
Hofmann
b,
Jolanda R.
Vetsch
b,
Benjamin W.
Thimm
b,
Ralph
Müller
b and
Wendelin J.
Stark
*a
aDepartment of Chemistry and Applied Biosciences, Institute for Chemical and Bioengineering, HCI E 107, ETH Zurich, Wolfgang-Pauli-Str. 10, CH-8093, Zurich, Switzerland. E-mail: wendelin.stark@chem.ethz.ch; Fax: +41 44 633 10 83; Tel: +41 44 632 09 80
bDepartment of Mechanical and Process Engineering, Institute for Biomechanics, ETH Zurich, 8093, Zurich, Switzerland
First published on 9th November 2010
Abstract
The present study evaluates the in vitro biomedical performance of an electrospun, flexible, anisotropic bilayer with one layer containing a collagen to mineral ratio similar to that in bone. The double membrane consists of a poly(lactide-co-glycolide) (PLGA) layer and an amorphous calcium phosphate (a-CaP)/collagen (Col)/PLGA layer. In vitro biomineralisation and a cell culture study with human mesenchymal stem cells (hMSC) were conducted to characterise such membranes for possible application as biomaterials. Nanofibres with different a-CaP/Col/PLGA compositions were synthesised by electrospinning to mimic the actual composition of bone tissue. Immersion in simulated body fluid and in cell culture medium resulted in the deposition of a hydroxyapatite layer. Incubation of hMSC for 4 weeks allowed for assessment of the proliferation and osteogenic differentiation of the cells on both sides of the double membrane. Confocal laser scanning microscopy was used to observe the proper adhesion of the cells. Calcium and collagen content was proven by Alizarin red S and Sirius red assays. Acute cytotoxic effects of the nanoparticles or the chemicals used in the scaffold preparation could be excluded based on viability assays (alamarBlue and alkaline phosphatase activity). The findings suggest possible application of such double membranes is in treatment of bone defects with complex geometries as wound dressing material.
Introduction
Biomaterials for bone substitution have to mimic or interact with the natural components of bone,1–10 namely collagen fibrils Type I (diameter: 80–100 nm11) and hydroxyapatite (HAp, Ca10(OH)2(PO4)6) as recently reviewed by Wahl and Czernuszka.12 An ideal material is close to an autologous implant and therefore needs to resemble the composition of natural bone and its structural information.13 In this regard, pioneering studies on “tapes” consisting of mineralised collagen fibrils have been reported14,15 and nanofibrils, made of collagen and HAp (40–80 wt% HAp), were prepared either by direct mineralisation of collagen or by dispersing HAp on self-assembling collagen fibres.16–19In most surgical applications, however, implant materials require some mechanical stability which demands an additional component since pure collagen/mineral composites are mechanically very sensitive. We therefore investigated the possibility to prepare triple component mineral/collagen/polymer fibres using electrospinning, a versatile method to synthesise fibres for wound dressing and tissue engineering.20 Two-component (mineral/polymer) fibres of highly bioactive and cotton wool-like poly(lactide-co-glycolide) (PLGA) fibres loaded with aerosol-derived amorphous calcium phosphate (a-CaP) nanoparticles were prepared earlier by Schneider et al.21 The base constituent polymer PLGA itself is approved by the Food and Drug Administration (FDA) for use in various clinical applications.22 Similarly, Jose et al.23 electrospun PLGA fibres with up to 20 wt% HAp.
The two-component system, collagen and HAp, was coelectrospun previously24–26 just as collagen combined with PLGA.27Membranes unifying the advantages of collagen, calcium phosphates and a synthetic polymer have been investigated.28–33 In a two-step procedure Ngiam et al.34 prepared PLGA and collagen blended PLGA fibres viaelectrospinning, these fibres were mineralised in a subsequent dipping procedure. In the present work a-CaP carrying PLGA blended collagen (Col) nanofibres were directly electrospun as triple component fibres. Besides their size, the increased dispersion stability of spherical a-CaP nanoparticles, a precursor of HAp crystallites,35,36 made the single step preparation of our nanocomposite possible at such high mineral loading. The mineral/collagen ratio in the resulting scaffold was similar to the one in bone.11,37,38
Anisotropic multi-layered membranes have been suggested as bone graft material for focal osteochondral defects39 or as functionally graded membrane for guided bone tissue regeneration.40–42 The present work uses triple component fibres with bone-like composition for the preparation of a bifunctional flexible dressing material for bone defects (a-CaP/Col/PLGA side) where a PLGA layer may protect the wound area against scar tissue ingrowth. The double membrane was investigated in terms of biomineralisation and ability to induce osteogenic differentiation with human mesenchymal stem cells (hMSC).
Experimental
Spherical, dispersible a-CaP nanoparticles
Amorphous calcium phosphate nanoparticles (Ca/P = 1.5) were prepared by flame spray synthesis according to a previously described procedure.43,44 Briefly, precursors of calcium-2-ethylhexanoic acid (Riedel de Haen, Ph. Eur. and Sigma-Aldrich) and tributyl phosphate (98% Sigma-Aldrich) were mixed accordingly. The mixture was diluted with xylene to a final metal concentration of 0.8 mol L−1, dispersed with oxygen (9 L min−1), and fed (9 mL min−1) through a capillary (diameter 0.4 mm) into a methane/oxygen flame. As formed a-CaP nanoparticles (production rate: 50 g h−1) were collected on bag-house filters and sieved (250 µm mesh) subsequently.Nanocomposite scaffold
Clinically approved PLGA (copolymer ratio of 85/15; Resomer Sample MD Type RG, Boehringer Ingelheim) and Type I collagen (calf skin, powder, cell culture tested, Sigma-Fine Chemicals, soluble part in HFIP (see below)) were purchased. Pure PLGA electrospinning solutions were prepared by dissolving PLGA with a concentration of 8 wt% in chloroform (Riedel de Haen, Ph. Eur.) containing Tween20 (Polysorbate, Fluka, Ph. Eur.) (5 wt% referred to PLGA). Collagen was dissolved (8 wt%) in hexafluoroisopropanol (HFIP, 99.0% Fluka) as suggested in the literature.45–48 The concentrations of all the solutions before any mixing are given in Table S1, ESI†. Collagen solutions were stored at room temperature (RT) overnight, three times centrifuged (21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500×g, 10 min) (himac CT15E Hitachi Koki Co., Ltd.) and the supernatant was collected. The a-CaP nanoparticles were dispersed in HFIP in an ultrasonic bath (Bandelin Sonorex Digitec) for 10 min with subsequent pauses (5 min). For the a-CaP/Col/PLGA scaffolds PLGA was added to the a-CaP solution before the latter was mixed with the collagen solution. The fibres were spun on a rotating coil (diameter = 8 cm, 130 rounds per minute (rpm)) covered by aluminium foil. The electrospinning conditions are listed in Table S1, ESI†. To electrospin the pure PLGA solution the needle tip was kept in a chloroform/air stream by a concentrically plugged sheath tube.49 Scaffolds were stored at RT under vacuum and further investigations were started within 2 weeks after preparation.
500×g, 10 min) (himac CT15E Hitachi Koki Co., Ltd.) and the supernatant was collected. The a-CaP nanoparticles were dispersed in HFIP in an ultrasonic bath (Bandelin Sonorex Digitec) for 10 min with subsequent pauses (5 min). For the a-CaP/Col/PLGA scaffolds PLGA was added to the a-CaP solution before the latter was mixed with the collagen solution. The fibres were spun on a rotating coil (diameter = 8 cm, 130 rounds per minute (rpm)) covered by aluminium foil. The electrospinning conditions are listed in Table S1, ESI†. To electrospin the pure PLGA solution the needle tip was kept in a chloroform/air stream by a concentrically plugged sheath tube.49 Scaffolds were stored at RT under vacuum and further investigations were started within 2 weeks after preparation.
An a-CaP/Col/PLGA–PLGA bilayer was produced by subsequently spinning first the a-CaP/Col/PLGA fibres and on top of it pure polymer or vice versa. The solutions were prepared as described above and a food grade dye (Coomassie Brilliant Blue, Merck, 0.8 wt% referred to a-CaP) was optionally added to the a-CaP/Col/PLGA solution to render both sides more easily distinguishable. Finally the double membrane was spun on a rotating sphere (diameter = 4.5 cm, 80 rpm) mimicking the shape of a femoral head.
a-CaP nanoparticles and scaffold characterisation
X-Ray disc centrifuge (XDC, Brookhaven Instruments, Holtsville, NY, USA), transmission electron microscopy (TEM, CM30 ST, LaB6 cathode, Philips Electron Optics, Eindhoven, the Netherlands) and Brunauer–Emmett–Teller (BET) surface area (Tristar 3000 Micrometrics, Norcross, GA, USA) were used to characterise the a-CaP nanoparticles. After sputtering the scaffolds with a 9 nm platinum layer, scanning electron microscopy (SEM, LEO 1530 Gemini) (voltage 3–5 kV) was performed to observe the morphology of the electrospun fibres and to follow any changes of the latter caused by further treatment. X-Ray diffraction (XRD) patterns were collected on an X'Pert PRO-MPD (Cu Kα radiation, X'Celerator linear detector system, step size of 0.033°, ambient conditions). Fourier transform infrared (FT-IR) spectroscopy measurements were done in KBr (Fluka puriss., 3 wt% sample) using a Tensor 27 Spectrometer (Bruker Optics, equipped with a diffuse reflectance accessory, DiffusIRTM, Pike technologies) (10 kHz, resolution 2 cm−1, 1000 scans). Attenuated total reflectance infrared spectroscopy (ATR-IR, Bruker Optics, Vertex 70) equipped with a commercial ZnSe ATR mirror unit (MVP, Harrick) was used to analyse the scaffolds (10 kHz, resolution 4 cm−1, 200 scans) after immersion in the simulated body fluid (SBF).Crosslinking
In order to render the scaffolds less soluble in SBF crosslinking was necessary. The effect of crosslinking the scaffolds for various times in vapours of glutaraldehyde (Sigma-Aldrich, 25 wt%)50 diluted with phosphate buffered saline (PBS, pH 7.4, Invitrogen) in a volume ratio of 1/3 was investigated. Different crosslinking times ranging from 5 to 60 min were tested.In vitro biomineralisation
The crosslinked a-CaP/Col and a-CaP/Col/PLGA scaffolds were irradiated for 15 min by a UV C lamp (50 W m−2) for sterilisation. The samples (∼1 mg crosslinked on aluminium foil) were placed in 10 mL SBF (pH 7.4) and incubated at 37 °C for 36 h and 120 h. SBF was prepared as described by Oyane et al.51 After immersion in SBF the samples were rinsed with Millipore water and dried at RT under vacuum.Cell culture study
Human mesenchymal stem cells (hMSC) were isolated from human bone marrow as previously described52 and cells in second passage were used. A cell culture study was conducted to investigate the a-CaP/Col/PLGA–PLGA double membrane. The electrospun samples were punched in discs of 22 mm in diameter (∼9.2 mg) and removed from the aluminium foil. The discs were crosslinked for 5 min as described above and sterilised under UV C light for 2 × 20 min subsequently. Samples with either PLGA or a-CaP/Col/PLGA on top were placed in 12-well polystyrene culture plates (Falcon, non-tissue-culture treated) and fixed with stainless steel metal rings sterilised in an autoclave (Varioklav 25 T, Sterico, Wangen, Switzerland).As a control medium a solution of DMEM, 10% FBS, 1% Pen–Strep, 1% antibiotic–antimycotic was taken. To induce differentiation of the hMSC towards the osteogenic lineage, an osteogenic medium composed of control medium with 50 µg mL−1 ascorbic acid-2-phosphate, 100 nM dexamethasone and 10 mM β-glycerophosphate (Sigma-Aldrich) was used. Cells were suspended in the corresponding medium at a concentration of 3 × 105cells mL−1 and 2 mL of the respective suspension were seeded into each well. Seeded scaffolds were incubated at 5% CO2 and 37 °C. For a period of 4 weeks the corresponding medium was changed 3 times a week. The cells' reaction to the scaffolds over time was assessed with biochemical assays after 1, 2 and 4 weeks, respectively. Scaffolds without seeded cells and empty wells with cells served as controls.
Cell viability
To determine the cell viability an alamarBlue assay (n = 4 per group) was used according to the manufacturer's instructions (Invitrogen). At the end of the respective culture period the cells were washed with PBS and 1 mL of a solution of alamarBlue reagent diluted in a ratio of 1/10 with the standard culture medium was added to each well and plates were incubated for 2 h at 37 °C. Immediately after incubation fluorescence was measured with a microplate reader (Tecan plate reader Infinite 200, Crailsheim, Germany, excitation wavelength 560 nm, emission wavelength 590 nm, auto cut-off 590 nm).Osteogenic differentiation capacity, mineral and collagen determination
The osteogenic differentiation phenotype of the cells was assessed by alkaline phosphatase (ALP) activity measurement based on the conversion of p-nitrophenyl phosphate to p-nitrophenol, which was measured spectrophotometrically at 405 nm as previously described.53 Briefly, scaffolds were washed three times with PBS after the alamarBlue assay, then treated with 0.2% Triton X-100 and 5 mM MgCl2 solution and disintegrated after the addition of 2 steel balls in the Mini-Beadbeater (BioSpec, Bartlesville, OK) set to 25 kHz for 10 s per cycle (all in all 3 cycles). ALP activity was tested right after sample collection.The other scaffolds were fixed with formalin (10% neutral buffered, Sigma-Aldrich) for 20 min and stored in PBS at 4 °C. An Alizarin red S assay54 followed by a Sirius red assay55,56 was done with 2 seeded bilayer scaffolds and as a control with a scaffold without cells seeded on it.
The scaffolds were incubated for 30 min at RT with 1 mL Alizarin red S solution (Fluka) (1 mg mL−1 in Millipore water, pH 5.5). The dye solution was removed and each scaffold was air-dried after it had been washed with Millipore water until no more colouring was eluted. After the staining, light microscopy images (Zeiss, Axio Imager.M2m) were recorded before the dyed scaffolds were destained with 5% perchloric acid in Millipore water. The absorbance of the supernatant at 405 nm was measured. In the following the scaffolds were washed once with 5% perchloric acid then three times with PBS and then incubated at RT for 18 h with 1 mL Sirius red solution (1 mg mL−1 Direct red 80 (ABCR) in saturated picric acid (Sigma-Aldrich)). Again after removing unbound dye by washing with Millipore water and air-drying the scaffolds microscopy pictures were taken. After destaining with Methanol:0.2 M NaOH (1:1) for 20 min absorbance at 490 nm was read.
Morphology of membranes and cells
For SEM the fixed scaffolds were rinsed for 10 min with 0.1 M Na–cacodylate buffer (sodium cacodylate trihydrate purum p.a., >98%, Fluka) then incubated with 0.04% aqueous OsO4 in 0.1 M Na–cacodylate at RT for 45 min. The scaffolds were again rinsed with Na–cacodylate buffer and then gradually dehydrated (10 min in EtOH 37%, 10 min in EtOH 67%, 3 × 10 min in EtOH 96%). Prior to SEM the samples were dried through lyophilisation (Christ Alpha 1-2, Osterode am Harz, Germany). Additionally confocal laser scanning micrographs (Leica SP2-AOBS) were recorded to determine the orientation and the location of the cells as well as their number. Prior to the confocal laser scanning microscopy fixed scaffolds were rinsed with PBS and then 0.1% Triton X-100 (in PBS) was added (PBT). At last the scaffolds were incubated in PBT at 37 °C for 1 h 30 min and then blocked with 2% bovine serum albumin in H2O for 30 min at 37 °C. Similar to Schuh et al.57phalloidin (Alexa Fluor 488, Invitrogen) staining (0.5% in PBT) was accomplished at 37 °C for 60 min followed by Hoechst 33258 (bisbenzimide) (0.1% in Millipore water) at 37 °C for 10 min and a final rinsing with PBS.Statistical analysis
Four samples (n = 4) in total were examined for biochemical analyses and expressed as average ± standard deviation of the mean. Statistical significance was evaluated by Student's t-test.Results and discussion
Spherical, dispersible a-CaP nanoparticles
The a-CaP nanoparticles were produced according to Loher et al.43 They exhibited a specific surface area of 87 ± 5 m2 g−1 as measured by BET and a corresponding primary particle diameter of 22 nm assuming spherical shape of the particles. TEM supported the assumption of a spherical shape of the a-CaP nanoparticles (Fig. 1a) and gave a primary particle diameter of 16 ± 4 nm (n = 50). A unimodal size distribution of the nanoparticles (hydrodynamic diameter = 40 nm) was revealed by X-ray disc centrifugation (XDC, Fig. 1b) and confirmed the possibility to well disperse the mineral component in the organic matrix. The diameter values were in accordance with the previously reported results.21 As additional controls, XRD patterns and FT-IR spectra of the as prepared a-CaP particles as well as the sintered particles (900 °C, 30 min; Fig. 1c and d) were in agreement with the literature21,43,44 and confirmed the identity and composition of the mineral phase. | ||
| Fig. 1 a-CaP nanoparticles were produced by flame spray synthesis. The as prepared nanoparticles were analysed by means of transmission electron microscopy (a) and X-ray disc centrifugation (b). X-Ray diffraction patterns (c) and Fourier transform infrared spectra (d) were recorded for as prepared particles and particles sintered for 30 min at 900 °C. | ||
Nanocomposite scaffolds
Electrospinning was used to prepare scaffolds of different compositions of a-CaP, collagen and PLGA. FT-IR spectra of the fibres and their respective ingredients are shown in the ESI (Fig. S1†) and were in agreement with the literature.34SEM micrographs showed a clear difference between the smooth surface of fibres without a-CaP particles (Fig. S2, ESI†) and the surface of fibres containing nanoparticles, the latter were more or less homogeneously distributed on the surface (Fig. 2). The diameters of the fibres (n = 100) are summarised in Table S2, ESI†. The presence of nanoparticles rendered the fibres thinner and PLGA amplified the average diameter of collagen fibres independently of the presence or absence of a-CaP. A new pliable anisotropic bilayer (Fig. 3a) was fabricated by electrospinning an a-CaP/Col/PLGA nanofibrous layer (∼10 µm thick) onto previously spun pure PLGA fibres (∼60 µm thick, support matrix). Fig. 3b and c show SEM micrographs of the two sides of the membrane. The PLGA fibres were slightly flattened and enlarged as they adhered to the aluminium foil. The interface of the two layers of the double membrane by means of SEM is shown in Fig. S3†. The double membrane was easier to handle as a-CaP/Col/PLGA scaffolds and more stress resistant. The two functionally different layers could be rendered distinguishable by selectively dying one side as shown in Fig. 3a. Fig. 3d–f illustrate a potential application of the flexible bone-like double membrane showing it wrapped around a femoral head (model). The advantages of this biomaterial are the structural and the compositional similarity to natural bone. Beyond that the fibrous meshes allow for cell ingrowth, which enhances bone regeneration.21 Furthermore the double membrane fortified the a-CaP/Col/PLGA fibres which by themselves were very difficult to handle in terms of mechanical stability. | ||
| Fig. 2 The structure and morphology of the fibres containing a-CaP nanoparticles depended on the mineral/collagen/polymer ratio as shown by scanning electron micrographs. The nanoparticles were not homogeneously distributed in scaffolds with a-CaP/Col (20/80) (a) and the fibrous structure was not uniform for the scaffold with a-CaP/Col (60/40) (b). In contrast to (a) and (b) the nanoparticles were homogeneously distributed in a-CaP/Col/PLGA (70/30/20) fibres (c). | ||
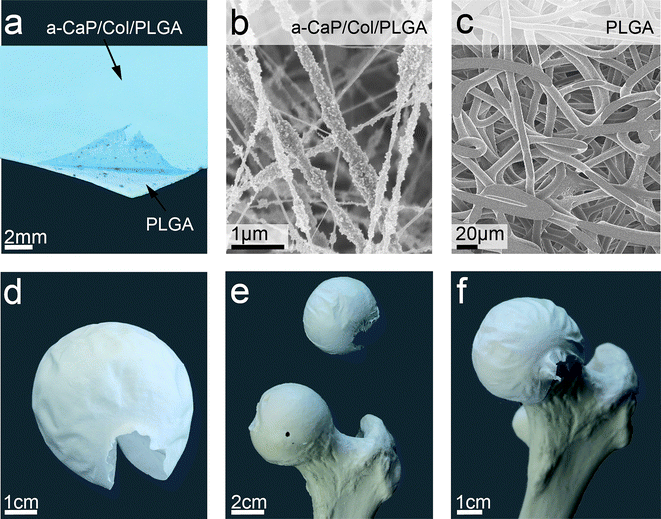 | ||
| Fig. 3 An anisotropic pliable bilayer was synthesised with a-CaP/Col/PLGA fibres dyed with Brilliant Blue as top layer on pure PLGA fibres serving as bottom layer (a). Scanning electron microscopy was used to characterise both sides of the double membrane (a-CaP/Col/PLGA (b) and PLGA (c)). With an adequate collector the bilayer can be electrospun into a spherical shape (d). The double membrane was positioned on a femur head model (e and f) illustrating the flexibility of the bilayer and indicating possible application as wrapping material. | ||
Crosslinking
Because as spun scaffolds dissolved in the simulated body fluid (note that scaffolds were partially soluble unless crosslinked), the scaffolds containing collagen were subjected to a crosslinking treatment. The effect of different exposure to glutaraldehyde/PBS was investigated (Fig. 4) and revealed an improved stability for fibres fortified with PLGA. Fibres containing no PLGA were strongly affected by the crosslinking procedure and lost their fibrous structure. As 5 min of crosslinking were enough for fibres to be stable in SBF, this crosslinking time was used throughout the remainder of the study. | ||
| Fig. 4 The effect of crosslinking the nanocomposite scaffolds in glutaraldehyde (25 wt%)/PBS (1/3) was observed by scanning electron microscopy. The fibres containing PLGA (a-CaP/Col/PLGA, 70/30/20) kept their as prepared structure and morphology (a) throughout the crosslinking times of 5 min (c) and 60 min (e). The morphology and structure of as prepared a-CaP/Col (60/40) (b) fibres were lost during crosslinking treatments, 5 min (d) and 60 min (f). | ||
In vitro biomineralisation
Biomineralisation experiments were conducted under sterile conditions in order to avoid bacterial or fungal degradation. After 36 h in SBF, the a-CaP/Col/PLGA fibres showed a dramatically modified surface in agreement with observations on mineralised fibres (scaffolds containing similar a-CaP in PLGA),10 as can be seen in Fig. 5b and c. The formation of a continuous HAp layer was observed, while the fibrous structure was not damaged. As controls (no PLGA), a-CaP/Col (60/40) fibres were also immersed in SBF for 36 h (Fig. 5a). The latter presented differences with the as prepared fibres (Fig. 2b) and with those crosslinked for 5 min (Fig. 4d). No comparably clear crystalline needles as for a-CaP/Col/PLGA fibres were revealed on the surface by SEM microscopy and the fibrous structure was entirely lost. This confirmed the importance of creating a triple component system and the mechanical support role of PLGA. The XRD patterns of the scaffolds after the immersion in SBF (ESI, Fig. S4†) confirmed that crystalline HAp was deposited on the a-CaP/Col/PLGA scaffolds. Both the β-TCP phase (26.0°, 28.1°, 31.4°, 32.8° and 34.6°) and the HAp phase (28.7°, 31.8°, 32.1° and 34.2°) were in agreement with studies on a two-component system based on a-CaP/PLGA.21 Attenuated total reflectance infrared spectra (ATR-IR, Fig. S5, ESI†) proved the formation of HAp58 (PO4− stretching bands at 1090 cm−1, at 1050 cm−1 and at 960 cm−1) and provided evidence that PLGA59 and collagen25,58 remained present in the samples after the biomineralisation study (for PLGA: C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching band at 1750 cm−1, C–H stretching at 1450 cm−1, O–C–O stretching at 1080 cm−1 and C–CH3 stretching at 1040 cm−1 and for collagen: amide I band at 1650 cm−1 and amide II band at 1530 cm−1).
O stretching band at 1750 cm−1, C–H stretching at 1450 cm−1, O–C–O stretching at 1080 cm−1 and C–CH3 stretching at 1040 cm−1 and for collagen: amide I band at 1650 cm−1 and amide II band at 1530 cm−1).
 | ||
| Fig. 5 In vitro biomineralisation was investigated by immersion of different scaffolds in the simulated body fluid (SBF). Scanning electron microscopy demonstrated that the fibrous structure was completely lost for a-CaP/Col (60/40) scaffolds containing no PLGA (a). The formation of hydroxyapatite crystals occurred on the surface of a-CaP/Col/PLGA (70/30/20) fibres after 36 h in SBF (b and c). | ||
Cell viability
An overview of the results obtained from the alamarBlue cell viability assay is shown in Fig. 6. The number of viable cells throughout 4 weeks of cell culture study confirmed the lack of cytotoxic effects. At the beginning, the values for scaffolds in control medium were as high as those in osteogenic medium. They later diminished slightly if compared to those in osteogenic medium with statistically significant difference after 4 weeks (p < 0.05). At the same time, the difference to the control measurements (cells not cultured on a scaffold but directly on the cell culture plate) followed a comparable trend. A possible explanation is that the scaffold promotes growth by offering a quasi three dimensional environment. The purely two dimensional cell culture plate surface is less attractive and provides little morphological stimulation. The results demonstrated that both sides of the double membrane are cytocompatible. | ||
| Fig. 6 Cell viability of human mesenchymal stem cells was assessed using an alamarBlue assay (n = 4; 0p ≥ 0.05; *p < 0.05) after 2 weeks (a and b) and after 4 weeks (c and d). Cell viability values for cells cultured on the PLGA side (a and c) or on the a-CaP/Col/PLGA side (b and d) proved the absence of any cytotoxic effects. Light grey bars specify the values obtained for cells cultured in osteogenic medium whereas dark grey bars are used for values of cells cultured in control medium. The control values (cells cultured in the same medium directly on the cell culture plate) are indicated as dotted lines. | ||
Osteogenic differentiation capacity, mineral and collagen determination
In order to analyse the osteogenic differentiation of hMSC during the cell culture ALP activity was determined (Fig. 7). As expected, the values for hMSC cultured in osteogenic medium were statistically significantly higher (p < 0.01) compared to those cultured in control medium after 2 and 4 weeks independent of what side was up. For example after 4 weeks of cell culture in osteogenic medium on the a-CaP/Col/PLGA side 40 ± 3 µg p-nitrophenol per scaffold were found compared to a non-measurable quantity of p-nitrophenol in the scaffold cultured in control medium. The low value for cells cultured in control medium indicated that neither side of the double membrane was osteoinductive by itself. In that case osteogenic differentiation of hMSC would have been observed without the additional contents in the osteogenic medium. As additional controls and to confirm that the scaffold material did not interfere with the assay, scaffolds without cells were analysed and showed indeed no ALP activity (data not shown). | ||
| Fig. 7 Osteogenic differentiation of human mesenchymal stem cells was assessed using an alkaline phosphatase (ALP) activity assay (n = 4; **p < 0.01) after 2 weeks (a and b) and after 4 weeks (c and d). ALP activity of cells cultured on the PLGA side (a and c) or on the a-CaP/Col/PLGA side (b and d) is given by means of p-nitrophenol content in the scaffold. Light grey bars specify the values obtained for cells cultured in osteogenic medium whereas dark grey bars are used for values of cells cultured in control medium. The control values (cells cultured in the same medium directly on the cell culture plate) are indicated as dotted lines. | ||
Fig. 8 summarises the main results of the Ca and collagen staining. For both tests and at all times the cell culture staining was more intense for those scaffolds with the a-CaP/Col/PLGA side up as compared to when only PLGA was on top. This was partly due to the initial presence of collagen and Ca in this layer. Generally speaking, scaffolds in non-osteogenic control medium showed weaker colouration and so did the scaffolds that had no cells seeded on them (data not shown). The absorbance of the solutions obtained after destaining the samples is presented in Table 1. The micrograph in Fig. 8c shows an inhomogeneous staining of the scaffold at larger scale. On the smaller scale it appeared that the colouration came from beneath (a-CaP/Col/PLGA layer) and that all the PLGA fibres showed a white surface (see inset in Fig. 8c). The assay also revealed that part of the a-CaP/Col/PLGA layer was removed during the course of the cell culture study as presented in the ESI (Fig. S6†). This may be due to an insufficient adhesion of the two layers, so that the a-CaP/Col/PLGA fibres were partly flushed away.
 | ||
| Fig. 8 Alizarin red S assay (a and b) stained the Ca present in the scaffolds, whereas Sirius red assay (c and d) was used to colour the collagen present in the scaffolds. Samples with their PLGA side turned up during 4 weeks of cell culture in osteogenic medium (a and c) presented by far less intense staining than scaffolds that had the a-CaP/Col/PLGA side turned up (b and d). | ||
Morphology of membranes and cells
A comparison of the SEM micrographs from the two sides of the membranes is shown in Fig. 9. Those scaffolds with the a-CaP containing layer turned up during the culture presented a tremendously different surface compared to those with the PLGA side up. HAp crystals similar to those observed in the in vitro biomineralisation assay conducted by Schneider et al.21 were observable. Furthermore, these images show that the fibrous structure was maintained for at least 4 weeks. Both membrane sides presented a film on the surface which may be attributed to extracellular matrix comparable to the one observed in the literature for comparable experiments.15,60,61 | ||
| Fig. 9 The scaffold morphology of the double membrane after cells were cultured for 1 week (a and c) or for 4 weeks (b and d) in osteogenic medium on the PLGA side (a and b) or on the a-CaP/Col/PLGA side (c and d). Scanning electron micrographs confirmed that a-CaP/Col/PLGA side presented cauliflower-like hydroxyapatite morphology on the preserved fibres upon mineralisation. | ||
As SEM is not the most suitable method to visualise the orientation of the cells, their locations as well as their number we performed confocal laser scanning microscopy at different time points of the cell culture (Fig. 10). In agreement with Fig. S6†, the a-CaP/Col/PLGA layer showed occasional fissures where the PLGA layer (underneath) became visible. Cells were widely spread and covered the whole surface for scaffolds cultured with the PLGA side up, whereas cells seeded on a-CaP/Col/PLGA layer preferred the intact (i.e.a-CaP containing) areas. This occasional peel-off may be related to significant mechanical stress in the fissure area. However, even in the fissure area, the cells adhered to the PLGA fibres as can be seen in Fig. S7, ESI†.
 | ||
| Fig. 10 Morphology of the cells cultured on the double membrane is illustrated by confocal laser scanning microscopy. Cells were seeded for 1 week (a and c) or for 4 weeks (b and d) in osteogenic medium on the PLGA side (a and b) or on the a-CaP/Col/PLGA side (c and d). Cell nuclei (blue spots) were stained with phalloidin and the actin skeleton was dyed with Hoechst 33258 (green fibrous network). Parts of the a-CaP/Col/PLGA layer were removed during the cell culture and in the fissure the PLGA fibres from below became exposed (c and d) (fissure area indicated by white dotted lines). | ||
Conclusion
Electrospinning was used to synthesise scaffolds with bone-like composition and fibrous structure. The scaffolds contained aerosol-derived a-CaP nanoparticles and collagen in a ratio of up to 70:30 strengthened with PLGA (20 wt%). The fibrous membranes presented a sufficiently high flexibility to be easily wrapped around complex shapes. Rapid formation of a HAp layer upon immersion of the nanocomposite in SBF revealed a high HAp forming potential of the material. In vitro proliferation of hMSC seeded on the double membrane was successful and neither side showed cytotoxicity. Differentiation of hMSC into the osteogenic lineage was better than in a 2D control. We could further confirm an augmented content of Ca and collagen in the ECM. Biomineralistaion on such anisotropic membrane was observed preferably on the a-CaP/Col/PLGA side. Further investigations are required to enhance the adhesion of the a-CaP/Col/PLGA fibre layer to the PLGA fibres. Potential applications of such anisotropic, triple component membranes may be in the treatment of arthritis on the femoral head or in bone wound healing.
Acknowledgements
The authors thank Joachim Hehl from the LMC of ETHZ for his assistance with the confocal laser scanning microscope and Erik Schmidt and Sven Reimann for their help with the ATR-IR measurements. Financial support by ETH Zurich and by the Swiss Commission for Technology and Innovation, CTI project 9141.1 is kindly acknowledged.References
- M. C. Chang, T. Ikoma, M. Kikuchi and J. Tanaka, J. Mater. Sci. Lett., 2001, 20(13), 1199–1201 CrossRef CAS.
- Y. Doi, T. Horiguchi, Y. Moriwaki, H. Kitago, T. Kajimoto and Y. Iwayama, J. Biomed. Mater. Res., 1996, 31(1), 43–49 CrossRef CAS.
- C. Du, F. Z. Cui, Q. L. Feng, X. D. Zhu and K. de Groot, J. Biomed. Mater. Res., 1998, 42(4), 540–548 CrossRef CAS.
- C. Du, F. Z. Cui, X. D. Zhu and K. de Groot, J. Biomed. Mater. Res., 1999, 44(4), 407–415 CrossRef CAS.
- B. Flautre, G. Pasquier, M. C. Blary, K. Anselme and P. Hardouin, J. Mater. Sci.: Mater. Med., 1996, 7(2), 63–67 CrossRef CAS.
- M. Kikuchi, S. Itoh, S. Ichinose, K. Shinomiya and J. Tanaka, Biomaterials, 2001, 22(13), 1705–1711 CrossRef CAS.
- M. Kikuchi, H. N. Matsumoto, T. Yamada, Y. Koyama, K. Takakuda and J. Tanaka, Biomaterials, 2004, 25(1), 63–69 CrossRef CAS.
- S. Liao, M. Ngiam, F. Watari, S. Ramakrishna and C. K. Chan, Bioinspiration Biomimetics, 2007, 2(3), 37–41 Search PubMed.
- K. Takaoka, H. Nakahara, H. Yoshikawa, K. Masuhara, T. Tsuda and K. Ono, Clin. Orthop. Relat. Res., 1988, 234, 250–254 CAS.
- K. S. Tenhuisen, R. I. Martin, M. Klimkiewicz and P. W. Brown, J. Biomed. Mater. Res., 1995, 29(7), 803–810 CrossRef CAS.
- S. Weiner and H. D. Wagner, Annu. Rev. Mater. Sci., 1998, 28, 271–298 CrossRef.
- D. A. Wahl and J. T. Czernuszka, Eur. Cells Mater., 2006, 11, 43–56 CAS.
- J. H. Bradt, M. Mertig, A. Teresiak and W. Pompe, Chem. Mater., 1999, 11(10), 2694–2701 CrossRef CAS.
- R. Burth, M. Gelinsky and W. Pompe, Tech. Textile, 1999, 8, 20–21 Search PubMed.
- A. Bernhardt, A. Lode, S. Boxberger, W. Pompe and M. Gelinsky, J. Mater. Sci.: Mater. Med., 2008, 19(1), 269–275 CrossRef CAS.
- M. Gelinsky, P. B. Welzel, P. Simon, A. Bernhardt and U. Konig, Chem. Eng. J., 2008, 137(1), 84–96 CrossRef CAS.
- S. Liao, F. Watari, M. Uo, S. Ohkawa, K. Tamura, W. Wang and F. Z. Cui, J. Biomed. Mater. Res., Part B, 2005, 74(2), 817–821 CrossRef.
- A. Tampieri, G. Celotti, E. Landi, M. Sandri, N. Roveri and G. Falini, J. Biomed. Mater. Res., Part A, 2003, 67(2), 618–625.
- W. Zhang, S. S. Liao and F. Z. Cui, Chem. Mater., 2003, 15(16), 3221–3226 CrossRef CAS.
- A. Greiner and J. H. Wendorff, Angew. Chem., Int. Ed., 2007, 46(30), 5670–5703 CrossRef CAS.
- O. D. Schneider, S. Loher, T. J. Brunner, L. Uebersax, M. Simonet, R. N. Grass and H. P. Merkle, et al., J. Biomed. Mater. Res., Part B, 2008, 84(2), 350–362 CrossRef.
- J. C. Middleton and A. J. Tipton, Biomaterials, 2000, 21(23), 2335–2346 CrossRef CAS.
- M. V. Jose, V. Thomas, K. T. Johnson, D. R. Dean and E. Nyalro, Acta Biomater., 2009, 5(1), 305–315 CrossRef CAS.
- S. H. Teng, E. J. Lee, P. Wang and H. E. Kim, Mater. Lett., 2008, 62(17–18), 3055–3058 CrossRef CAS.
- V. Thomas, D. R. Dean, M. V. Jose, B. Mathew, S. Chowdhury and Y. K. Vohra, Biomacromolecules, 2007, 8(2), 631–637 CrossRef CAS.
- J. Venugopal, S. Low, A. T. Choon, T. S. S. Kumar and S. Ramakrishna, J. Mater. Sci.: Mater. Med., 2008, 19(5), 2039–2046 CrossRef CAS.
- M. V. Jose, V. Thomas, D. R. Dean and E. Nyairo, Polymer, 2009, 50(15), 3778–3785 CrossRef CAS.
- S. S. Liao, F. Z. Cui, W. Zhang and Q. L. Feng, J. Biomed. Mater. Res., Part B, 2004, 69(2), 158–165 CAS.
- S. S. Liao, F. Z. Cui and Y. Zhu, J. Bioact. Compat. Polym., 2004, 19(2), 117–130 CrossRef CAS.
- M. Ngiam, S. Liao, A. J. Patil, Z. Y. Cheng, F. Y. Yang, M. J. Gubler and S. Ramakrishna, et al., Tissue Eng., Part A, 2009, 15(3), 535–546 Search PubMed.
- A. A. Sawyer, S. J. Song, E. Susanto, P. Chuan, C. X. F. Lam, M. A. Woodruff and D. W. Hutmacher, et al., Biomaterials, 2009, 30(13), 2479–2488 CrossRef CAS.
- H. N. Wang, Y. B. Li, Y. Zuo, J. H. Li, S. S. Ma and L. Cheng, Biomaterials, 2007, 28(22), 3338–3348 CrossRef CAS.
- S. S. Liao and F. Z. Cui, Tissue Eng., 2004, 10(1–2), 73–80 CrossRef CAS.
- M. Ngiam, S. S. Liao, A. J. Patil, Z. Y. Cheng, C. K. Chan and S. Ramakrishna, Bone, 2009, 45(1), 4–16 CrossRef CAS.
- E. D. Eanes, in Calcium Phosphates in Biological and Industrial Systems, ed. Z. Amjad, Kluwer, Boston, MA, 1st edn, 1998, ch. 2, pp. 21–40 Search PubMed.
- M. S. Tung, in Calcium Phosphates in Biological and Industrial Systems, ed. Z. Amjad, Kluwer, Boston, MA, 1st edn, 1998, ch. 1, pp. 1–20 Search PubMed.
- W. Suchanek and M. Yoshimura, J. Biomed. Mater. Res., 1998, 13(1), 94–117 CAS.
- R. Z. LeGeros, Clin. Orthop. Relat. Res., 2002, 395, 81–98 Search PubMed.
- T. Gotterbarm, W. Richter, M. Jung, S. B. Vilei, P. Mainil-Varlet, T. Yamashita and S. J. Breusch, Biomaterials, 2006, 27(18), 3387–3395 CrossRef CAS.
- S. Liao, W. Wang, M. Uo, S. Ohkawa, T. Akasaka, K. Tamura and F. Z. Cui, et al., Biomaterials, 2005, 26(36), 7564–7571 CrossRef CAS.
- A. Tampieri, M. Sandri, E. Landi, D. Pressato, S. Francioli, R. Quarto and I. Martin, Biomaterials, 2008, 29(26), 3539–3546 CrossRef CAS.
- S. H. Teng, E. J. Lee, P. Wang, D. S. Shin and H. E. Kim, J. Biomed. Mater. Res., Part B, 2008, 87(1), 132–138 CrossRef.
- S. Loher, W. J. Stark, M. Maciejewski, A. Baiker, S. E. Pratsinis, D. Reichardt and F. Maspero, et al., Chem. Mater., 2005, 17(1), 36–42 CrossRef CAS.
- D. Mohn, D. Ege, K. Feldman, O. D. Schneider, T. Imfeld, A. R. Boccaccini and W. J. Stark, Polym. Eng. Sci., 2010, 50(5), 952–960 CAS.
- J. A. Matthews, G. E. Wnek, D. G. Simpson and G. L. Bowlin, Biomacromolecules, 2002, 3(2), 232–238 CrossRef CAS.
- K. S. Rho, L. Jeong, G. Lee, B. M. Seo, Y. J. Park, S. D. Hong and S. Roh, et al., Biomaterials, 2006, 27(8), 1452–1461 CrossRef CAS.
- L. Yang, C. F. C. Fitie, K. O. van der Werf, M. L. Bennink, P. J. Dijkstra and J. Feijen, Biomaterials, 2008, 29(8), 955–962 CrossRef CAS.
- L. Yang, K. O. Van der Werf, C. F. C. Fitie, M. L. Bennink, P. J. Dijkstra and J. Feijen, Biophys. J., 2008, 94(6), 2204–2211 CrossRef CAS.
- G. Larsen, R. Spretz and R. Velarde-Ortiz, Adv. Mater., 2004, 16(2), 166–169 CrossRef CAS.
- E. D. Boland, J. A. Matthews, K. J. Pawlowski, D. G. Simpson, G. E. Wnek and G. L. Bowlin, Front. Biosci., 2004, 9, 1422–1432 CrossRef CAS.
- A. Oyane, H. M. Kim, T. Furuya, T. Kokubo, T. Miyazaki and T. Nakamura, J. Biomed. Mater. Res., Part A, 2003, 65(2), 188–195.
- L. Meinel, V. Karageorgiou, R. Fajardo, B. Snyder, V. Shinde-Patil, L. Zichner and D. Kaplan, et al., Ann. Biomed. Eng., 2004, 32(1), 112–122 CrossRef.
- S. Hofmann, H. Hagenmuller, A. M. Koch, R. Muller, G. Vunjak-Novakovic, D. L. Kaplan and H. P. Merkle, et al., Biomaterials, 2007, 28(6), 1152–1162 CrossRef CAS.
- C. A. Gregory, W. G. Gunn, A. Peister and D. J. Prockop, Anal. Biochem., 2004, 329(1), 77–84 CrossRef CAS.
- A. Sittichockechaiwut, A. M. Scutt, A. J. Ryan, L. F. Bonewald and G. C. Reilly, Bone, 2009, 44(5), 822–829 CrossRef.
- H. Tullberg-Reinert and G. Jundt, Histochem. Cell Biol., 1999, 112(4), 271–276 CrossRef CAS.
- E. Schuh, J. Kramer, J. Rohwedel, H. Notbohm, R. Muller, T. Gutsmann and N. Rotter, Tissue Eng., Part A, 2010, 16(4), 1281–1290 Search PubMed.
- H. Furedi and A. G. Walton, Appl. Spectrosc., 1968, 22(1), 23–26 CrossRef CAS.
- N. T. Paragkumar, E. Dellacherie and J. L. Six, Appl. Surf. Sci., 2006, 253(5), 2758–2764 CrossRef.
- X. J. Xin, M. Hussain and J. J. Mao, Biomaterials, 2007, 28(2), 316–325 CrossRef CAS.
- A. John, H. K. Varma, S. Vijayan, A. Bernhardt, A. Lode, A. Vogel and B. Burmeister, et al., Biomed. Mater., 2009, 4(1), 1–9 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional FT-IR spectra, electron micrographs, XRD patterns, ATR-IR spectra, light microscopy images, confocal laser scanning micrographs, electrospinning parameters and fibre diameters. See DOI: 10.1039/c0nr00615g |
| This journal is © The Royal Society of Chemistry 2011 |