Phosphorus and phosphorus–nitrogen doped carbon nanotubes for ultrasensitive and selective molecular detection
Eduardo
Cruz-Silva
*a,
Florentino
Lopez-Urias
b,
Emilio
Munoz-Sandoval
b,
Bobby G.
Sumpter
a,
Humberto
Terrones
ac,
Jean-Christophe
Charlier
c,
Vincent
Meunier
ad and
Mauricio
Terrones
e
aOak Ridge National Laboratory, P.O. Box 2008, MS6367, Oak Ridge, Tennessee 37831-6367, USA. E-mail: cruzsilvae@ornl.gov
bAdvanced Materials Department, IPICyT, Camino a la Presa Sn. Jose 2055, San Luis Potosi, Mexico 78216
cUniversité Catholique de Louvain, Institut de la Matière Condensée et des Nanosciences, Place Croix du Sud 1 (PCPM-Boltzmann), B-1348, Louvain-la-Neuve, Belgium
dDepartment of Physics, Applied Physics, and Astronomy, Rensselaer Polytechnic Institute, Troy, NY 12180-3590, USA
eResearch Center for Exotic Nanocarbons (JST), Shinshu University, 4-17-1 Wakasato, Nagano City, 380-8553, Nagano Japan
First published on 9th December 2010
Abstract
A first-principles approach is used to establish that substitutional phosphorus atoms within carbon nanotubes strongly modify the chemical properties of the surface, thus creating highly localized sites with specific affinity towards acceptor molecules. Phosphorus–nitrogen co-dopants within the tubes have a similar effect for acceptor molecules, but the P–N bond can also accept charge, resulting in affinity towards donor molecules. This molecular selectivity is illustrated in CO and NH3 adsorbed on PN-doped nanotubes, O2 on P-doped nanotubes, and NO2 and SO2 on both P- and PN-doped nanotubes. The adsorption of different chemical species onto the doped nanotubes modifies the dopant-induced localized states, which subsequently alter the electronic conductance. Although SO2 and CO adsorptions cause minor shifts in electronic conductance, NH3, NO2, and O2adsorptions induce the suppression of a conductance dip. Conversely, the adsorption of NO2 on PN-doped nanotubes is accompanied with the appearance of an additional dip in conductance, correlated with a shift of the existing ones. Overall these changes in electric conductance provide an efficient way to detect selectively the presence of specific molecules. Additionally, the high oxidation potential of the P-doped nanotubes makes them good candidates for electrode materials in hydrogen fuel cells.
1. Introduction
It is well known that the intrinsic electronic properties of single-walled carbon nanotubes (SWCNTs) can be modified by the adsorption of molecules on their surface.1 The adsorption usually proceeds in parallel with a significant modification of the nanotube electronic structure. For instance, the exposure of SWCNTs to NO2 and NH3 is known to induce two orders of magnitude changes in the nanotube conductivity, namely a conductivity increase when exposed to NO2 and a conductivity decrease when exposed to NH3.2 Molecular oxygen can also increase the conductivity of SWCNTs,3 due to charge transfer and strong interactions between the nanotube and the substrate.2 Chemical and structural modifications of nanotubes can therefore improve the nanotube sensitivity and selectivity, as shown for chemical functionalization,4 structural defects,5,6 and chemical doping.7,8It was recently found that phosphorus can effectively dope carbon nanotubes, either as a single substitutional dopant9 or as a co-dopant with nitrogen.10 Theoretical investigations showed that both P and PN defects are characterized by the presence of a highly localized state close to the Fermi level, a promising premise for notorious chemical reactivity and sensing capabilities.11 In this paper, we analyze in detail how phosphorus (P) and phosphorus–nitrogen (PN) doped carbon nanotubes can be used as ultrasensitive molecular sensors, based on the chemical reactivity of the phosphorus atoms and the localized states it induces in P- and PN-doped carbon nanotubes. Additionally, we note that these doped nanotubes could hold tremendous potential for applications for cathode materials in fuel cells, where the oxygen contamination of the Pt based cathodes currently used is a major problem.12 Specifically, doped carbon nanotubes could therefore catalyze a four-electron oxygen reduction reaction (ORR) process with a much higher electrocatalytic activity, more resistance to CO-poisoning, and better long-term operation stability even than those of commercially available or similar platinum-based electrodes.13 As demonstrated in this paper, P-doped nanotubes provide a viable material for fuel cells, thanks to the presence of a highly localized electronic state that should allow for even more efficient electrocatalytic activity than N-doped nanotubes.
2. Computational details
Ab initio calculations were performed using the density functional theory14,15 implementation of the SIESTA code16 within the local spin density approximation (LSDA). Norm-conserving pseudopotentials are used to represent the core electrons,17 and the wave functions are expanded in terms of a numeric pseudo-atomic double-ζ basis set with polarization orbitals, as described by Junquera et al.,18 using an energy shift of 25 meV to confine the basis functions, and a cutoff radii in the 5–8 a.u. range. A real space mesh equivalent to an energy cutoff of 200 Ry was used for the electrostatic potential integrals. In order to simulate the doping of the nanotube, a single P or PN doping site was created on a 8 × 1 × 1 supercell of a (6,6) carbon nanotube; which comprised a vacuum region of at least 15 Å between nanotubes in neighboring supercells, and a separation of 19.8 Å between doping sites. Given the system size, γ point calculations to sample the Brillouin zone were found accurate and numerically converged. To simulate adsorption, the molecules of interest are initially positioned about 2.5 Å from the doped site, and the structure is then relaxed by conjugate gradient (CG) geometry optimization until residual forces are lower than 0.04 eV Å−1. The thermal stability of the adsorption process is verified by molecular dynamics (MD) using a time step of 1 fs for the integration of the equations of motion, for a minimum of 1000 steps. The temperature of 300 K is controlled with the Nosé thermostat, using a Nosé mass of 10.0 Ry·fs2,19Charge transfer and adsorption energies were corrected using the basis set superposition error (BSSE) numerical technique. Charge redistribution after adsorption is calculated by subtracting the sum of the charge densities of the individual systems from the final charge density. Finally, chemical sensing properties are studied by computing the electronic transport using equilibrium Green functions20–22 for adsorbed species displaying the strongest interactions. Local variations in the electrostatic potential could result in screening of the impurity potential and in turn to a reduced efficiency of the P- or PN-doped nanotube system as a molecular sensor. The use of non-equilibrium Green's function methodology could be used in order to take these variations into account. However, the use of equilibrium conditions is justified by considering that for a real system, most of the potential drop occurs at the contacts, which together with the difference in the length scale between an experimental device (on the order of micrometres) and our model (ca. 2 nm) results in a very soft potential variation over the radius of action of the dopant at the nanometre scale.
The bonding environment of doping atoms in the nanotubes was studied using a Natural Bond Orbital (NBO) analysis23 using the NBO 5.0 program.24 This analysis uses the one-electron density matrix to derive molecular bonding information from the electronic density between the atoms. For this analysis, the density matrix was calculated for a finite segment of a pristine and doped nanotubes, using the NWChem25 suite of programs, with a 6-31G* basis set and the B3LYP exchange-correlation functional.26
3. Results and discussion
The molecules of interest for chemically specific detection studied in this paper include CO, NH3, NO2, SO2, and O2. The results of the adsorption energy are summarized in Table 1. We find that CO and NH3 have a marked preference toward PN-doped sites, while no adsorption is seen for P-doped nanotubes. After relaxation, CO adsorbs on PN-doped CNTs (binding energy of 0.203 eV), while a minimal adsorption energy is observed for P-doped CNTs (0.088 eV). The equilibrium distance between the adsorbate and the nanotube was 2.38 Å for PN doping and 3.01 Å for P doping, while the charge transfer was very low in both cases. For NH3, it has a higher adsorption energy on the PN-doped nanotube (0.444 eV) than on P-doped system, which is negligible (0.06 eV). In the PN case, NH3 was found to loose 0.16 electrons while the equilibrium distance was 2.41 Å. For the P-doped systems, the equilibrium distance of 3.53 Å, accompanied with a negligible charge transfer and low binding energy makes clear that the molecule was not adsorbed.| Molecule | P doped CNT | PN doped CNT | ||||
|---|---|---|---|---|---|---|
| d Eq | E Bind | Δqmol | d Eq | E Bind | Δqmol | |
| CO | 3.01 | 0.088 | 0.02 | 2.38 | 0.203 | −0.04 |
| NH3 | 3.53 | 0.064 | 0.00 | 2.41 | 0.444 | −0.16 |
| NO2 | 1.93 | 1.545 | 0.07 | 2.65 | 0.232 | 0.15 |
| O2 | 1.7 | 0.756 | 0.27 | 3.07 | 0.061 | 0.03 |
| SO2 | 2.62 | 0.435 | 0.27 | 2.73 | 0.335 | 0.25 |
In contrast to CO and NH3, NO2 and SO2 are adsorbed in both doping cases. NO2 has a very strong interaction with a P-doped site, with a binding energy of 1.55 eV, very low charge increase (0.07 electrons), and an equilibrium distance of 1.93 Å, which denotes a chemical bond rather than physisorption. Conversely, the binding energy to a PN-doped site was 0.23 eV, with a charge transfer of 0.27 electrons, and a longer equilibrium distance of 2.65 Å. SO2 had similar interactions with both P- and PN-doping sites. The equilibrium distances for P and PN doping sites were 2.62 Å and 2.73 Å, respectively and the binding energies were 0.44 and 0.34 eV, while the charge increase was also very similar (0.27 and 0.25 electrons).
Finally, O2 has affinity only for the P-doped site, with an equilibrium distance of 1.7 Å, a binding energy of 0.76 eV, and a charge increase of 0.27 electrons. For the PN-doped case the interaction was very low, with an equilibrium distance of 3.07 Å, a binding energy of 0.06 eV and a very low charge transfer.
The results described above are summarized in Table 1. From these data, it is evident that phosphorus doping creates an affinity to acceptor molecules (i.e.NO2) in the doped carbon nanotube, around the localized state previously described in ref. 9, and 11. In Fig. 1, the equilibrium geometries are shown for the molecules that have a non-negligible binding energy with the P-doped nanotubes (NO2, O2, and SO2). The charge density redistribution is also depicted in Fig. 1 with an isosurface at ±0.02 e− Å−3 for the aforementioned molecules. In all cases, an increase in the electron density is observed between the adsorbed molecule and the phosphorus atom, as well as an electron density reduction in the vicinity of the phosphorus atom. This result is consistent with the presence of a localized state around the phosphorus atom as being an active part of the adsorption process.
 | ||
| Fig. 1 Equilibrium geometries and charge density variations for (a) NO2; (b) O2 and (c) SO2 molecules adsorbed on phosphorus doped carbon nanotubes. The white (grey) clouds represent an increase (decrease) in charge density, with isosurfaces plotted at ±0.02 e− Å−3. | ||
Following NO2 adsorption, the increase in electronic density observed between the nitrogen and phosphorus atoms (see Fig. 1a), along with a short equilibrium distance, the reduced charge transfer, and the high binding energy, all indicate the formation of a covalent bond. Additionally, a bond length reduction of 4% is observed in the P–C bond in the nanotube, confirming a change in the chemical environment for the phosphorus atom. A similar effect is observed for oxygen adsorption (Fig. 1b), which shows a density increase coupled with a high binding energy. The P–C bond length is also reduced by about 4%, but in this case, significant charge transfer takes place. Fig. 1c shows the final geometry for the adsorption of SO2, which has both a lower binding energy and also smaller reductions in bond lengths in the nanotube.
Fig. 2 shows the equilibrium geometries for molecules adsorbed on a PN-doped nanotube. The charge density increases between the absorbed CO and the P atom (Fig. 2a), without disturbing the bond lengths and with a small binding energy. For NH3 (Fig. 2b), the electronic density increases significantly in the P–N bond. This indicates a charge transfer from the adsorbed molecule, resulting in an increase of ca. 4% in the length of this bond, while the P–C bonds remain unchanged.
 | ||
| Fig. 2 Equilibrium geometries and charge density variations for (a) CO, (b) NH3, (c) NO2; and (d) SO2 molecules adsorbed on phosphorus nitrogen doped carbon nanotubes. The white (grey) clouds represent an increase (decrease) in charge density, with isosurfaces plotted at ±0.02 e− Å−3. | ||
Fig. 2c shows the final geometry for NO2 absorbed on a PN-doped nanotube, which differs from the P-doped case in that there is an effective charge transfer of 0.15 electrons, while the binding energy is of 0.23 eV. This suggests a physical adsorption process, instead of the chemical adsorption seen for P-doped nanotubes. Fig. 2d shows the adsorption geometry for SO2, which has similar behavior to the adsorption on P-doped nanotubes.
Thermal stability was evaluated for the adsorbed molecules. After thermalization at 300 K, both CO and NH3 molecules unbind from the doping site on PN-doped nanotubes, although the adsorption energies are 0.2 and 0.44 eV. This confirms that for these molecules, the adsorption site is not thermally stable. For NO2 and SO2, only small changes in their position were observed during thermalization, and the molecules remained bound to the phosphorus atoms in the doped nanotubes. For the O2, a similar behavior was observed, in which the O2 molecule in P-doped nanotubes remains strongly bonded, whereas the non-adsorbed O2 molecule in PN-doped nanotubes remains at a distance above 2.8 Å.
It may seem surprising that P- and PN-doped nanotubes behave in such different ways. A P-doped nanotube presents affinity only towards acceptor molecules, and creates strong covalent bonds, while a PN-doped nanotube displays adsorption properties varying broadly with the type of adsorbate and forms mainly electrostatic interactions. In order to better understand the origins of these differences, we complemented the total energy calculations presented above with a natural bond orbital (NBO) analysis. The results are summarized in Fig. 3 and Table 2.
| α | β | |||
|---|---|---|---|---|
| P-doped | X | C | X | C |
| P-Ca | sp4.81d0.06 | sp2.43 | sp5.01d0.06 | sp2.48 |
| P-Cb | sp5.62d0.07 | sp2.57 | sp5.09d0.05 | sp2.76 |
| P (LP) | sp0.96 | — | sp0.97 | — |
| PN-doped | X | C | ||
| N–C | sp1.56 | sp2.54 | Degenerate | |
| P–C | sp4.6d0.06 | sp2.45 | ||
| N (LP1) | sp5.07 | — | ||
| N (LP2) | sp17.13 | — | ||
| P (LP) | sp0.68 | — | ||
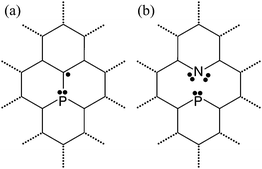 | ||
| Fig. 3 Schematic representation of the bonding environment of the P and PN doped carbon nanotubes. The phosphorus doped case is similar to the doped fullerene shown in ref. 27. The PN doped case is characterized by the charge transfer from P to N and the highly polarized PN bond. | ||
We found that the P-doping is associated with the presence of a lone electron pair located at the phosphorus atom with mixed s and p characters, as well as a p-like radical located at one of the neighboring carbon atoms, as seen in Fig. 3a. This result is compatible with previous observations made for the phospho-fullerene.27 As seen in Table 2, the hybridizations of α and β spins in P-doped nanotubes have slight variations due to the different number of electrons in the α and β spin populations.
Turning to PN-doped SWCNTs, we find that the bonding environment is very different to the P-doped counterpart (Fig. 3b). There is a considerable charge transfer from phosphorus to nitrogen, as their natural charges derived from NBO theory are +1.12 |e| and −0.58 |e|, respectively. Other atomic population theories, such as Bader analysis of the charge density, confirm that the phosphorus atom has a partial positive charge whereas the nitrogen atom has a partial negative charge, although the actual values are different. This charge transfer contributes to the formation of a highly polar bond between phosphorus and nitrogen, which is out of the detection threshold of the NBO but is evidenced by the presence of a short interatomic distance of the PN dimer and its thermal stability. Both N and P remain with a lone pair of electrons that, along with their bonds to carbon atoms, completes the full Lewis octet for both species. The partial positive charge of the phosphorus atom in PN-doped nanotubes accounts for the lower affinity from the P localized state towards acceptor molecules, and a higher affinity towards the donor ones. The partial negative charge of the lone pairs from the nitrogen atom, which have a strong p character, is likely to be screened by the carbon network. It follows that the N site is likely to be less chemically active than the P site.
Quantum transport calculations indicate that molecule adsorption perturbs the conductance of doped nanotubes, either by shifting the energy of the localized states or by creating new states. Fig. 4 displays the calculated conductance for P-doped nanotubes. We observe that NO2 and O2 adsorption have very similar behaviors: they induce a shift of the bound state at the Fermi level on the phosphorus atom located at lower energies (ca. −1.2 eV), while in the SO2 case, this energy level is only slightly shifted by −0.3 eV. The first two cases correspond to chemisorptions whereas the latter corresponds to physisorption, as explained above.
 | ||
| Fig. 4 Conductance for a P-doped carbon nanotube with an adsorbed molecule. From left to right: NO2, O2, and SO2. The dashed lines are the base conductance of a nanotube, the green lines are the conductance of P-doped nanotubes, and the red lines are the conductance after the molecule has been adsorbed. | ||
Fig. 5 shows the conductance for PN-doped nanotubes. CO has little effect on the conductance, only resulting in minor shifts. NH3 adsorption causes the suppression of a conductance dip located ca. 1 eV, and the shift of ca. 0.2 eV of the conductance dip located at −0.8 eV. These two dips are originally due to the presence of localized states around the P–N doping site, which are affected by the presence of NH3.
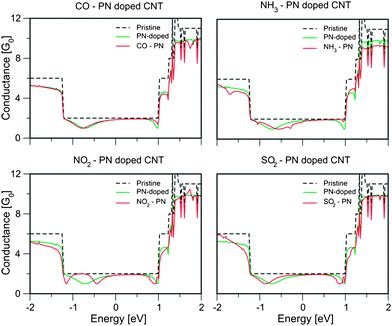 | ||
| Fig. 5 Conductance of a PN doped nanotube with an adsorbed molecule at the doping site. From top left, clockwise: CO, NH3, SO2 and NO2. CO has very mild effects due to its small interaction with the doped nanotube. | ||
For the NO2 adsorption on PN-doped nanotubes, the conductance dip located at ca −0.86 eV is split in two different dips, suggesting that this state was split in two nearby states (or that there is a shift plus a new state), located at −1.2 eV and −0.45 eV. SO2 has similar effects as in P-doped nanotubes, only causing a small downshift in the energy of the localized states and their corresponding dips.
For these doped nanotubes to be of practical use as selective molecular sensors, adsorbed molecules need to be identified. As discussed in ref. 6, changes of about 10% of the conductance at EF can be detected experimentally; and modifications of the conductance slope near EF can be an alternative for experimental measurements. Fig. 6 shows the conductance for the adsorbed molecules for P- and PN-doped nanotubes. It is observed that P-doped nanotubes exhibit the most significant changes in the conductance near the Fermi energy when a molecule is adsorbed. In Table 3, we show the calculated conductance changes and the difference in the linear response currents at −0.5 V for the molecules shown in Fig. 4 and 5. As can be seen in Fig. 6, both NO2 and O2 have a very similar effect in the conductance of a P-doped nanotube, with changes in both ΔG and ΔI that are not easily distinguished from each other. SO2 has a milder effect, with ΔG = 41% at EF and ΔI in the 10–15% range.
| P-doped | ΔG | ΔI (−0.5 V) | ΔI (0.5 V) |
| NO2 | 57% | 60% | 21% |
| O2 | 57% | 59% | 22% |
| SO2 | 41% | 11% | 15% |
| PN-doped | |||
| CO | 0% | 2% | 0% |
| NH3 | −10% | −21% | −3% |
| NO2 | −1% | −10% | 0% |
| SO2 | 4% | 12% | 2% |
 | ||
| Fig. 6 Comparison of the conductance near the Fermi energy for different adsorbed molecules on a doped site, for (a) a P-doped carbon nanotube; and (b) a PN-doped carbon nanotube. | ||
For PN-doped nanotubes, it is confirmed that CO has a very weak interaction, as the conductance and current changes are below detection thresholds. NH3 can be identified by a ΔG of −10% at Fermi energy, while ΔI (−0.5 V) is −21%. NO2 and SO2 have weak effects at EF, but ΔI (−0.5 V) values of −10% and 12%, respectively, could help in identification of the adsorbed molecules.
Recent developments in transport theory have allowed the calculation of quantum electronic transport properties along modular systems that could reach lengths in the order of micrometres,28,29 and have been successfully implemented using ab initio calculations.30 Under this approach, it has been found that as the nanotubes become longer, different transport regimes are developed depending on the energy of the carriers. While carriers with energies closer to those of the impurity bound states become strongly localized and have an exponential decay in conductance as a function of the nanotube length, those with energies far from these states have weak localization and their conductance decays linearly with the nanotube length.28,29 It follows that we can only infer that for a nanotube with lengths in the mesoscopic scale and with chemical disorder, the molecular species with strong chemical binding to the doped nanotubes would be easier to identify, since these interactions cause shifts in the energy of the localized states around the impurities. Molecular species that show only minor shifts in the conductance dips will likely be harder to identify since these changes will probably be screened by an exponentially decaying conductance.
4. Conclusions
In conclusion, substitutional P atoms modify the chemical properties of the surface of carbon nanotubes, thus creating sites with affinity towards acceptor molecules due to the presence of highly localized states around them. We found that P–N co-dopants have a reduced affinity for acceptor molecules, but the P–N bond can also take up charge, resulting in affinity towards donor molecules due to a partial positive charge at the phosphorus atom. Among the molecules studied here, CO and NH3 were found to adsorb only on PN-doped nanotubes, O2 was adsorbed only on P-doped nanotubes, while NO2 and SO2 were adsorbed on both P- and PN-doped nanotubes. Molecular dynamics studies revealed that CO and NH3 molecules unbind at room temperature, while the adsorption of the other molecules seem to be thermally stable. A NBO analysis showed that the P atom has a very different chemical environment in P- and PN-doped nanotubes, as a result of variations in charge and local bonding conditions.Chemical changes at the localized electronic states affect its energy, resulting in different positions for the conductance dips that are associated with them. SO2 and CO cause only minor shifts in the conductance, while NH3 causes the suppression of a conductance dip, and NO2 and O2 had similar effects in the conductance of P-doped carbon nanotubes. Finally, the adsorption of NO2 in PN doped nanotubes caused the appearance of a new dip in conductance, and at the same time the shift of the existing ones. Since changes in the conductance at Fermi energy of about 10% can be detected experimentally, and in particular since modifications of the conductance slope near EF can be an alternative for experimental measurements, the results of this study demonstrate the potential for a molecular sensor with identifiable selectivity that is based on P- and PN-doped carbon nanotubes. Diverse variations in realistic experimental setups could produce even different results that are out of the scope of the present report. While a complete assessment of the transport properties of the P and PN doped nanotubes and their potential as molecular sensors cannot be achieved only by theoretical methods, our results provide valuable information that, in conjunction with experimental data, could be further used to design realistic molecular sensors.
While the oxidation of P-doped nanotubes implies the poisoning of the adsorption sites and would limit their applications to anaerobic environments, the absence of interaction between oxygen and the PN-doped site allows the use of these sensors in ambient conditions. The fact that P-doped and PN-doped SWCNTs can be efficiently synthesized9,10 and provide a localized electronic state with a high oxidation potential should also be particularly attractive for applications in electrochemical redox applications. Currently, platinum nanoparticles are widely used as the cathode material in hydrogen/oxygen fuel cells (like polymer electrolyte fuel cells) due to their efficiency in catalyzing the oxygen reduction reaction (ORR). However platinum does not exhibit good stability and the catalytic performance begins degrading as soon as it is introduced into a fuel cell and continues until it is no longer active.12 This, combined with the high cost of platinum, constitute significant disadvantages to producing large-scale quantities of fuel cells for commercial applications. The use of N-doped CNTs has recently shown tremendous promise for providing a solution to this important problem.13 Here we note that the low affinity of PN-doped nanotubes to being poisoned by oxidation or carbon monoxide contamination indicates that these new materials should be even better for this purpose.
Acknowledgements
The authors at ORNL acknowledge support from the Laboratory Directed Research and Development Program of ORNL and from US Department of Energy under contract no. DEAC05-00OR22725 with UT-Battelle, LLC at ORNL. Part of this work was supported by the Center for Nanophase Materials Sciences (CNMS), sponsored by the Division of Scientific User Facilities, US Department of Energy and by the Division of Materials Science and Engineering, Basic Energy Sciences, US Department of Energy. The authors acknowledge the support from the National Center for Computational Sciences. H.T. acknowledges support as a visiting professor from the Ecole Polytechnique of Louvain and from Oak Ridge National Laboratory. J.-C.C. acknowledges financial support from the F.R.S.-FNRS of Belgium. Parts of this work are directly connected to the Belgian Program on Interuniversity Attraction Poles (PAI6) on “Quantum Effects in Clusters and Nanowires,” and to the ARC on “Hybrid metal/organic nanosystems” sponsored by the Communauté Française de Belgique. M.T. thanks JST—Japan for funding the Research Center for Exotic NanoCarbons, under the Japanese regional Innovation Strategy Program by the Excellence.References
- A. Goldoni, L. Petaccia, S. Lizzit and R. Larciprete, J. Phys.: Condens. Matter, 2010, 22, 013001 CrossRef.
- J. Kong, N. R. Franklin, C. W. Zhou, M. G. Chapline, S. Peng, K. J. Cho and H. J. Dai, Science, 2000, 287, 622–625 CrossRef CAS.
- P. G. Collins, K. Bradley, M. Ishigami and A. Zettl, Science, 2000, 287, 1801–1804 CrossRef CAS.
- S. S. Wong, E. Joselevich, A. T. Woolley, C. L. Cheung and C. M. Lieber, Nature, 1998, 394, 52–55 CrossRef CAS.
- F. Mercuri, A. Sgamellotti, L. Valentini, I. Armentano and J. M. Kenny, J. Phys. Chem. B, 2005, 109, 13175–13179 CrossRef CAS.
- Z. Zanolli and J.-C. Charlier, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 155447 CrossRef.
- F. Villalpando-Paez, A. H. Romero, E. Munoz-Sandoval, L. M. Martinez, H. Terrones and M. Terrones, Chem. Phys. Lett., 2004, 386, 137–143 CrossRef CAS.
- P. Young, Y. J. Lu, R. Terrill and J. Li, J. Nanosci. Nanotechnol., 2005, 5, 1509–1513 CrossRef CAS.
- I. O. Maciel, J. Campos-Delgado, E. Cruz-Silva, M. A. Pimenta, B. G. Sumpter, V. Meunier, F. Lopez-Urias, E. Munoz-Sandoval, H. Terrones, M. Terrones and A. Jorio, Nano Lett., 2009, 9, 2267–2272 CrossRef CAS.
- E. Cruz-Silva, D. A. Cullen, L. Gu, J. M. Romo-Herrera, E. Munoz-Sandoval, F. Lopez-Urias, B. G. Sumpter, V. Meunier, J.-C. Charlier, D. J. Smith, H. Terrones and M. Terrones, ACS Nano, 2008, 2, 441–448 CrossRef CAS.
- E. Cruz-Silva, F. Lopez-Urias, E. Munoz-Sandoval, B. G. Sumpter, H. Terrones, J.-C. Charlier, V. Meunier and M. Terrones, ACS Nano, 2009, 3, 1913–1921 CrossRef CAS.
- N. Yousfi-Steiner, P. Mocoteguy, D. Candusso and D. Hissel, J. Power Sources, 2009, 194, 130–145 CrossRef CAS.
- K. P. Gong, F. Du, Z. H. Xia, M. Durstock and L. M. Dai, Science, 2009, 323, 760–764 CrossRef CAS.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864–B871 CrossRef.
- W. Kohnand and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- J. M. Soler, E. Artacho, J. D. Gale, A. Garcia, J. Junquera, P. Ordejon and D. Sanchez-Portal, J. Phys.: Condens. Matter, 2002, 14, 2745–2779 CrossRef CAS.
- N. Troullier and J. L. Martins, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 43, 1993–2006 CrossRef CAS.
- J. Junquera, O. Paz, D. Sanchez-Portal and E. Artacho, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64, 235111 CrossRef.
- S. Nose, J. Chem. Phys., 1984, 81, 511–519 CrossRef CAS.
- S. Datta, Electronic Transport in Mesoscopic Systems, Cambridge University Press, 1995 Search PubMed.
- M. B. Nardelli, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 7828–7833 CrossRef.
- V. Meunier and B. G. Sumpter, J. Chem. Phys., 2005, 123, 024705 CrossRef.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev. (Washington, DC, U. S.), 1988, 88, 899–926 CrossRef CAS.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, and F. Weinhold, NBO 5.0, Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, 2001 Search PubMed.
- R. A. Kendall, E. Aprá, D. E. Bernholdt, E. J. Bylaska, M. Dupuis, G. I. Fann, R. J. Harrison, J. Ju, J. A. Nichols, J. Nieplocha, T. P. Straatsma, T. L. Windus and A. T. Wong, Comput. Phys. Commun., 2000, 128, 260–283 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- B. G. Sumpter, J. Huang, V. Meunier, J. M. Romo-Herrera, E. Cruz-Silva, H. Terrones and M. Terrones, A theoretical and experimental study on manipulating the structure and properties of carbon nanotubes using substitutional dopants, Int. J. Quantum Chem., 2009, 109, 97–118 CrossRef CAS.
- R. Avriller, S. Roche, S. Latil, F. Triozon and X. Blase, Low dimensional quantum transport properties of chemically disordered carbon nanotubes: from weak to strong localization regimes, Mod. Phys. Lett. B, 2007, 21, 1955–1982 CrossRef CAS.
- R. Avriller, S. Latil, F. Triozon, X. Blase and S. Roche, Chemical disorder strength in carbon nanotubes: magnetic tuning of quantum transport regimes, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 121406 CrossRef.
- A. Lopez-Bezanilla, F. Triozon, S. Latil, X. Blase and S. Roche, Effect of the chemical functionalization on charge transport in carbon nanotubes at the mesoscopic scale, Nano Lett., 2009, 9, 940–944 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |