Bringing natural products into the fold – exploring the therapeutic lead potential of secondary metabolites for the treatment of protein-misfolding-related neurodegenerative diseases
P. Matthew
Joyner
a and
Robert H.
Cichewicz
*ab
aNatural Products Discovery Group, Department of Chemistry and Biochemistry, Stephenson Life Sciences Research Center, University of Oklahoma, 101 Stephenson Parkway, Room 1000, Norman, OK, USA 73019-5251. E-mail: rhcichewicz@ou.edu; Tel: +1 (405) 325-6969
bCellular and Behavioral Neurobiology Program, University of Oklahoma, Norman, OK, USA 73019
First published on 7th October 2010
Abstract
Covering: up to June 2010
This review, citing 187 references, is an account of natural products exhibiting bioactive properties relevant to the treatment of protein aggregation-related neurodegenerative diseases (e.g., Huntington's, Parkinson's, and prion diseases). We have compared the chemical properties of secondary metabolite leads to compounds that are in current clinical use for treating central nervous system targets. This comparison revealed that natural products possess many of the desirable traits found in central nervous system-acting agents. It was also observed that the discovery of secondary metabolite leads from marine and microbial sources lags far behind the substantial discoveries emerging from plants. We have concluded this review by providing details concerning many of the most promising secondary metabolite leads that have been identified with therapeutic applications specific to the treatment of Huntington's, Parkinson's and prion diseases. We have also summarized compounds that have more generalized bioactivities that are favorable for treating a broad range of neurodegenerative disorders (i.e., anti-inflammatory, prevention of glutamate-induced neurotoxicity, neurotrophic activity, and inhibition of monoamine oxidase activity).
 Matthew Joyner | Matthew Joyner completed B.S. degrees in Chemistry and Mathematics at Lubbock Christian University in 2005 and then went on to complete his Ph.D. in Chemistry and Biochemistry at the University of Oklahoma in 2010. Under the guidance of Professor Robert Cichewicz at the University of Oklahoma, he developed a metabolomics-based approach to identifying new drug targets for Huntington's disease and also isolated and characterized new natural products from marine, fungal and bacterial sources. Matthew is currently pursuing postdoctoral research in the lab of Professor Robert Cichewicz, and is interested in continuing his work with natural products and their applications to human diseases. |
 Robert H. Cichewicz | Robert H. Cichewicz received B.S. degrees in biology and anthropology from Grand Valley State University and an M.S. degree in Pharmaceutical Sciences from the University of Louisiana at Monroe in 1999. He received his Ph.D. degree in 2002 under Muraleedharan G. Nair at Michigan State University and conducted postdoctoral training with Phil Crews at the University of California, Santa Cruz. In 2005, he joined the Department of Chemistry and Biochemistry at the University of Oklahoma as an assistant professor. His research interests include developing therapeutics for neurodegenerative and infectious diseases, as well as creating new approaches for enhancing natural products research. |
1 Introduction
There has been a worldwide increase in the number of reported cases of neurodegenerative disease over the last century, including Alzheimer's, Parkinson's, Huntington's, and prion diseases. While a substantial portion of the newly reported cases are attributable to increasing human lifespan, other factors such as enhanced medical diagnostic tools and improved awareness of neurodegenerative disease features have added to this increase. Remarkably, Alzheimer's, Parkinson's, Huntington's, and prion diseases are united by a shared pathological feature: the accumulation of aggregation-prone misfolded proteins in central nervous system (CNS) tissues. These misfolded proteins are ultimately responsible for the assemblage of symptoms (e.g., behavioral and movement disorders, cognitive decline, memory loss, etc.) that are the hallmarks of these diseases. Although the mechanisms through which aggregation-prone proteins cause neurodegeneration remain unclear, there are well-established links demonstrating their roles in the dysregulation of cell signaling, oxidative stress, inflammation, apoptosis, and other cellular abnormalities.1–3 Currently there are no cures for any of these diseases, so the development of new therapeutic leads aimed at halting or reversing the progression of these ailments is urgently needed.4Natural products are anticipated to play a significant role in the development of new therapeutic leads for neurodegenerative disease. Secondary metabolites have long served as an important resource for the development of small-molecule therapeutics,5–7 due in part to their combination of unique chemical features and potent bioactivities. The promising applications of natural products for the development of new neurodegenerative disease therapeutics is exemplified by galanthamine (1) and huperzine A (2), which have both proven to be potent inhibitors of acetylcholinesterase and are currently in clinical use for the treatment of Alzheimer's disease.8,9
Previous reports have summarized sub-sets of natural products that exhibit promising activities for the treatment of Alzheimer's and Parkinson's diseases.10–20 We have undertaken this review to evaluate the diverse range of studies aimed at applying natural products to the field of neurodegenerative disease drug discovery. We anticipate that this review will provide a unique perspective on the broad potential for natural products to help combat protein aggregation-related neurodegenerative disorders. In order to maintain our focus on natural products that are directly pertinent to protein aggregation-related neurodegenerative diseases, we have omitted compounds with biological activities that are non-specific to CNS disorders (e.g., antioxidants). We have also omitted discussion of most polyphenols such as flavonoids due to the extensive number of reviews already written specifically about these types of compounds.21–27 As part of this review, we have investigated the physiochemical properties of natural product leads and compared their features to drugs that are known to interact with CNS targets. Although our analysis does provide broad coverage of the natural products that are being examined for the full spectrum of protein aggregation-related neurodegenerative diseases, we have omitted further detailed coverage of Alzheimer's disease-specific leads. Instead, we refer the interested reader to the companion review on Alzheimer's disease by Williams in this issue of Natural Product Reports (see DOI: 10.1039/c0np00027b).
2 Natural products as lead compounds for the treatment of protein aggregation-related neurodegenerative disorders
Before discussing the specific natural products that have emerged as leads for treating Alzheimer's, Parkinson's, Huntington's, and prion diseases, we have summarized the relative impact that secondary metabolites have had on the drug development process for neurodegenerative disease. Our analysis reveals that Alzheimer's disease has been the subject of the most intense scrutiny, with 71.9% of the described leads having been studied in relationship to this disorder (Fig. 1). Both Huntington's and Parkinson's diseases have received more modest research effort, accounting for 2.2% and 2.4%, respectively, of the described natural product leads. Prion diseases account for only 1.7% of natural product leads. Drug leads not specific to any particular disease (e.g., modulators of neuroinflammation) that could potentially be used for any or all protein aggregation-related neurodegenerative disorders accounted for a substantial portion of the reported leads (21.8%).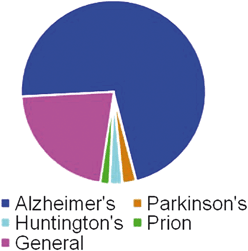 | ||
| Fig. 1 Number of natural products identified as having bioactivity relevant to specific neurodegenerative diseases. Data are presented as a percentage of the total number of compounds identified with bioactivity specifically pertaining to neurodegeneration (n = 537; Alzheimer's disease = 71.9%, Parkinson's disease = 2.4%, Huntington's disease = 2.2%, prion diseases = 1.7%, general neurodegeneration = 21.8%). | ||
It is estimated that in the United States alone, approximately four million people suffer from Alzheimer's disease, five hundred thousand from Parkinson's disease, one hundred thousand from Huntington's disease and three hundred from prion diseases. It is certain that there are many more people worldwide suffering from these diseases as well. Considering the relative proportion of people suffering from each of these disorders, it is not surprising that the development of Alzheimer's disease leads has dominated previous research endeavors. However, the disproportionate effort aimed at creating new drugs for combating Alzheimer's compared to other neurodegenerative ailments suggests that other major disorders like Huntington's and Parkinson's diseases represent understudied but important areas for further investigation.
A necessary part of the process for identifying neurodegenerative disease-relevant drug leads is a consideration of each compound's chemical features (i.e., molecular weight, C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logP, topological polar surface area, ClogD, number of hydrogen bond donors, and the strongest basic pKa). Specifically, it is important to recognize whether a lead compound bears the proper combination of physical properties that will enable it to penetrate the blood–brain barrier. Since central nervous system (CNS)-active drugs must possess a more stringently refined set of chemical properties compared to orally available systemic drugs, we examined how the natural product-derived leads described in this review compared to approved CNS-active agents. This point of concern is highly relevant because the frequently espoused chemical diversity of natural products could potentially serve as a liability if their assorted structural features drove secondary metabolites outside the realm of requirements for acceptable bioavailability. Scatter plots generated from a principle components analysis of natural product leads and approved CNS-active agents showed remarkable overlap for the chemical features of these two groups of compounds (Fig. 2a). Due to the substantial integration of the hundreds of compounds included in this review, additional scatter plots of the same data showing only the approved CNS-active agents (Fig. 2b) or natural product leads (Fig. 2c) are also provided. While natural products as a whole do exhibit somewhat greater scatter in terms of their structural features, it is important to recognize that the lead compounds included in this analysis have not undergone any type of optimization. Therefore, the significant similarity of secondary metabolite leads to CNS-active agents is even more extraordinary.
logP, topological polar surface area, ClogD, number of hydrogen bond donors, and the strongest basic pKa). Specifically, it is important to recognize whether a lead compound bears the proper combination of physical properties that will enable it to penetrate the blood–brain barrier. Since central nervous system (CNS)-active drugs must possess a more stringently refined set of chemical properties compared to orally available systemic drugs, we examined how the natural product-derived leads described in this review compared to approved CNS-active agents. This point of concern is highly relevant because the frequently espoused chemical diversity of natural products could potentially serve as a liability if their assorted structural features drove secondary metabolites outside the realm of requirements for acceptable bioavailability. Scatter plots generated from a principle components analysis of natural product leads and approved CNS-active agents showed remarkable overlap for the chemical features of these two groups of compounds (Fig. 2a). Due to the substantial integration of the hundreds of compounds included in this review, additional scatter plots of the same data showing only the approved CNS-active agents (Fig. 2b) or natural product leads (Fig. 2c) are also provided. While natural products as a whole do exhibit somewhat greater scatter in terms of their structural features, it is important to recognize that the lead compounds included in this analysis have not undergone any type of optimization. Therefore, the significant similarity of secondary metabolite leads to CNS-active agents is even more extraordinary.
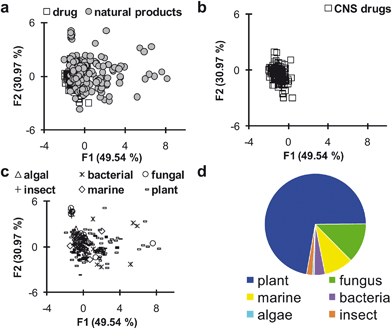 | ||
| Fig. 2 a–c) Scatter plots from principal components analysis of the chemical properties exhibited by CNS-active drugs and natural products with bioactivity relevant to neurodegenerative disease. Data points that cluster together have similar chemical properties. Chemical properties analyzed are molecular weight, ClogP, topological polar surface area, ClogD, number of hydrogen bond donors and the strongest basic pKa. a) Data points for CNS drugs and natural products, showing the large amount of overlap between the two groups. b) Data points representing only CNS drugs. c) Data points representing natural products with bioactivity relevant to neurodegenerative disease grouped by their biological source. d) Number of natural products with bioactivity relevant to neurodegeneration divided by type of source organism. Data presented as percentage of total compounds identified with bioactivity pertaining to neurodegeneration (n = 204; plant = 72.1%, fungus = 12.7%, marine = 9.3%, bacterial = 3.4%, insect = 2.0%, algae = 0.5%). | ||
A detailed breakdown of the six major chemical features that are of greatest concern for developing CNS-active drugs is illustrated in Fig. 3. Scatter plots comparing CNS-active drugs against natural products show that important properties such as ClogP (Fig. 3b), ClogD (Fig. 3d), and pKa properties (Fig. 3f) are within the desired ranges. Some deviation is observed concerning molecular weight (i.e., natural products tend to be larger) (Fig. 3a) and total polar surface area (i.e., natural products tend to have greater polar surface area) (Fig. 3c), although these parameters are not substantially divergent. In general, natural products tend to possess a larger number of H-bond donor groups compared to CNS-active drugs (Fig. 3e), but the unoptimized nature of the secondary metabolite lead pool may be the primary reason for this discrepancy. Overall, natural products meet many of the basic requirements for CNS-active drugs and therefore secondary metabolite leads should be regarded as generally compatible with the blood–brain-barrier penetration needs of CNS-active agents.
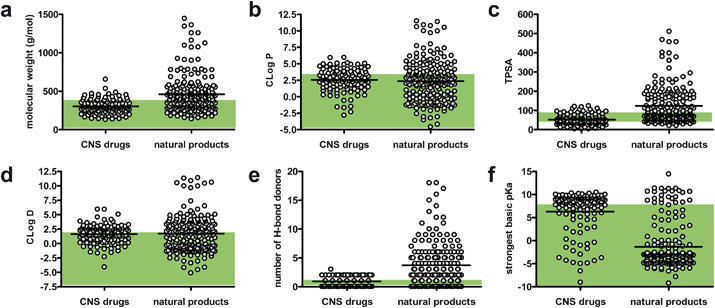 | ||
| Fig. 3 Scatter plots showing the distribution of values of chemical properties for CNS drugs and natural products with bioactivity relevant to neurodegenerative disease: a) molecular weight, b) ClogP, c) topological surface area (TPSA), d) ClogD, e) number of H-bond donors, and f) strongest basic pKa. Bars represent the mean value of the data points in each column. Shaded regions show the “ideal” values for each property.28 | ||
We also considered the biogenic sources of the reported natural product leads (Fig. 2c and 2d). We observed that a disproportionate number of compounds have been identified from plants, while other important groups of organisms are significantly under-represented. We were especially surprised that marine organisms and microbes (fungi and bacteria) have both been particularly under-utilized despite their incredible drug discovery potentials. We anticipate that focused efforts to screen new marine and microbial sources for Alzheimer's, Parkinson's, Huntington's, and prion disease leads will be particularly fruitful.
3 Alzheimer's disease
One of the characteristic features associated with Alzheimer's disease is the presence of insoluble extracellular amyloid plaques. These plaques consist primarily of the aggregation-prone peptide β-amyloid (Aβ), which is essential to Alzheimer's disease neuropathology.29 The misfolding and aggregation of Aβ contributes to the disruption of normal neuronal functions, including decreased production and altered regulation of neurotransmitters and neurotrophins, mitochondrial dysfunction, oxidative stress, inflammation, and tau hyperphosphorylation.29,30 The combined effects of these problems lead to the degeneration of cholinergic neurons, especially in the hippocampus, which results in the development of the primary symptoms of Alzheimer's disease.29,30 A variety of biological targets have been proposed for attempting to modulate Alzheimer's disease, including acetylcholinesterase, β-secretase, protein kinases such as Gsk3 and Cdk5 and protection of cells against Aβ toxicity. Natural products that modulate these targets have been identified, and the interested reader is encouraged to refer to the companion review by Williams for further details.4 Parkinson's disease
The primary symptoms of Parkinson's disease (loss of motor function and coordination) are largely due to the deterioration of dopaminergic neurons.31 The affected regions of the brains of Parkinson's disease patients accumulate protein aggregates called Lewy bodies that consist primarily of misfolded α-synuclein.32 Although the native function of α-synuclein is still not clear,32,33 several cellular features of Parkinson's disease have been identified, including mitochondrial dysfunction, inflammation, oxidative damage, and apoptosis.34Nearly all of the current therapies approved for Parkinson's disease focus exclusively on enhancing dopaminergic activity in the brain. Unfortunately, this approach is only effective for modifying the symptoms of Parkinson's disease rather than its cause.31 The lack of a clear understanding of the underlying biochemical basis of Parkinson's disease has made the development of effective neuroprotective treatments for this condition very difficult.35 The following section describes the limited number of natural products that have been identified as having activities related to the treatment of Parkinson's disease.
The neurotoxic effects of 6-hydroxydopamine (6-OHDA) mimic many of the pathological features of Parkinson's disease, making the use of 6-OHDA toxicity the most widespread model for identifying therapeutic leads for Parkinson's disease.36 This compound specifically targets catecholaminergic (dopaminergic and noradrenalinergic) neurons in animals, making it a highly useful tool for identifying new therapeutic leads that ameliorate the symptoms of Parkinson's disease.37 Unfortunately, investigations using a 6-OHDA model of Parkinson's disease lack the ability to identify compounds targeting the non-dopaminergic features of Parkinson's disease. However, useful alternatives to the 6-OHDA model have not yet emerged.37,38
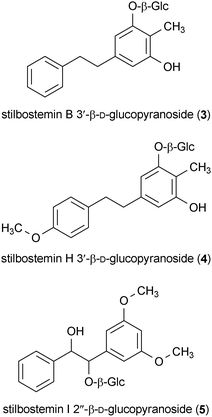
Three compounds isolated from the roots of Stemona tuberosa protected SH-SY5Y cells against 6-OHDA toxicity.39 Stilbostemin B 3′-β-D-glucopyranoside (3), stilbostemin H 3′-β-D-glucopyranoside (4), and stilbostemin I 2′′-β-D-glucopyranoside (5) all protected cells from 6-OHDA-induced toxicity at concentrations of 1 μM.39
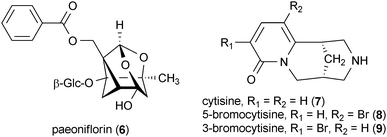
Paeoniflorin (6), a compound isolated from the roots of Paeonia lactiflora with a very unique cage-like pinane moiety, decreased behavioral abnormalities in rats induced by 6-OHDA when administered at doses as low as 5 mg/kg.40 Interestingly, this report also showed that 6 is not a dopamine receptor agonist, which may indicate the potential for this compound as a non-dopaminergic therapy for Parkinson's disease.
Cytisine (7) is a quinolizidine alkaloid that was originally isolated from the seeds of Sophora secundiflora.41 Compound 7 and its derivative 5-bromocytisine (8) significantly reduced striatal dopamine loss induced by 6-OHDA in rats at a dose of 2 mg/kg. However, another derivative of 7, 3-bromocytisine (9), had no effect on striatal dopamine concentrations.42 These results are significant since it has been shown that a decrease in striatal dopamine levels is a key pathological feature of the 6-OHDA model of Parkinson's disease.37
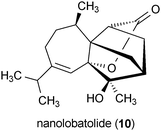
Nanolobatolide (10) is a metabolite isolated from the marine soft coral Sinularia nanolobata.43 Treatment of SH-SY5Y neuronal cells with 10 at a concentration of 0.1 μM significantly reduced the toxicity of 6-OHDA in these cells.
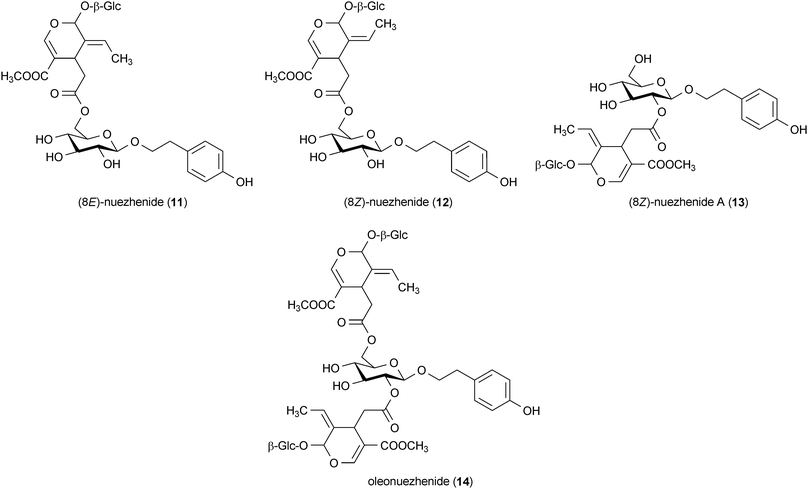
A series of secoiridoid glycosides were isolated from the plant Ligustrum japonicum that attenuated the toxicity of 6-OHDA in human neuroblastoma cells. The metabolites (8E)-nuezhenide (11), (8Z)-nuezhenide (12), (8Z)-nuezhenide A (13) and oleonuezhenide (14) all protected SH-SY5Y cells against 6-OHDA-induced toxicity at concentrations of 10 μM.44
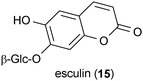
In addition to 6-OHDA-induced toxicity, dopamine-induced toxicity has also been used to investigate the therapeutic potential of small molecules.45 Esculin (15), a metabolite from Fraxinus sieboldiana, protected SH-SY5Y cells from dopamine-induced toxicity when applied to the cells at a concentration of 0.1 μM.45
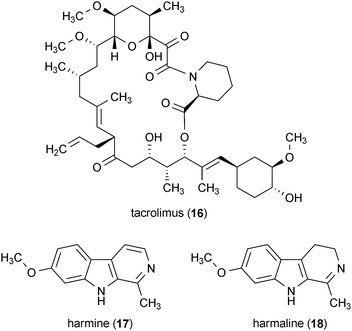
Several compounds that are described in more detail in other sections of this review have demonstrated promising biological activity for Parkinson's disease as well. The bacterial metabolite tacrolimus (16) protected mice from the degeneration of dopaminergic neurons due to 6-OHDA toxicity when administered at a dose of 1.5 mg/kg.46 Further description of the activity of 16 can be found in section 5.4. The metabolites harmine (17) and harmaline (18) enhanced the release of dopamine in striatal brain slices, indicating that these compounds may have therapeutic potential for Parkinson's disease treatment.47 Further description of the activity of 17 and 18 can be found in section 7.4.
5 Huntington's disease
Huntington's disease is a progressive neurodegenerative disorder arising from a CAG trinucleotide repeat expansion mutation in the huntingtin (Htt) gene.48,49 Individuals carrying mutant forms of huntingtin (mHtt) encoding for ≥35 glutamine repeats are at risk of developing Huntington's disease. The number of polyglutamine-encoding CAG repeats in mHtt strongly influences the age of disease onset, symptom severity, and rate of Huntington's disease progression.50 Mutant forms of the huntingtin protein are highly prone to aggregation, resulting in the formation of microscopic plaques that are distributed throughout affected regions of patients' brains. Unfortunately, the mechanism by which mutant huntingtin protein causes Huntington's disease is not known. However, oxidative stress, mitochondrial dysfunction, and apoptosis are some of the cellular processes that are believed to be induced as part of the disease process.48 Additional druggable targets for Huntington's disease have been proposed (e.g., inhibition of protein kinases, modulation of mutant huntingtin protein aggregation, and enhancement of the clearance of mutant huntingtin protein from cells), but the efficacy of these treatment methods have not yet been fully validated.515.1 Gsk3 as a target for Huntington's disease
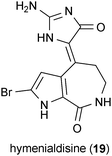
Glycogen synthase kinase 3 (Gsk3) has been identified as a promising target for the development of Huntington's disease therapeutics. Although Gsk3 fulfills a variety of cellular functions, its role in enhancing apoptotic signaling associated with mutant huntingtin-affected regions of the brain represents a possible target for therapeutic modulation.52,53 Supporting this view are data demonstrating that inhibition of Gsk3 can protect cells against mHtt toxicity.54 As evidence for the enthusiasm surrounding the development of drugs that target Gsk3, we have noted that more than 25 patents have been issued concerning the pharmacological management of this kinase. Although the compounds described in this section were originally identified as having therapeutic relevance to Alzheimer's disease, we have listed them here in order to draw attention to their potential applications to Huntington's disease.Hymenialdisine (19), a marine sponge metabolite, was identified as a potent inhibitor of Gsk3 (IC50 = 10 nM).55 A panel of 15 structural analogues of 19 were also tested in this study for their inhibitory activity against Gsk3, but 19 was much more active (10–10,000-fold greater) than any of the analogues.
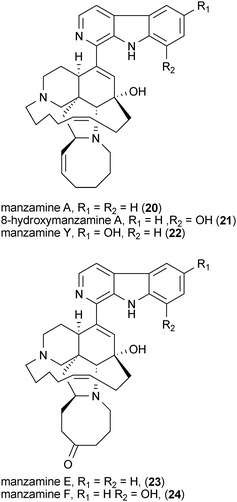
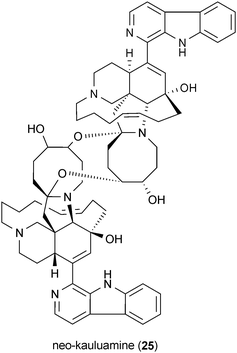
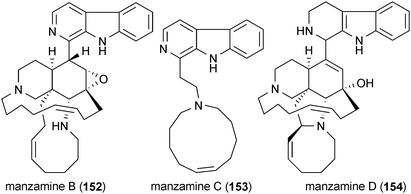
Manzamines are marine sponge-derived alkaloids that exhibit modest Gsk3 inhibitory activities.56 In studies utilizing an in vitro cell-free assay, manzamine A (20), 8-hydroxymanzamine A (21), manzamine Y (22), manzamine E (23), manzamine F (24), and the manzamine dimer neo-kauluamine (25), all inhibit Gsk3 at a concentration of 25 μM.56,57 However, only compounds 20 and 21 were shown to retain their Gsk3 inhibitory activities in a cell-based model system.57 A recent molecular docking study suggests that 21 binds to the ATP-noncompetitive pocket of Gsk3.58
5.2 Caspase inhibitors
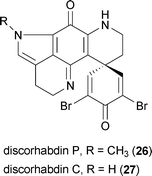
Apoptosis has been implicated as playing an important role in Huntington's disease. Accordingly, the blockade of apoptotic signals through inhibition of caspases has been proposed as an important therapeutic target for Huntington's disease intervention.59 Caspases 1 and 3 are upregulated in Huntington's disease patients and in mouse models of the disease. In both humans and rodents, caspases enzymatically cleave the huntingtin protein, resulting in an increase in the amount of toxic mutant huntingtin fragments in diseased cells.59,60The marine sponge Batzella sp. yielded the metabolite discorhabdin P (26), which inhibits caspase CPP32 with an IC50 value of 0.78 μM.61 It is interesting to note that the related metabolite discorhabdin C (27) does not inhibit caspase CPP32, even though the only difference between these two compounds is the presence of the N-methylpyrroline in 27 as opposed to the unsubstituted pyrroline in 26.
5.3 Decreasing mutant huntingtin aggregation
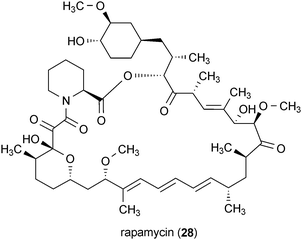
Misfolding and aggregation of the mutant huntingtin protein have been closely linked with toxicity and the pathogenesis of Huntington's disease.48 Possible methods for combating the toxic effects of the misfolded mutant huntingtin include inhibiting the formation of mutant huntingtin aggregates or enhancing the ability of cells to remove protein aggregates through autophagy. Autophagy is the process by which cells degrade and recycle large protein bodies and organelles.62 It has been shown that increasing the autophagic capacity of cells containing misfolded proteins and protein fragments can enhance cell survival.62 Accordingly, it has been proposed that increasing autophagy could serve as a means for combating Huntington's disease, as well as other neurodegenerative disorders.63–65Rapamycin (28) is a well-known secondary metabolite that was first isolated from Streptomyces hygroscopicus.66 The compound interacts with the protein FKB12, resulting in inhibition of the mTOR complex and upregulation of autophagy.67,68 It was recently shown that 28 increased autophagy further when administered in combination with lithium, which activates autophagy through an mTOR-independent pathway.69 The potential of this combination approach was demonstrated in a Drosophila model of mHtt-induced neurodegeneration.69 Broader applications of 28 in treating other protein aggregation associated diseases, including Alzheimer's disease, have been proposed as well.70
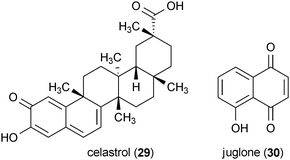
Two secondary metabolites were identified in a screening of pure compounds for the ability to reduce in vitro aggregation of the mutant huntingtin protein.71 Celastrol (29), a metabolite isolated from the plant Tripterygium wilfordii, and juglone (30), a well-known natural product from Juglans nigra, both exhibited potent activity in this assay (IC50 = 3.55 μM and 2.67 μM, respectively). Further investigation of these compounds in striatal cells expressing mHtt showed that the application of 29 and 30 at a concentration of 10 μM significantly increased the number of cells with cytoplasmic localization of mutant huntingtin protein (note that normal huntingtin protein is typically localized in both the nucleus and the cytoplasm in this cell line, but mutant huntingtin is localized exclusively to the nucleus).71
5.4 Protection against mutant huntingtin toxicity
Tacrolimus (16), also known as FK506, is a bacterial metabolite produced by Streptomyces tsukubaensis.72 When administered in a Huntington's disease mouse model at a dose of 4 mg/kg, 16 delayed the onset of disease-related symptoms, but it did not prolong lifespan.73 However, a separate study reported that administration of 16 to mice in a Huntington's disease rodent model at a dose of 0.5 mg/kg accelerated the onset of the neurological phenotype of the disease. It was proposed that this deleterious effect was due to the calcineurin inhibitory properties of 16.74 In yet another report, 16 was shown to restore normal transport of brain-derived neurotrophic factor (BDNF), which is typically disrupted by mutant huntingtin toxicity. However, the latter activity was also ascribed to the calcineurin-inhibitory effects of 16.75 At first glance these reports appear to be in conflict. However, closer inspection reveals that the study by Ditzler et al. used a dose of 16 that was 8-fold greater than the study by Hernández-Espinosa and Morton. The two studies also utilized different methods of measuring the symptoms of mutant huntingtin toxicity, with Ditzler et al. using only behavioral changes to determine the effect of the mutant huntingtin toxicity, while Hernández-Espinosa and Morton used a combination of behavioral observations and biochemical markers. Since these two studies used such dissimilar doses and metrics of the progression of Huntington's disease symptoms, it is not surprising that they found different results employing the same mouse model of Huntington's disease.Resveratrol (31) is a stilbene metabolite produced by a variety of plants, including grapes.76 In a Drosophila model of Huntington's disease, treatment of the flies with 31 at a concentration of 10 μM reduced the amount of neurodegeneration caused by mutant huntingtin; however, it did not increase the lifespan of the flies.77 The mechanism by which 31 elicited its protective effects was speculated to arise from the compound's potential antioxidant activity78 and its sirtuin-activating properties.79
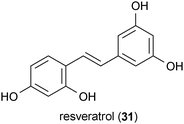
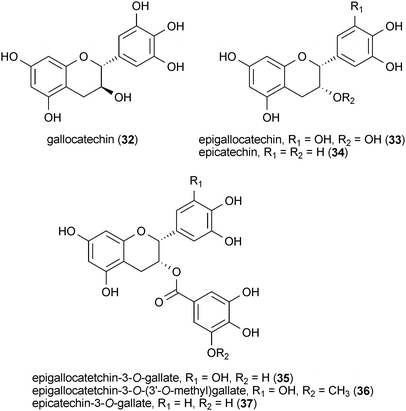
A series of green-tea catechins were reported to protect a yeast model of Huntington's disease from the toxic effects of a mutant huntingtin protein fragment.80 Gallocatechin (32), epigallocatechin (33), epicatechin (34), epigallocatetchin-3-O-gallate (35), epigallocatetchin-3-O-(3′-O-methyl)gallate (36) and epicatechin-3-O-gallate (37) protected cells from mutant huntingtin toxicity at concentrations ranging from 0.1 nM to 10 μM. A separate study showed that 35 also modulated mutant huntingtin aggregation in vitro and that it reduced the detrimental effects of mutant huntingtin in a Drosophila model.81 For a more complete summary of the reported neuroprotective effects of green tea catechins, the reader is directed to a review of the subject by Mandel et al.82
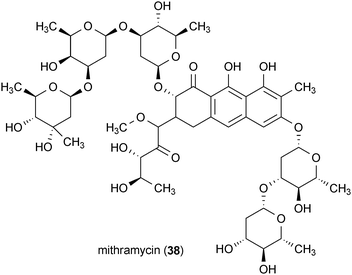
The chemotherapeutic agent mithramycin (38) was reported to enhance the survival of mice in a transgenic model of Huntington's disease.83 This metabolite was isolated from the bacteria Streptomyces agrillaceus and exhibited the ability to significantly increase the lifespan of the mice in this disease model. This report suggested that the prevention of mutant huntingtin-induced hypermethylation of histone H3 by 38 may be the mechanism by which it protects mice against mutant huntingtin's toxicity.83 Compound 38 is an attractive therapeutic lead for Huntington's disease since it is already an FDA approved drug.
6 Prion diseases
Prion diseases, also known as transmissible spongiform encephalopathies, are a rare but fatal group of neurodegenerative diseases observed in humans and other mammals.84,85 These diseases are caused by an infectious prion, which is a misfolded protein that forms amyloid plaques and is able to transfer its misfolded conformation to other proteins.86 Approximately 300 cases of prion diseases are diagnosed in the U.S. each year and they are particularly devastating to patients since they are often fatal within one year of diagnosis.87 The most common prion disease in humans is Creutzfeldt–Jakob disease, but other forms of the disease include kuru, fatal familial insomnia (FFI), and Gerstmann–Straussler–Scheinker disease.87Several cellular models of prion diseases have been developed that have been useful in the search for drug leads.86 Almost all of the reported assays have targeted the inhibition of the prion misfolding process, reduction of the total amount of prion protein in cells, and stabilization of the natively folded prion protein.88
6.1 Prion amyloidogenesis inhibition
The polyene macrocycle amphotericin B (39) was originally isolated from Streptomyces nodosus in 1955 and has been widely used as an antifungal drug over the past 50 years.89–93 It was reported that 4.5 μg/mL of compound 39 is sufficient to reduce the generation of misfolded prion protein in infected GT1-7 and S12 cells.94 Treatment of these cell lines with 39 at this concentration induced no observed toxicity.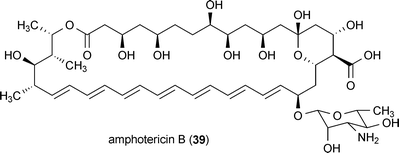
The Cannabis sativa metabolite cannabidiol (40) reduced the accumulation of prion protein in infected cells at a concentration of 5 μM.95 The major psychoactive constituent of C. sativa, Δ9-tetrahydrocannabinol (41), also exhibited activity, but its effects were limited to only a subset of the investigated cell lines. Compound 40 significantly increased the survival of prion infected mice when administered at a dose of 20 mg/kg.95 Interestingly, a variety of other potential protective activities pertaining to neurodegeneration have been reported for 40 including anti-inflammatory activity, antioxidant activity, and protection against glutamate-induced toxicity.96
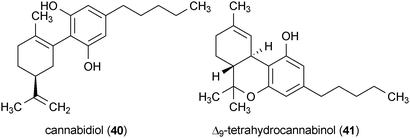
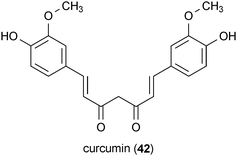
Curcumin (42) is found in Curcuma longa and has been reported to exhibit a wide range of biological activities, including several that are related to other neurodegenerative diseases.97,98 In regard to prion diseases, 42 potently inhibited prion protein accumulation in a neuroblastoma cell model (IC50 ∼10 nM).99
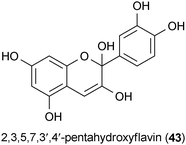
A screen of 2000 pure compounds consisting of natural products and drugs identified several substances that inhibit the accumulation of the misfolded prion protein in infected cells.100 A group of 17 compounds were obtained that exhibited IC50 values of ≤1 μM. Most of the active natural products consisted of polyphenols, such as epigallocatetchin-3-O-gallate (35), epicatechin 3-O-gallate (37), and 2,3,5,7,3′,4′-pentahydroxyflavin (43). The activities of these compounds were confirmed in both in vitro and in vivo models.100 The activity of 35 pertaining to Huntington's disease is described in section 5.4.
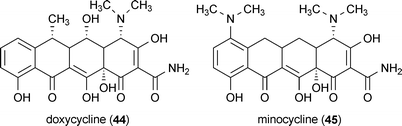
A group of tetracycline derivatives have also been reported to exhibit anti-prion activity.101 Doxycycline (44) and minocycline (45) were particularly effective, with in vitro and in vivo activity reported for both compounds. These compounds are attractive targets for therapeutic development since they are already US-FDA-approved antibiotics.101 There is an extensive body of literature available summarizing the therapeutic potential of 45 for many neurological disorders, including Alzheimer's, Parkinson's and Huntington's disease, to which the interested reader is directed for more information.102–105
7 General neurodegeneration
A substantial number of cellular dysfunctions are shared among neurons experiencing any one of the four major protein aggregation-related neurodegenerative diseases. Accordingly, many research groups have examined these non-specific disease targets in the hope of finding compounds that have broad applications to more than one neurodegenerative disorder. Several assays have been described for identifying generalized neuroprotective agents, including preventing glutamate-induced neurotoxicity,8,106,107 modulating neurotrophic activity,18,19,108,109 providing protection against inflammation,110,111 and inhibiting monoamine oxidases.112 A wide variety of secondary metabolites have emerged from these assays, and they are summarized in the following sections based on their biological targets.7.1 Glutamate-induced neurotoxicity
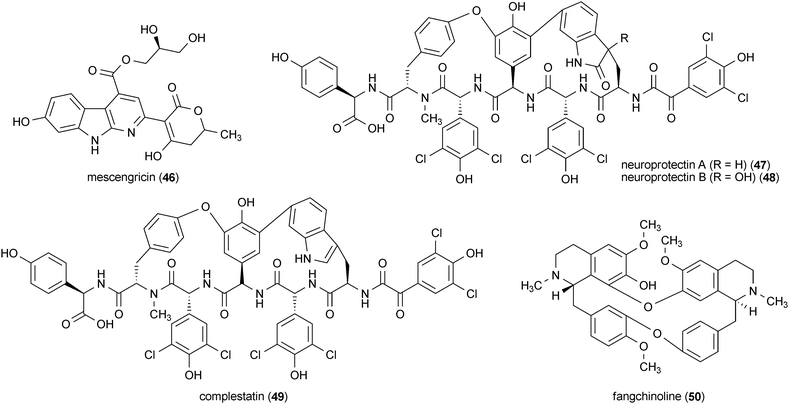
Glutamate is an important neurotransmitter; however, excessive glutamate exposure leads to neuronal cell death as a result of unbridled excitotoxicity.8 Much of the damage caused by glutamate excitoxicity is attributable to excessive Ca2+ efflux in neurons, which triggers a signaling cascade that leads to apoptosis and oxidative stress.8,107 Evidence has emerged suggesting a probable link between glutamate excitotoxicity and neuronal cell death in Alzheimer's, Parkinson's, and Huntington's diseases.106 These shared features make prevention of glutamate-induced toxicity an appealing target for preventing neurodegeneration.Mescengricin (46) was isolated from Streptomyces griseoflavus 2853-SVS4. This bacterial metabolite exhibited potent protective effects against glutamate toxicity in chick primary mescenephalic neurons (EC50 = 6 nM).113 Further experiments in the N18-RE-105 neuronal cell line demonstrated that the mechanism of protection of 46 against glutamate toxicity did not involve antioxidant activity.113
Three compounds isolated from the bacteria Streptomyces sp. Q27107, neuroprotectin A (47) and B (48) and complestatin (49), all protected cells from glutamate toxicity.114 The three compounds protected chick telencephalic neurons from 20 mM glutamate toxicity with IC50 values of ∼0.44 μM.
Fangchinoline (50) is a bis-benzylisoquinoline alkaloid from Stephania tetrandra.115 Compound 50 protected cerebellar granule cells from glutamate-induced toxicity at a concentration of 1 μM. Treatment of the cells with 50 also reduced Ca2+ influx, which is significant because disruption of Ca2+ homeostasis is an important factor contributing to glutamate toxicity in neurons.8,107
Seven iridoid glycosides, 8-O-E-p-methoxycinnamoylharpagide (51), 8-O-Z-p-methoxycinnamoylharpagide (52), 6′-O-E-p-methoxycinnamoylharpagide (53), 6′-O-Z-p-methoxycinnamoylharpagide (54), E-harpagoside (55), Z-harpagoside (56), and harpagide (57), were isolated from the roots of Scrophularia buergeriana and protected rat cortical cells against glutamate toxicity. The compounds exhibited moderate to good potency with activity noted at concentrations ranging from 0.1–10.0 μM.116

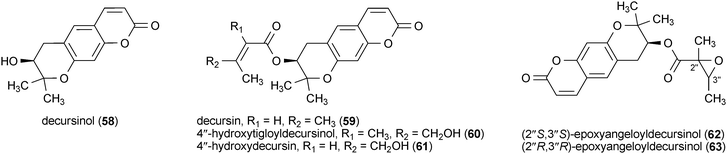
Six dihydropyranocoumarin analogues showed the ability to enhance the viability of rat cortical cells treated with a toxic concentration of glutamate (100 μM).117 Decursinol (58) and decursin (59) were isolated from Angelica decursiva, while 4′′-hydroxytigloyldecursinol (60), 4′′-hydroxydecursin (61), (2′′S,3′′S)-epoxyangeloyldecursinol (62) and (2′′R,3′′R)-epoxyangeloyldecursinol (63) were isolated from Angelica gigas. All six compounds (58–63) significantly increased viability of the glutamate-treated cells at concentrations of 0.1–1.0 μM.117
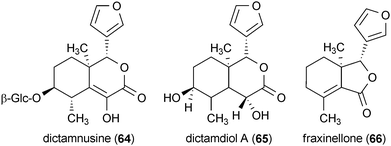
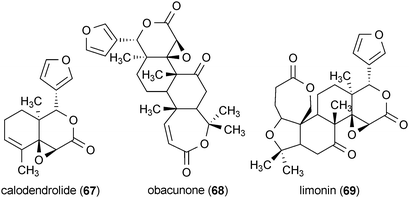
The root bark of Dictamnus dasycarpus yielded the liminoid analogues dictamnusine (64), dictamdiol A (65), fraxinellone (66), calodendrolide (67), obacunone (68), and limonin (69).118 All of the liminoids provided significant protection against glutamate-induced neurotoxicity in primary cultures of rat cortical cells. Whereas the positive control dizocilpine maleate (a noncompetitive agonist of the NMDA receptor) was active at 10.0 μM, compounds 64–69 exhibited greater potency, with activity reported at a concentration of 0.1 μM.118
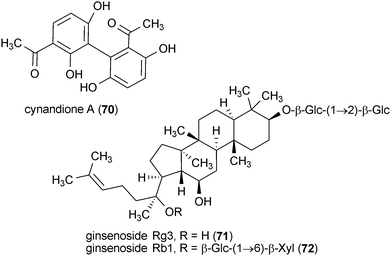
Cynandione A (70) is a metabolite isolated from the roots of Cyanchum wilfordii.119 Rat cortical cells treated with 50 μM 70 were protected against glutamate-induced toxicity. However, cells were still susceptible to excitotoxicity induced by treatment with N-methyl-D-aspartate.119
Ginsenoside Rg3 (71) and Rb1 (72) protected rat cortical cell cultures from glutamate-induced toxicity.120 Both compounds exhibited maximum protection at a concentration of 0.10 μM. Further activity of these compounds is described in more detail in section 7.2.
It is worth noting that a significant number of natural products have been reported to protect N18-RE-105 neuronal cells against glutamate toxicity.121–131 However, it is likely that this protection was achieved through an antioxidant mechanism and that these compounds do not confer any specific protection against glutamate toxicity.125,132 Therefore, the potential application of these natural products is not discussed further.
7.2 Neurotrophic activity
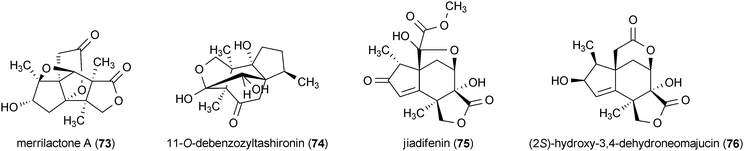
Healthy neuronal activity is promoted and maintained by neurotrophins, small proteins that participate in the regulation of several neuronal processes including survival, neurite outgrowth, and neural stem cell differentiation.109,133 Due to the array of therapeutically relevant modulatory effects exhibited by neurotrophins, small molecules that mimic or regulate the activities of these proteins have been sought for the treatment of neurodegenerative diseases,109,133,134 In particular, natural product effectors of endogenous neurotrophins such as nerve growth factor (NGF), as well as modulators of neurotrophic targets like TrkA (the cellular receptor for NGF), hold great promise as therapeutics for treating neurodegenerative conditions.135 Related to the neurotrophin-focused approach is the attempt to develop stem-cell-based therapies for neurodegenerative diseases.136 Stem-cell approaches to treating neurodegeneration could be greatly enhanced by the availability of small molecules that can effectively control and direct the differentiation of neural stem cells. Although there have been a small number of reviews concerning natural products with neurotrophic activity,18,19,108 the most promising results in this area of research, as well as highlights of more recent discoveries, are presented here.Merrilactone A137 (73) and 11-O-debenzozyltashironin138 (74) are sesquiterpenes isolated from the pericarps of Illicium merrillianum. Treatment of fetal rat cortical neurons with 73 and 74 at concentrations of 0.1 μM resulted in substantial enhancement in cellular neurite outgrowth.137,138 Two additional sesquiterpene metabolites from Illicium jiadifengpi, jiadifenin (75) and (2S)-hydroxy-3,4-dehydroneomajucin (76), are structurally very similar to 73 and 74 and exhibited comparable neurotrophic acitivity.139 Both 75 and 76 significantly enhanced neurite outgrowth in rat cortical neurons at concentrations of 0.1 μM and 0.01 μM, respectively. Neurite outgrowth induced by 75 and 76 was similar to the enhancement observed using basic fibroblast growth factor.139
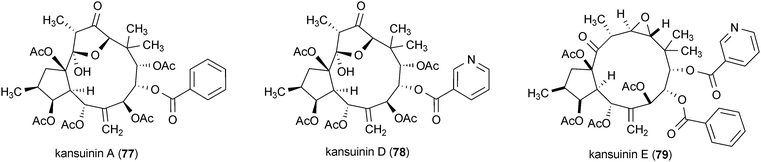
Three diterpenes, kansuinin A (77), D (78) and E (79), isolated from the roots of Euphorbia kansui, were tested for their stimulatory effects on TrkA, the cellular binding target of NGF.140 Survival of TrkA expressing fibroblasts was specifically promoted by 79 (ED50 = 0.2 μg/mL), while 77 and 78 promoted the survival of fibroblasts expressing both TrkA and TrkB (ED50 = 7.9 and 0.6 μg/mL, respectively, for TrkA expressing fibroblasts). TrkB is the receptor for brain-derived neurotrophic factor (BDNF), a neurotrophin that has a variety of important functions for cholinergic and dopaminergic neurons.133
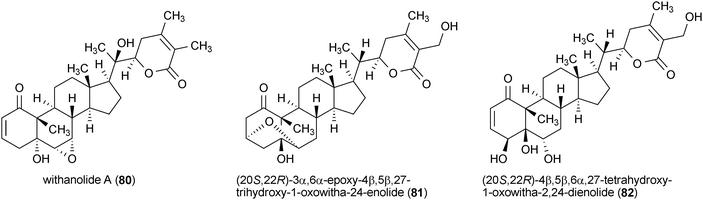
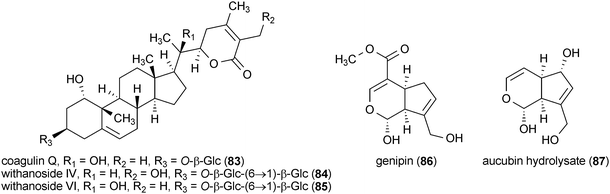
A series of withanolides were isolated from the roots of an Ayurvedic medicinal plant, Withania somnifera, that enhanced neurite outgrowth in the human neuroblastoma cell line SH-SY5Y.141 Treatment of SH-SY5Y cells with withanolide A (80), (20S,22R)-3α,6α-epoxy-4β,5β,27-trihydroxy-1-oxowitha-24-enolide (81), (20S,22R)-4β,5β,6α,27-tetrahydroxy-1-oxowitha-2,24-dienolide (82), coagulin Q (83), withanoside IV (84) and withanoside VI (85) (each at a concentration of 1 μM) resulted in significantly increased neurite outgrowth. However, the activities of these compounds were somewhat distinct from one another with 81, promoting the growth of axonal neurites, while 84 and 85 increased the growth of dendritic neurites.141
Genipin (86) and aucubin hydrolysate (87) are the products of enzymatic hydrolysis of two iridoid glycosides isolated from Gardenia jasminoides and Aucuba japonica, respectively.142 Both compounds exhibited the ability to enhance neurite outgrowth in PC12h cells at a concentration of 1 μg/mL. Interestingly, the glycoside of 87 is inactive in the neurite outgrowth assay, while the glycoside of 86 exhibited significantly less activity than the hydrolysate, indicating that removal of the glucose moiety is important for conveying activity.142
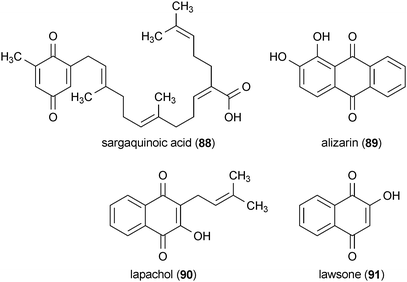
Several quinone-containing compounds promoted NGF-mediated neurite outgrowth in PC12D cells.143 Sargaquinoic acid (88), isolated from Sargassum spp., and alizarin (89), isolated from Rubia tinctorum, were the most potent of these compounds. Both significantly increased the number of cells with neurites at concentrations of 6.25 μg/mL and 12.5 μg/mL, respectively. The plant metabolites lapachol (90), from Tabebuia avellandedae, and lawsone (91), from Lawsonia inermis, also enhanced neurite outgrowth, but at much higher concentrations of 50 μg/mL and 100 μg/mL, respectively.143
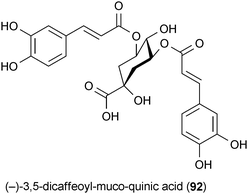
Aster scaber is traditionally used in Korea for the treatment of bruises, headaches and dizziness.144 The quinic acid metabolite (−)-3,5-dicaffeoyl-muco-quinic acid (92) isolated from this plant has been shown to enhance neurite outgrowth in PC12 cells at a concentration of 5 μM. Experimental evidence indicates that 92 activates a cascade of signaling pathways similar to NGF, which suggests that this secondary metabolite specifically activates TrkA.144
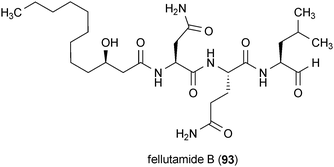
A compound isolated from the fungus Penicillium fellutanum, fellutamide B (93), has been reported to possess unique neurotrophic acitivty.145 Treatment of glioblastoma cells with 93 at a concentration of 10 μM resulted in a large increase in the production and secretion of NGF. The mechanism of action of 93 has been shown to be through inhibition of the proteasome, which represents a novel target in the search for neurotrophic compounds.145
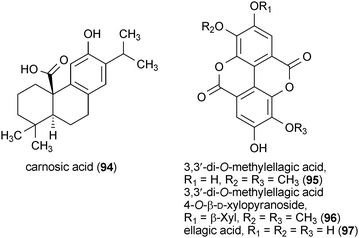
Carnosic acid (94) is a diterpene from rosemary (Rosmarinus officinalis) that has been identified as having neurotrophic activity.146 Treatment of PC12h cells with 94 at a concentration of 15 μM resulted in enhanced neurite outgrowth comparable to the outgrowth observed upon treatment of cells with NGF. Gene transcription analysis indicated that 94 activated the differentiation of the PC12h cells via a mechanism that is distinct from the activity of NGF.146
Both 3,3′-di-O-methylellagic acid (95) and 3,3′-di-O-methylellagic acid 4-O-β-D-xylopyranoside (96) were isolated from the African tree Terminalia superba.147 Treatment of neural stem cells with 95 and 96 at a concentration of 10 μM for 24 h followed by treatment at a concentration of 1 μM for an additional 72 h resulted in a significant increase in the number of differentiated neuronal cells with no apparent toxicity. Interestingly, the related compound ellagic acid (97), which differs primarily from 95 and 96 by the presence of hydroxyl groups rather than methoxy groups at the C-3 and C-3′ positions, was less active and somewhat toxic to the neural stem cells.147
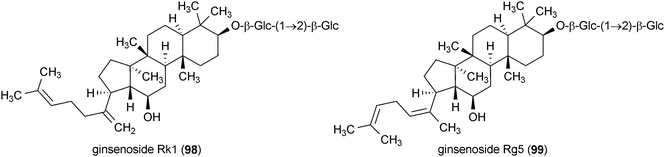
Ginsenoside Rg3 (71), Rk1 (98) and Rg5 (99), isolated from the roots of Panax sanchi-ginseng, enhanced the differentiation of neural stem cells.148 The most active of the three compounds was 99. Neural stem cell differentiation required transient exposure of the cells to 8 μM 99 for 24 h followed by 72 h incubation with the same compound at 1 μM.148 This treatment also led to the increased proliferation of neurons in the culture. Ginsenoside Rb1 (72), ginsenoside Rb3 (100), notoginsenoside R4 (101) and notoginsenoside Fa (102) are saponins isolated from Panax japonicus.149 All four compounds enhanced neurite outgrowth in SK–N–SH cells when applied to the cells at concentrations of 100 μM each. Immunostaining experiments showed that 72 and 100–102 promoted the extension of both axonal and dendritic neurites.149

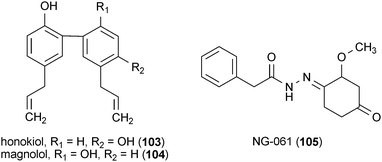
The neolignans honokiol (103) and magnolol (104), found in the plant Magnolia obovata, enhanced neurite outgrowth in rat cortical neurons when applied to cells at a concentration of 10 μM.150 It was observed that compound 103 was much more potent than 104. Neurites in cells treated with 103 were almost 50% longer than those found in cells treated with 104. Additionally, 103 also promoted the survival of neurons in cell cultures, resulting in a 40% increase in cell viability.150
The fungal metabolite NG-061 (105), isolated from Penicillium minioluteum, increased neurite outgrowth in PC12 cells through an NGF-independent mechanism.151 Treatment of cells with 10 μg/mL of 105 substantially enhanced neurite outgrowth compared to NGF-positive controls. A combination treatment consisting of both 105 and NGF resulted in an even greater neurite growth enhancement.151
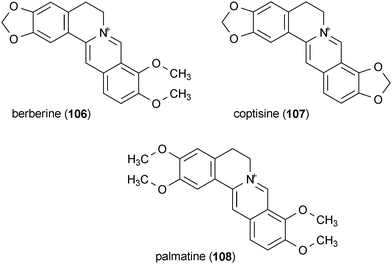
A series of alkaloids were isolated from the rhizome of Coptis chinensis which potentiated neurite outgrowth in PC12 cells by NGF.152 Berberine (106), coptisine (107) and palmatine (108) were active at concentrations of 5 μg/mL, 5 μg/mL and 25 μg/mL, respectively, when applied to cells in combination with 10 ng/mL NGF.
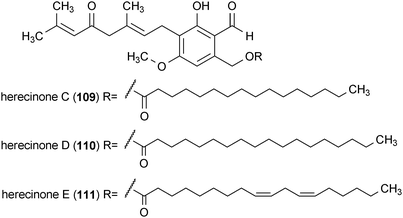
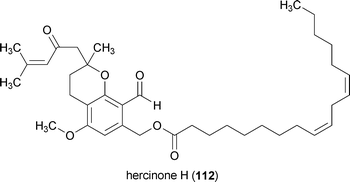
Hercinone C (109), D (110), E (111) and H (112) are neurotrophic metabolites from the mushroom Hericium erinaceum.153,154 All four compounds stimulated the secretion of NGF into the extracellular medium by mouse astroglial cells when applied at a concentration of 33 μg/mL. The amount of NGF which these compounds caused to be secreted by the astroglial cells decreases in the order 112 > 110 > 111 > 109.153,154
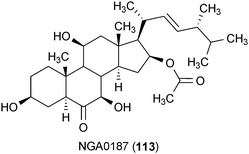
The fungal metabolite NGA0187 (113) was isolated from Acremonium sp. TF-0356.155 This compound exhibited neurotrophic activity through an enhancement of neurite outgrowth in PC12 cells at a concentration of 30 μg/mL. The activity of 113 that was observed at this concentration was equivalent to the positive control, NGF.155
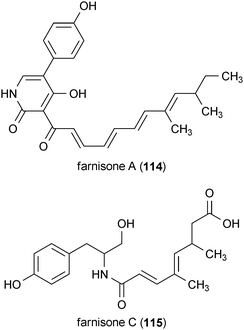
Farnisone A (114) and C (115), isolated from the fungus Paecylomyces farinosus RCEF 0101, enhanced neurite outgrowth in PC12 cells at concentrations of 20 and 50 μM, respectively.156 A third compound, farnisone B, did not enhance neurite growth, even though it only differed from 114 by the presence of a hydroxylamine in the pyridone ring.156 Although both 114 and 115 induced neurite growth independent of NGF, neither compound was as effective as NGF by itself.156
A metabolite isolated from coffee beans, trigonelline (116), exhibited neurotrophic activity in SK-N-SH human neuroblastoma cells.157 Treatment of the cells with 116 at a concentration of 30 μM resulted in a significant increase in the number of cells with neurites. Cells treated with 116 exhibited positive staining for the axonal marker phosphorylated NF-H, indicating that the compound induced the growth of axons from neuroblastoma cells.157
Two iridoid metabolites isolated from the plant Picrorhiza scrophulariiflora, picroside I (117) and II (118), potentiated the neurotrophic effects of NGF in PC12D cells.158 Neither 117 nor 118 were able to enhance neurite outgrowth alone, but when applied to the cells at a concentration of 60 μM in combination with NGF (2 ng/mL), neurite outgrowth was substantially increased compared to the positive control alone (NGF, 30 ng/mL). The potentiating neurotrophic activity of 117 and 118 was observed to act in a dose-dependent manner.158
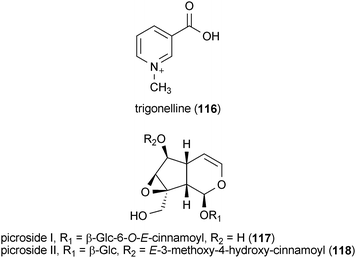
Nardosinone (119) is a plant metabolite isolated from Nardostachys chinensis.159 Similar to 117 and 118, 119 did not exhibit any neurotrophic activity by itself. However, when applied to PC12D cells at a concentration of 30 μM in combination with NGF (2 ng/mL), 119 significantly increased the number of neurite-bearing cells and it was found to be more effective than treatment with NGF (30 ng/mL) alone.159
Neurotrophic activity has been reported for panaxytriol (120), which was isolated from Panax ginseng.160 Treatment of PC12 cells with 120 at a concentration of 60 μM in the presence of NGF (50 ng/mL) enhanced neurite outgrowth.160 However, due to the unusually high concentration of NGF used in this study and the lack of quantitative analysis of the neurite growth, the significance of these results remains uncertain.
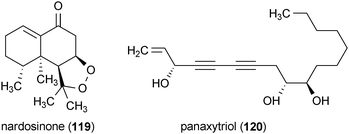
A series of sesterterpenes were reported from a marine sponge, Spongia sp., with neurotrophic activity.161 Scalarolide acetate (121), 12-epi-scalarin (122), 12-O-deacetyl-12-epi-scalarin (123), and two additional scalarane sesterterpenes, (124) and (125), all enhanced the outgrowth of neurites in PC12 cells when applied to the cells at a concentration of 50 μg/mL. The activity of these compounds decreased in the order 121 > 124 > 122 > 123 > 125.161
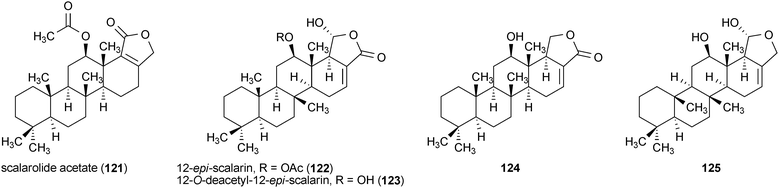
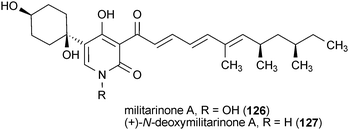
The militarinones are neurotrophic alkaloids from the fungus Paecilomyces militaris RCEF 0095.162–164 Militarinone A (126) exhibited the most pronounced activity of the four metabolites, as well as no observable toxicity, resulting in a significant enhancement of neurite outgrowth in PC12 cells at a concentration of 33 μM. The dehydroxy analog of 126, (+)-N-deoxymilitarinone (127), showed greatly reduced potency, requiring a dose of 100 μM to elicit a similar effect. In contrast, treatment of human neurons (IMR-32) with 127 at 100 μM resulted in cell toxicity.162,164 The effects of militarinones B (128) and C (129) on PC12 cells were rather modest, with doses of 100 μM resulting in only marginal enhancement of neurite outgrowth.163
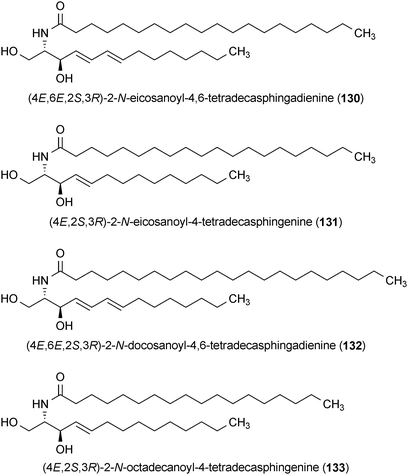
A series of sphingolipids were isolated from Bombycis Corpus 101A, which is made from infecting Bombyx mori (silk moth) larvae with the fungus Beauveria bassiana 101A.165 The compounds, (4E,6E,2S,3R)-2-N-eicosanoyl-4,6-tetradecasphingadienine (130), (4E,2S,3R)-2-N-eicosanoyl-4-tetradecasphingenine (131), (4E,6E,2S,3R)-2-N-docosanoyl-4,6-tetradecasphingadienine (132) and (4E,2S,3R)-2-N-octadecanoyl-4-tetradecasphingenine (133) obtained from this mixture potentiated the effects of nerve growth factor (NGF), leading to the enhancement of PC12 cell neurite outgrowth at concentrations of 10 μM.165 Compounds 130 and 133 were the most active and 131 and 133, which lack the C-6 double bond present in the former compounds, were much less active.165
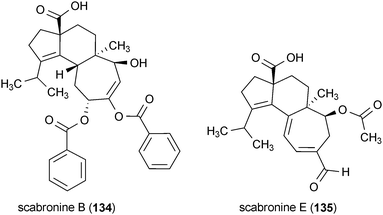
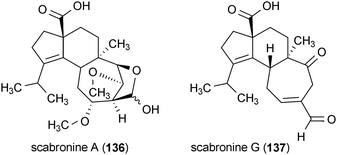
Four diterpenes from the fruiting bodies of mushrooms were found to stimulate the production of NGF in rat astroglial cells.166,167 Scabronine B (134) and E (135) were isolated from Hericium erinaceum, while scabronine A (136) and G (137) were obtained from Sarcodon scabrosus. Treatment of rat astroglial cells with 134 and 135 at concentrations of 60 μM and 90 μM, respectively, increased the production and secretion of NGF into the extracellular medium. Both 136 and 137 exhibit similar activity; however, they required a slightly higher dose (100 μM) to elicit these effects.166,167
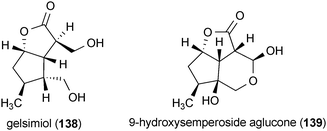
Two compounds isolated from Verbena littoralis, gelsimiol (138) and 9-hydroxysemperoside aglucone (139), potentiated the neurotrophic effects of NGF in PC12D cells when applied at concentrations of 100 μM.168 In the presence of NGF (2 ng/mL), 138 and 139 increased the number of neurite-bearing cells by more than 50% and increased the length of neurites by more than 100% compared to treatment with NGF (30 ng/mL) alone.168
7.3 Anti-inflammatory activity
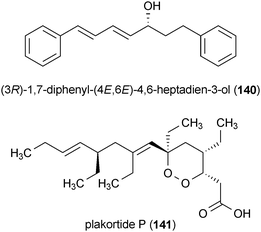
Chronic inflammation is associated with a variety of neurodegenerative diseases and it has been shown to be a contributing factor to neuronal cell death.1 Activated microglia are one of the prime participants in neuroinflammation.110 These immune system cells are thought to contribute to neuronal death through production of reactive nitrogen species (e.g., nitric oxide) and reactive oxygen species (e.g., hydrogen peroxide).169 Combating these inflammatory processes has been proposed as an important means for blocking the development and progression of neurodegenerative diseases, and a selected group of anti-inflammatory lead compounds have already entered clinical trials.111The diarylheptanoid (3R)-1,7-diphenyl-(4E,6E)-4,6-heptadien-3-ol (140), isolated from the plant Curcuma comosa, has been shown to significantly reduce the expression of inducible nitric oxide synthase (iNOS) and cyclooxygenase 2 (COX-2) in lipopolysaccharide (LPS) treated HAPI microglial cells at a concentration of 1 μM.170 Compound 140 also decreased the production of nitric oxide (NO) and prostaglandin E2 (PGE2), which are the respective products of iNOS and COX-2.
Plakortide P (141), isolated from the marine sponge Plakortis angulospiculatus, reduced the amount of thromboxane B2 (TXB2), a biochemical marker of the inflammatory response, released by LPS treated rat microglia (IC50 = 0.93 μM).171 Moreover, no cytotoxicity was observed from this metabolite in a variety of mammalian cell lines.
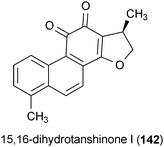
The plant metabolite 15,16-dihydrotanshinone I (142), isolated from Salvia miltiorrhiza, exhibited anti-inflammatory activity in LPS-activated microglia.172 Treatment of activated microglia with 142 at a concentration of 5 μM significantly reduced the expression of several inflammatory response biomarkers including iNOS, interleukin-1B, tumor necrosis factor-A, and TNF-A converting enzyme.
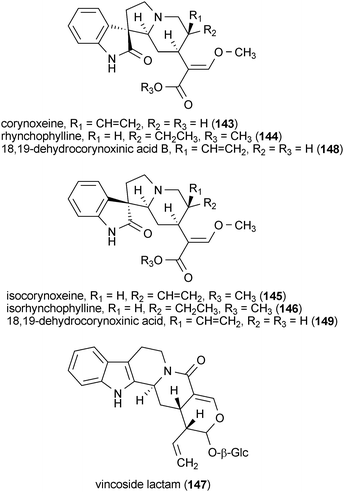
Four oxindole alkaloids and one indole alkaloid glycoside reduced the amount of NO released by LPS-treated rat microglial cells.173 Corynoxeine (143), rhynchophylline (144), isocorynoxeine (145), isorhynchophylline (146) and vincoside lactam (147), isolated from the leaves of Uncaria rhynchophylla, were active with IC50 values of 15.7, 18.5, 13.7, 19.0 and 16.4 μM, respectively. These values compare favorably with the activity of resveratrol (IC50 = 11.5 μM) in the same assay. Interestingly, two other oxindole alkaloids, 18,19-dehydrocorynoxinic acid B (148) and 18,19-dehydrocorynoxinic acid (149), were not active even though the only structural difference between them and compounds 143 and 145 was the configuration of the C-20 stereocenter and the presence of a carboxylic acid in 148 and 149 rather than the methyl ester that was present in 143 and 145.173
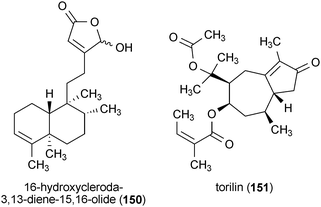
16-Hydroxycleroda-3,13-diene-15,16-olide (150), a diterpene from the bark of Polyalthia longifolia var. pendula, significantly reduced the inflammatory response of LPS-activated microglia when applied to the cells at a concentration of 10 μM.174 The expression of iNOS, COX-2 and other markers of the inflammatory response was also reduced. Additionally, 150 enhanced neuronal survival in neuronal cells (SH-SY5Y) co-cultured with microglia-like HAPI cells, indicating its therapeutic potential for protecting neurons from toxicity associated with the inflammatory response of microglia.174
A sesquiterpene from the stem and root bark of Ulmus davidiana var. japonica, torilin (151), was reported to inhibit the inflammatory enzymes iNOS, COX-2, and interleukin-1β in LPS-activated microglia when applied to cells at a concentration of 30 μM.175 Compound 151 also reduced the release of the products of iNOS and COX-2 (NO and PGE2, respectively) and interleukin-1β into the culture medium.
Several of the compounds described in previous sections of this review have been demonstrated to possess anti-inflammatory activity as well. Nanolobatolide (10) demonstrated significant anti-inflammatory activity and was reported to reduce the expression of iNOS in activated microglia when applied at a concentration of 10 μM.43 Manzamine A (20), B (152), C (153) and D (154) exhibited low micromolar anti-inflammatory activity, but manzamine E (23) and manzamine F (24) did not.176 Treatment of activated microglia with celastrol (29) at a concentration of 100 nM significantly inhibited the production of NO, TNF-α and IL-1β.177
7.4 Monoamine oxidase inhibitors
Monoamine oxidases (MAO) are enzymes that deaminate neurotransmitters.112 Both the MAOA and B isoforms have been implicated as targets for treating neurodegenerative disease.112 Recent evidence suggests that inhibition of both MAO A and B can confer neuroprotection through antiapoptotic mechanisms, reduction of reactive oxygen species production, and stabilization of the mitochondrial membrane.178 MAO inhibitors have a long history of use for the treatment of Parkinson's disease, but they have more recently demonstrated potential as therapeutics for a broader range of neurodegenerative diseases (i.e., Alzheimer's and Huntington's disease).112
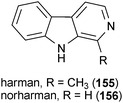
Two β-carbolines were isolated from brewed coffee that inhibit MAOA and B.179 Harman (155) selectively inhibited MAOA (IC50 = 0.34 μM) in vitro while norharman (156) was active against both MAOA (IC50 = 6.50 μM) and B (IC50 = 4.70 μM) in vitro. The related compounds harmine (17) and harmaline (18) from Banisteriopsis caapi also inhibited MAO with IC50 values of 12.6 nM and 15.8 nM, respectively.180 Further experiments showed that 17 is a selective inhibitor MAOA (IC50 = 4.54 nM), but not MAOB.47
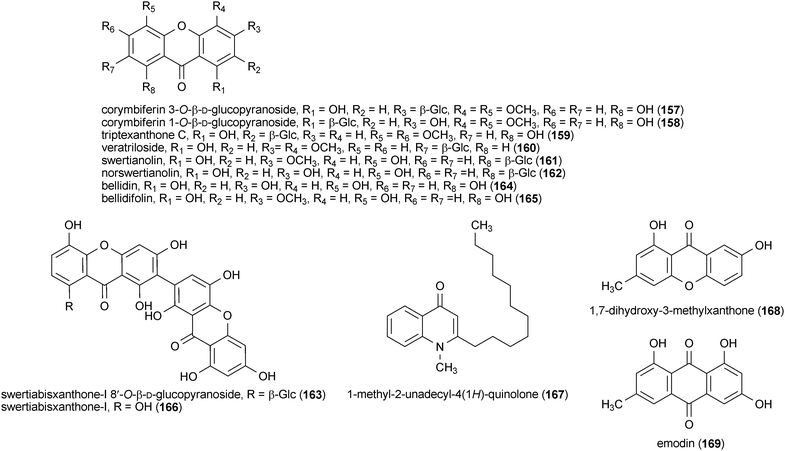
Seven xanthone glycosides, corymbiferin 3-O-β-D-glucopyranoside (157), corymbiferin 1-O-β-D-glucopyranoside (158), triptexanthone C (159), veratriloside (160), swertianolin (161), norswertianolin (162) and swertiabisxanthone-I 8′-O-β-D-glucopyranoside (163), and three xanthone aglycones, bellidin (164), bellidifolin (165) and swertiabisxanthone-I (166), were isolated from Gentianella amarella ssp. acuta and examined for their inhibitory activity against MAOA and B.181 Compounds 164 and 165 show the greatest inhibition of MAOA (91% and 99%, respectively) when tested at a concentration of 10 μM, and these values were greater than that of the positive control pargyline (60% at a concentration of 10 μM), a known inhibitor of MAOA and B. All ten compounds inhibited MAOB at a concentration of 10 μM, but 158 and 161 show the greatest activity (94% and 71%, respectively).181
The fruits of Evodia rutaecarpa yielded the metabolite 1-methyl-2-unadecyl-4(1H)-quinolone (167).182 This compound selectively inhibited MAOB with an IC50 value of 15.3 μM, but was inactive against MAOA. Determination of the kinetic pattern of MAOB inhibition by 152 show that it is an irreversible inhibitor of MAOB.182
A xanthone, 1,7-dihydroxy-3-methylxanthone (168), and the anthraquinone emodin (169) were isolated from the fungus Anixiella micropertusa.183 Both 168 and 169 inhibit MAO activity in vitro with IC50 values of 20.6 μM and 37.0 μM, respectively.
Lemuninol A (170) is a naphthoquinone–naphthalene dimer isolated from Lemuni Hitam (a Malaysian herbal medicine containing the heartwood of Diospyros species).184 MAO was inhibited 62% by 170 at a concentration of 25 μM.
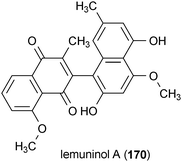
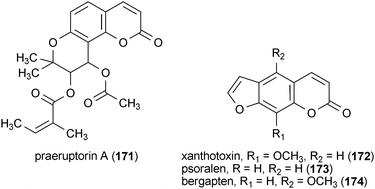
Four coumarin analogues from the roots of Peucedanum japonicum, praeruptorin A (171), xanthotoxin (172), psoralen (173) and bergapten (174), exhibited in vitro MAO inhibitory activity.185 The most potent of these compounds was 174 (IC50 = 13.8 μM), followed by 171 (IC50 = 27.4 μM), 173 (IC50 = 35.8 μM), and 172 (IC50 = 40.7 μM).
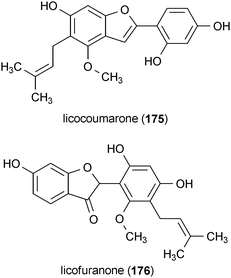
Licocoumarone (175) and licofuranone (176) are two benzofurans isolated from licorice (Glycrrhiza spp.).186 These compounds demonstrated modest potency against MAO activity, with IC50 values of 60 μM (175) and 87 μM (176).
Coptisine (107), described in section 7.2, selectively inhibited MAOA with an IC50 value of 1.8 μM but did not inhibit MAOB.187 This compound was significantly more potent than the structurally similar berberine (106) and palmatine (108), which have been reported to inhibit MAOA with IC50 values of 98.2 μM and 90.6 μM, respectively.187
8 Summary and conclusions
Currently there are no effective therapies for treating Alzheimer's, Huntington's, Parkinson's and prion diseases. However, given the large number of promising secondary metabolites discussed in this review, it is evident that natural products are well-suited to serve as a leading resource for the discovery of unique drug leads. We anticipate that screening programs focused on testing natural products have a considerable likelihood of uncovering new molecules that are capable of modulating targets related to neurodegenerative disease in novel ways that are certain to rapidly advance the drug discovery process.Our analysis of the natural products literature suggests that many sources of secondary metabolites such as bacteria, fungi, and marine organisms remain under-utilized as resources for discovering new neurodegenerative disease drug leads. We have also observed that natural products are highly compatible with currently accepted chemical features that are desirable for CNS-active drugs. Further enhancements of lead pharmacophores driven by medicinal chemistry will serve to optimize drug leads and provide a wealth of important scaffolds upon which to design a new generation of therapeutics for neurodegenerative diseases.
The neurodegeneration in Alzheimer's, Huntington's, Parkinson's and prion diseases is linked to misfolded proteins. Although the exact mechanism of the toxicity associated with these aggregation-prone proteins is yet to be determined, inflammation, apoptosis and oxidative stress are among the cellular abnormalities that have been linked to these diseases. The many pathological features that are shared among these diseases provides a strong rationale for the search for natural product leads which modulate targets applicable to many neurodegenerative diseases, including compounds that reduce glutamate-induced toxicity, neurotrophic compounds, anti-inflammatory compounds and inhibitors of monoamine oxidases. In addition to these general targets, there are also a small number of druggable targets that have been identified for specific diseases such as caspase and Gsk3 inhibitors for Huntington's disease and inhibitors of prion propagation and amyloidogenesis for prion diseases. The combination of the attractive chemical properties of natural product leads with the variety of druggable targets that have been identified for neurodegeneration associated with protein misfolding makes it clear that many opportunities remain for new discoveries in the area of natural products relevant to neurodegenerative diseases. Considering that millions of people currently suffer from these diseases, the work summarized here provides hope that new, effective therapies for curing Alzheimer's, Huntington's, Parkinson's and prion diseases should be within our grasp.
9 Acknowledgements
This work was made possible in part by a grant from the National Institutes of Health (1R21NS064313-01A1).10 References
- S. Amor, F. Puentes, D. Baker and P. van der Valk, Immunology, 2010, 129, 154–169 CrossRef CAS.
- C. G. Glabe, Neurobiol. Aging, 2006, 27, 570–575 CrossRef CAS.
- D. M. Skovronsky, V. M. Lee and J. Q. Trojanowski, Annu. Rev. Pathol.: Mech. Dis., 2006, 1, 151–170 Search PubMed.
- R. Scatena, G. E. Martorana, P. Bottoni, G. Botta, P. Pastore and B. Giardina, Expert Opin. Invest. Drugs, 2007, 16, 59–72 Search PubMed.
- M. S. Butler, J. Nat. Prod., 2004, 67, 2141–2153 CrossRef CAS.
- D. J. Newman, J. Med. Chem., 2008, 51, 2589–2599 CrossRef CAS.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2007, 70, 461–477 CrossRef CAS.
- X.-x. Dong, Y. Wang and Z.-h. Qin, Acta Pharmacol. Sin., 2009, 30, 379–387 Search PubMed.
- M. Heinrich and H. Lee Teoh, J. Ethnopharmacol., 2004, 92, 147–162 CrossRef CAS.
- P. J. Houghton and M. J. Howes, Neurosignals, 2005, 14, 6–22 CrossRef CAS.
- J. A. Clement, B. J. Yoder and D. G. I. Kingston, Mini-Rev. Org. Chem., 2004, 1, 183–208 Search PubMed.
- N. G. Gomes, M. G. Campos, J. M. Orfao and C. A. Ribeiro, Progress Neuro-Psychopharmacol. Biol. Psychiatry, 2009, 33, 1372–1389 Search PubMed.
- C. Viegas Jr., V. d. S. Bolzani, E. J. Barreiro and C. A. M. Fraga, Mini-Rev. Med. Chem., 2005, 5, 915–926 CrossRef.
- J. M. Barbosa Filho, K. C. P. Medeiros, M. d. F. F. M. Diniz, L. M. Batista, P. F. Athayde-Filho, M. S. Silva, E. V. L. d. Cunha, J. R. G. S. Almeida and L. J. Quintans-Júnior, Rev. Bras. Farmacogn., 2006, 16, 258–285 Search PubMed.
- M. Jung and M. Park, Molecules, 2007, 12, 2130–2139 Search PubMed.
- M. R. Loizzo, R. Tundis and F. Menichini, Curr. Med. Chem., 2008, 15, 1209–1228 CrossRef CAS.
- G. Orhan, I. Orhan, N. Subutay-Oztekin, F. Ak and B. Sener, Recent Pat. CNS Drug Discovery, 2009, 4, 43–51 Search PubMed.
- Y. Ohizumi, T. Yamakuni and Y. Li, J. Appl. Pharmacol., 2004, 12, 195–197 Search PubMed.
- C. Tohda, T. Kuboyama and K. Komatsu, Neurosignals, 2005, 14, 34–45 CrossRef CAS.
- S. Bastianetto, J. Brouillette and R. Quirion, Neurochem. Res., 2007, 32, 1720–1725 CrossRef CAS.
- J. G. Frade, N. R. Ferreira, R. M. Barbosa and J. Laranjinha, Curr. Med. Chem.: Cent. Nerv. Syst. Agents, 2005, 5, 307–318 Search PubMed.
- S. Mandel and M. B. H. Youdim, Free Radical Biol. Med., 2004, 37, 304–317 CrossRef CAS.
- C. Ramassamy, Eur. J. Pharmacol., 2006, 545, 51–64 CrossRef CAS.
- O. Weinreb, T. Amit, S. Mandel and M. Youdim, Genes Nutr., 2009, 4, 283–296 Search PubMed.
- K. Aquilano, S. Baldelli, G. Rotilio and M. R. Ciriolo, Neurochem. Res., 2008, 33, 2416–2426 CrossRef CAS.
- A. K. Patel, J. T. Rogers and X. Huang, Int. J. Clin. Exp. Med., 2008, 1, 181–191 Search PubMed.
- K. Vafeiadou, D. Vauzour and J. P. E. Spencer, Endocr., Metab. Immune Disord.: Drug Targets, 2007, 7, 211–224 Search PubMed.
- T. T. Wager, X. Hou, P. R. Verhoest and A. Villalobos, ACS Chem. Neurosci., 2010, 1, 435–449 Search PubMed.
- M. Pallas and A. Camins, Curr. Pharm. Des., 2006, 12, 4389–4408 CrossRef CAS.
- H. W. Querfurth and F. M. LaFerla, N. Engl. J. Med., 362, pp. 329–344 Search PubMed.
- A. J. Lees, J. Hardy and T. Revesz, Lancet, 2009, 373, 2055–2066 CrossRef CAS.
- P. Brundin, L. Jia-Yi, J. L. Holton, O. Lindvall and T. Revesz, Nat. Rev. Neurosci., 2008, 9, 741–745 CrossRef CAS.
- V. N. Uversky, J. Neurochem., 2007, 103, 17–37 CAS.
- H. Allain, D. Bentué-Ferrer and Y. Akwa, Prog. Neurobiol., 2008, 84, 25–39 CrossRef CAS.
- A. H. V. Schapira, E. Bezard, J. Brotchie, F. Calon, G. L. Collingridge, B. Ferger, B. Hengerer, E. Hirsch, P. Jenner, N. Le Novere, J. A. Obeso, M. A. Schwarzschild, U. Spampinato and G. Davidai, Nat. Rev. Drug Discovery, 2006, 5, 845–854 CrossRef CAS.
- P. Jenner, Ann. Neurol., 2008, 64, S16–S29 CAS.
- N. Simola, M. Morelli and A. Carta, Neurotoxic. Res., 2007, 11, 151–167 Search PubMed.
- C. Colosimo, G. Fabbrini and A. Berardelli, Nat. Clin. Pract. Neurol., 2006, 2, 600–610 Search PubMed.
- K. Y. Lee, S. H. Sung and Y. C. Kim, J. Nat. Prod., 2006, 69, 679–681 CrossRef CAS.
- D. Z. Liu, J. Zhu, D. Z. Jin, L. M. Zhang, X. Q. Ji, Y. Ye, C. P. Tang and X. Z. Zhu, J. Ethnopharmacol., 2007, 112, 327–332 CrossRef CAS.
- K. Gorter, Arch. Pharm., 1897, 235, 321–332 Search PubMed.
- J. A. Abin-Carriquiry, G. Costa, J. Urbanavicius, B. K. Cassels, M. Rebolledo-Fuentes, S. Wonnacott and F. Dajas, Eur. J. Pharmacol., 2008, 589, 80–84 CrossRef CAS.
- Y.-J. Tseng, Z.-H. Wen, C.-F. Dai, M. Y. Chiang and J.-H. Sheu, Org. Lett., 2009, 11, 5030–5032 CrossRef CAS.
- S. H. Sung, E. S. Kim, K. Y. Lee, M. K. Lee and Y. C. Kim, Planta Med., 2006, 72, 62–64 CrossRef CAS.
- D.-L. Zhao, L.-B. Zou, S. Lin, J.-G. Shi and H.-B. Zhu, Neuropharmacology, 2007, 53, 724–732 CrossRef CAS.
- A. Nitta, R. Murai, M. Keiko and S. Furukawa, Adv. Behav. Biol., 2002, 51, 463–467 Search PubMed.
- M. J. Schwarz, P. J. Houghton, S. Rose, P. Jenner and A. D. Lees, Pharmacol., Biochem. Behav., 2003, 75, 627–633 CrossRef CAS.
- S. Imarisio, J. Carmichael, V. Korolchuk, C.-W. Chen, S. Saiki, C. Rose, G. Krishna, J. E. Davies, E. Ttofi, B. R. Underwood and D. C. Rubinsztein, Biochem. J., 2008, 412, 191–209 CrossRef CAS.
- F. O. Walker, Lancet, 2007, 369, 218–228 CrossRef CAS.
- D. R. Langbehn, M. R. Hayden and J. S. Paulsen, Am. J. Med. Genet. B: Neuropsychiatr. Genet., 2010, 153B, 397–408 Search PubMed.
- W. Fecke, M. Gianfriddo, G. Gaviraghi, G. C. Terstappen and F. Heitz, Drug Discovery Today, 2009, 14, 453–464 CrossRef CAS.
- G. N. Bijur, P. De Sarno and R. S. Jope, J. Biol. Chem., 2000, 275, 7583–7590 CrossRef CAS.
- T. Takadera, R. Yoshikawa and T. Ohyashiki, Neurosci. Lett., 2006, 408, 124–128 CrossRef CAS.
- J. Carmichael, K. L. Sugars, Y. P. Bao and D. C. Rubinsztein, J. Biol. Chem., 2002, 277, 33791–33798 CrossRef CAS.
- L. Meijer, A. M. Thunnissen, A. W. White, M. Garnier, M. Nikolic, L. H. Tsai, J. Walter, K. E. Cleverley, P. C. Salinas, Y. Z. Wu, J. Biernat, E. M. Mandelkow, S. H. Kim and G. R. Pettit, Chem. Biol., 2000, 7, 51–63 CrossRef CAS.
- M. Hamann, D. Alonso, E. Martin-Aparicio, A. Fuertes, M. J. Perez-Puerto, A. Castro, S. Morales, M. L. Navarro, M. Del Monte-Millan, M. Medina, H. Pennaka, A. Balaiah, J. Peng, J. Cook, S. Wahyuono and A. Martinez, J. Nat. Prod., 2007, 70, 1397–1405 CrossRef CAS.
- K. A. El Sayed, M. Kelly, U. A. K. Kara, K. K. H. Ang, I. Katsuyama, D. C. Dunbar, A. A. Khan and M. T. Hamann, J. Am. Chem. Soc., 2001, 123, 1804–1808 CrossRef CAS.
- J. Peng, S. Kudrimoti, S. Prasanna, S. Odde, R. J. Doerksen, H. K. Pennaka, Y.-M. Choo, K. V. Rao, B. L. Tekwani, V. Madgula, S. I. Khan, B. Wang, A. M. S. Mayer, M. R. Jacob, L. C. Tu, J. r. Gertsch and M. T. Hamann, J. Med. Chem., 2010, 53, 61–76 CrossRef CAS.
- L. R. Pattison, M. R. Kotter, D. Fraga and R. M. Bonelli, J. Neurol., 2006, 253, 1137–1142 CrossRef CAS.
- S. Cornelis, K. Kersse, N. Festjens, M. Lamkanfi and P. Vandenabeele, Curr. Pharm. Des., 2007, 13, 367–385 CrossRef CAS.
- S. P. Gunasekera, P. J. McCarthy, R. E. Longley, S. A. Pomponi, A. E. Wright, E. Lobkovsky and J. Clardy, J. Nat. Prod., 1999, 62, 173–175 CrossRef CAS.
- S. Sarkar, B. Ravikumar, R. A. Floto and D. C. Rubinsztein, Cell Death Differ., 2009, 16, 46–56 CrossRef CAS.
- J. Davies, S. Sarkar and D. Rubinsztein, BMC Biochem., 2007, 8, S2–S2 CrossRef.
- B. Ravikumar and D. C. Rubinsztein, Mol. Aspects Med., 2006, 27, 520–527 CrossRef CAS.
- E. Roze, F. Saudou and J. Caboche, Curr. Opin. Neurol., 2008, 21, 497–503 CrossRef CAS.
- S. N. Sehgal, H. Baker and C. Vezina, J. Antibiot., 1975, 28, 727–732 CAS.
- E. I. Graziani, Nat. Prod. Rep., 2009, 26, 602–609 RSC.
- C. B. Kang, Y. Hong, S. Dhe-Paganon and H. S. Yoon, Neurosignals, 2008, 16, 318–325 CrossRef CAS.
- S. Sarkar, G. Krishna, S. Imarisio, S. Saiki, C. J. O'Kane and D. C. Rubinsztein, Hum. Mol. Genet., 2008, 17, 170–178 CAS.
- Z. Berger, B. Ravikumar, F. M. Menzies, L. G. Oroz, B. R. Underwood, M. N. Pangalos, I. Schmitt, U. Wullner, B. O. Evert, C. J. O'Kane and D. C. Rubinsztein, Hum. Mol. Genet., 2006, 15, 433–442 CAS.
- J. Wang, S. Gines, M. MacDonald and J. Gusella, BMC Neurosci., 2005, 6, 1 CrossRef.
- A. Kuroda, H. Hatanaka, T. Kino, T. Goto, H. Tanaka, S. Tada and T. Taga, Tennen Yuki Kagobutsu Toronkai Koen Yoshishu, 1986, 28, 97–104 Search PubMed.
- S. Ditzler, J. Stoeck, M. LeBlanc, C. Kooperberg, S. Hansen, L. Coppin and J. M. Olson, Preclinica, 2003, 1, 115–126 Search PubMed.
- D. Hernández-Espinosa and A. Jennifer Morton, Brain Res. Bull., 2006, 69, 669–679 CrossRef CAS.
- J. Pineda, R. Pardo, D. Zala, H. Yu, S. Humbert and F. Saudou, Mol. Brain, 2009, 2, 33 Search PubMed.
- H. I. Rocha-González, M. Ambriz-Tututi and V. Granados-Soto, CNS Neuroscience & Therapeutics, 2008, 14, 234–247 Search PubMed.
- J. Pallos, L. Bodai, T. Lukacsovich, J. M. Purcell, J. S. Steffan, L. M. Thompson and J. L. Marsh, Hum. Mol. Genet., 2008, 17, 3767–3775 CrossRef CAS.
- A. Solans, A. Zambrano, M. Rodriguez and A. Barrientos, Hum. Mol. Genet., 2006, 15, 3063–3081 CrossRef CAS.
- J. A. Parker, M. Arango, S. Abderrahmane, E. Lambert, C. Tourette, H. Catoire and C. Neri, Nat. Genet., 2005, 37, 349–350 CrossRef CAS.
- R. B. Williams, W. R. Gutekunst, P. M. Joyner, W. Duan, Q. Li, C. A. Ross, T. D. Williams and R. H. Cichewicz, J. Agric. Food Chem., 2007, 55, 9450–9456 CrossRef CAS.
- D. E. Ehrnhoefer, M. Duennwald, P. Markovic, J. L. Wacker, S. Engemann, M. Roark, J. Legleiter, J. L. Marsh, L. M. Thompson, S. Lindquist, P. J. Muchowski and E. E. Wanker, Hum. Mol. Genet., 2006, 15, 2743–2751 CrossRef CAS.
- S. A. Mandel, Y. Avramovich-Tirosh, L. Reznichenko, H. Zheng, O. Weinreb, T. Amit and M. B. Youdim, Neurosignals, 2005, 14, 46–60 CrossRef CAS.
- R. J. Ferrante, H. Ryu, J. K. Kubilus, S. D'Mello, K. L. Sugars, J. Lee, P. Lu, K. Smith, S. Browne, M. F. Beal, B. S. Kristal, I. G. Stavrovskaya, S. Hewett, D. C. Rubinsztein, B. Langley and R. R. Ratan, J. Neurosci., 2004, 24, 10335–10342 CrossRef CAS.
- M. Geissen, S. Krasemann, J. Matschke and M. Glatzel, Vaccine, 2007, 25, 5631–5636 CrossRef CAS.
- S. N. Fontaine and D. R. Brown, Protein Pept. Lett., 2009, 16, 14–26 CrossRef CAS.
- C. Krammer, I. Vorberg, H. M. Schatzl and S. Gilch, Infect. Disord. Drug Targets, 2009, 9, 3–14 Search PubMed.
- National Institute of Neurological Disorders and Stroke, National Institutes of Health, http://www.ninds.nih.gov/index.htm, Accessed February 2010.
- A. J. Nicoll and J. Collinge, Infect. Disord. Drug Targets, 2009, 9, 48–57 Search PubMed.
- E. T. Stiller, J. Vandeputte and J. L. Wachtel, Antibiot. Annu., 1955, 3, 587–591 Search PubMed.
- M. Oura, T. H. Sternberg and E. T. Wright, Antibiot. Annu., 1955, 3, 566–573 Search PubMed.
- W. Mechlinski, C. P. Schaffner, P. Ganis and G. Avitabile, Tetrahedron Lett., 1970, 11, 3873–3876 CrossRef.
- K. M. Abu-Salah, Br. J. Biomed. Sci., 1996, 53, 122–133 Search PubMed.
- A. Lemke, A. F. Kiderlen and O. Kayser, Appl. Microbiol. Biotechnol., 2005, 68, 151–162 CrossRef.
- A. Mange, N. Nishida, O. Milhavet, H. E. M. McMahon, D. Casanova and S. Lehmann, J. Virol., 2000, 74, 3135–3140 CrossRef CAS.
- S. Dirikoc, S. A. Priola, M. Marella, N. Zsurger and J. Chabry, J. Neurosci., 2007, 27, 9537–9544 CrossRef CAS.
- T. Iuvone, G. Esposito, D. De Filippis, C. Scuderi and L. Steardo, CNS Neuroscience & Therapeutics, 2009, 15, 65–75 Search PubMed.
- P. Anand, S. G. Thomas, A. B. Kunnumakkara, C. Sundaram, K. B. Harikumar, B. Sung, S. T. Tharakan, K. Misra, I. K. Priyadarsini, K. N. Rajasekharan and B. B. Aggarwal, Biochem. Pharmacol., 2008, 76, 1590–1611 CrossRef CAS.
- L. Lin and K.-H. Lee, in Studies in Natural Products Chemistry, ed. A. Rahman, Elsevier, Amsterdam, 2006, vol. 33, pp. 785–812 Search PubMed.
- B. Caughey, L. D. Raymond, G. J. Raymond, L. Maxson, J. Silveira and G. S. Baron, J. Virol., 2003, 77, 5499–5502 CrossRef CAS.
- D. A. Kocisko, G. S. Baron, R. Rubenstein, J. Chen, S. Kuizon and B. Caughey, J. Virol., 2003, 77, 10288–10294 CrossRef CAS.
- G. Forloni, M. Salmona, G. Marcon and F. Tagliavini, Infect. Disord. Drug Targets, 2009, 9, 23–30 Search PubMed.
- D. Blum, A. Chtarto, L. Tenenbaum, J. Brotchi and M. Levivier, Neurobiol. Dis., 2004, 17, 359–366 CrossRef CAS.
- H.-S. Kim and Y.-H. Suh, Behav. Brain Res., 2009, 196, 168–179 CrossRef CAS.
- D. Orsucci, V. Calsolaro, M. Mancuso and G. Siciliano, CNS Neurol. Disord. Drug Targets, 2009, 8, 222–231 CAS.
- D. P. Stirling, K. M. Koochesfahani, J. D. Steeves and W. Tetzlaff, Neuroscientist, 2005, 11, 308–322 CrossRef CAS.
- R. Planells-Cases, J. Lerma and A. Ferrer-Montiel, Curr. Pharm. Des., 2006, 12, 3583–3596 CrossRef CAS.
- A. L. Sheldon and M. B. Robinson, Neurochem. Int., 2007, 51, 333–355 CrossRef CAS.
- B. G. Gold and J. E. Villafranca, Curr. Top. Med. Chem., 2003, 3, 1368–1375 CrossRef CAS.
- R. D. Price, S. A. Milne, J. Sharkey and N. Matsuoka, Pharmacol. Ther., 2007, 115, 292–306 CrossRef CAS.
- S. T. Dheen, C. Kaur and E. A. Ling, Curr. Med. Chem., 2007, 14, 1189–1197 CrossRef CAS.
- Y. Gilgun-Sherki, E. Melamed and D. Offen, Curr. Pharm. Des., 2006, 12, 3509–3519 CrossRef CAS.
- C. J. Van der Schyf, S. Gal, W. J. Geldenhuys and M. B. Youdim, Expert Opin. Invest. Drugs, 2006, 15, 873–886 Search PubMed.
- J.-S. Kim, K. Shin-ya, K. Furihata, Y. Hayakawa and H. Seto, Tetrahedron Lett., 1997, 38, 3431–3434 CrossRef CAS.
- H. Kobayashi, K. Shin-Ya, K. Nagai, K. Suzuki, Y. Hayakawa, H. Seto, B. S. Yun, I. J. Ryoo, J. S. Kim, C. J. Kim and I. D. Yoo, J. Antibiot., 2001, 54, 1013–1018 CAS.
- S. D. Kim, S. K. Oh, H. S. Kim and Y. H. Seong, Arch. Pharmacal Res., 2001, 24, 164–170 Search PubMed.
- S. R. Kim, K. Y. Lee, K. A. Koo, S. H. Sung, N. G. Lee, J. Kim and Y. C. Kim, J. Nat. Prod., 2002, 65, 1696–1699 CrossRef CAS.
- S. Y. Kang, K. Y. Lee, S. H. Sung and Y. C. Kim, J. Nat. Prod., 2005, 68, 56–59 CrossRef CAS.
- J. S. Yoon, S. H. Sung and Y. C. Kim, J. Nat. Prod., 2008, 71, 208–211 CrossRef CAS.
- M. K. Lee, H. Yeo, J. Kim, G. J. Markelonis, T. H. Oh and Y. C. Kim, J. Neurosci. Res., 2000, 59, 259–264 CrossRef CAS.
- Y. C. Kim, S. R. Kim, G. J. Markelonis and T. H. Oh, J. Neurosci. Res., 1998, 53, 426–432 CrossRef CAS.
- J. S. Kim, K. Shin-ya, J. Eishima, K. Furihata and H. Seto, J. Antibiot., 1996, 49, 947–948 CAS.
- J. S. Kim, K. Shin-ya, J. Eishima, K. Furihata and H. Seto, J. Antibiot., 1996, 49, 1172–1174 CAS.
- W. G. Kim, J. P. Kim, C. J. Kim, K. H. Lee and I. D. Yoo, J. Antibiot., 1996, 49, 20–25 CAS.
- W.-G. Kim, J.-P. Kim, H. Koshino, K. Shin-Ya, H. Seto and I.-D. Yoo, Tetrahedron, 1997, 53, 4309–4316 CrossRef CAS.
- K. Shin-ya, T. Kunigami, J. S. Kim, K. Furihata, Y. Hayakawa and H. Seto, Biosci., Biotechnol., Biochem., 1997, 61, 1768–1769 CrossRef CAS.
- K. Shin-ya, S. Shimizu, T. Kunigami, K. Furihata, Y. Hayakawa and H. Seto, J. Antibiot., 1995, 48, 1378–1381 CAS.
- K. Shin-Ya, S. Shimizu, T. Kunigami, K. Furihata and H. Seto, J. Antibiot., 1995, 48, 574–578 CAS.
- W. G. Kim, I. J. Ryoo, B. S. Yun, K. Shin-ya, H. Seto and I. D. Yoo, J. Antibiot., 1999, 52, 758–761 CAS.
- N. Kotoda, K. Shin-ya, K. Furihata, Y. Hayakawa and H. Seto, J. Antibiot., 1997, 50, 770–772 CAS.
- T. Kunigami, K. Shin-Ya, K. Furihata, Y. Hayakawa and H. Seto, J. Antibiot., 1998, 51, 880–882 CAS.
- Y. Murakami, S. Kato, M. Nakajima, M. Matsuoka, H. Kawai, K. Shin-Ya and H. Seto, J. Antibiot., 1996, 49, 839–845 CAS.
- T. Murphy, R. Schnaar and J. Coyle, FASEB J., 1990, 4, 1624–1633 CAS.
- B. Connor and M. Dragunow, Brain Res. Rev., 1998, 27, 1–39 CrossRef CAS.
- E. C. Yuen and W. C. Mobley, Ann. Neurol., 1996, 40, 346–354 CrossRef CAS.
- N. Webster and M. Pirrung, BMC Neurosci., 2008, 9, S1 CrossRef.
- S. U. Kim and J. de Vellis, J. Neurosci. Res., 2009, 87, 2183–2200 CrossRef CAS.
- J.-m. Huang, R. Yokoyama, C.-s. Yang and Y. Fukuyama, Tetrahedron Lett., 2000, 41, 6111–6114 CrossRef CAS.
- J.-m. Huang, R. Yokoyama, C.-s. Yang and Y. Fukuyama, J. Nat. Prod., 2001, 64, 428–431 CrossRef CAS.
- R. Yokoyama, J.-M. Huang, C.-S. Yang and Y. Fukuyama, J. Nat. Prod., 2002, 65, 527–531 CrossRef CAS.
- Q. Pan, F. C. Ip, N. Y. Ip, H. X. Zhu and Z. D. Min, J. Nat. Prod., 2004, 67, 1548–1551 CrossRef CAS.
- T. Kuboyama, C. Tohda, J. Zhao, N. Nakamura, M. Hattori and K. Komatsu, NeuroReport, 2002, 13, 1715–1720 CrossRef CAS.
- M. Yamazaki, K. Chiba and T. Mohri, Biol. Pharm. Bull., 1996, 19, 791–795 CAS.
- C. K. Tsang, S. Atsuko and Y. Kamei, J. Appl. Phycol., 2001, 13, 349–357 Search PubMed.
- J. Y. Hur, P. Lee, H. Kim, I. Kang, K. R. Lee and S. Y. Kim, Biochem. Biophys. Res. Commun., 2004, 313, 948–953 CrossRef CAS.
- J. Hines, M. Groll, M. Fahnestock and C. M. Crews, Chem. Biol., 2008, 15, 501–512 CrossRef CAS.
- K. Kosaka, J. Mimura, K. Itoh, T. Satoh, Y. Shimojo, C. Kitajima, A. Maruyama, M. Yamamoto and T. Shirasawa, J. Biochem., 2010, 147, 73–81 CrossRef CAS.
- T. K. Tabopda, J. Ngoupayo, J. W. Liu, A. C. Mitaine-Offer, B. T. Ngadjui, M. A. Lacaille-Dubois and B. Luu, Nat. Prod. Commun., 2009, 4, 517–520 CAS.
- J. W. Liu, S. J. Tian, J. de Barry and B. Luu, J. Nat. Prod., 2007, 70, 1329–1334 CrossRef CAS.
- C. Tohda, N. Matsumoto, K. Zou, M. R. Meselhy and K. Komatsu, Jpn. J. Pharmacol., 2002, 90, 254–262 CrossRef CAS.
- Y. Fukuyama, K. Nakade, Y. Minoshima, R. Yokoyama, H. Zhai and Y. Mitsumoto, Bioorg. Med. Chem. Lett., 2002, 12, 1163–1166 CrossRef CAS.
- M. Ito, N. Sakai, K. Ito, F. Mizobe, K. Hanada and K. Mizoue, J. Antibiot., 1999, 52, 224–230 CAS.
- K. Shigeta, K. Ootaki, H. Tatemoto, T. Nakanishi, A. Inada and N. Muto, Biosci., Biotechnol., Biochem., 2002, 66, 2491–2494 CrossRef CAS.
- H. Kawagishi, M. Ando, H. Sakamoto, S. Yoshida, F. Ojima, Y. Ishiguro, N. Ukai and S. Furukawa, Tetrahedron Lett., 1991, 32, 4561–4564 CrossRef.
- H. Kawagishi, M. Ando, K. Shinba, H. Sakamoto, S. Yoshida, F. Ojima, Y. Ishiguro, N. Ukai and S. Furukawa, Phytochemistry, 1993, 32, 175–178.
- Y. Nozawa, N. Sakai, K. Matsumoto and K. Mizoue, J. Antibiot., 2002, 55, 629–634 CAS.
- Y. Cheng, B. Schneider, U. Riese, B. Schubert, Z. Li and M. Hamburger, J. Nat. Prod., 2004, 67, 1854–1858 CrossRef CAS.
- C. Tohda, N. Nakamura, K. Komatsu and M. Hattori, Biol. Pharm. Bull., 1999, 22, 679–682 CAS.
- P. Li, K. Matsunaga, T. Yamakuni and Y. Ohizumi, Eur. J. Pharmacol., 2000, 406, 203–208 CrossRef CAS.
- P. Li, K. Matsunaga, K. Yamamoto, R. Yoshikawa, K. Kawashima and Y. Ohizumi, Neurosci. Lett., 1999, 273, 53–56 CrossRef CAS.
- H. Yun, T.-C. Chou, H. Dong, Y. Tian, Y.-m. Li and S. J. Danishefsky, J. Org. Chem., 2005, 70, 10375–10380 CrossRef CAS.
- T. Tokue, S. Miura, H. Kato, H. Hirota, T. Ohta and S. Tsukamoto, Heterocycles, 2006, 69, 521–526 CrossRef CAS.
- K. Schmidt, W. Gunther, S. Stoyanova, B. Schubert, Z. Li and M. Hamburger, Org. Lett., 2002, 4, 197–199 CrossRef CAS.
- K. Schmidt, U. Riese, Z. Li and M. Hamburger, J. Nat. Prod., 2003, 66, 378–383 CrossRef CAS.
- Y. Cheng, B. Schneider, U. Riese, B. Schubert, Z. Li and M. Hamburger, J. Nat. Prod., 2006, 69, 436–438 CrossRef CAS.
- H. C. Kwon, K. C. Lee, O. R. Cho, I. Y. Jung, S. Y. Cho, S. Y. Kim and K. R. Lee, J. Nat. Prod., 2003, 66, 466–469 CrossRef CAS.
- T. Kita, Y. Takaya, Y. Oshima, T. Ohta, K. Aizawa, T. Hirano and T. Inakuma, Tetrahedron, 1998, 54, 11877–11886 CrossRef CAS.
- Y. Obara, N. Nakahata, T. Kita, Y. Takaya, H. Kobayashi, S. Hosoi, F. Kiuchi, T. Ohta, Y. Oshima and Y. Ohizumi, Eur. J. Pharmacol., 1999, 370, 79–84 CrossRef CAS.
- Y. S. Li, K. Matsunaga, R. Kato and Y. Ohizumi, J. Pharm. Pharmacol., 2001, 53, 915–919 CrossRef CAS.
- G. C. Brown, Biochem. Soc. Trans., 2007, 35, 1119–1121 CAS.
- A. Thampithak, Y. Jaisin, B. Meesarapee, S. Chongthammakun, P. Piyachaturawat, P. Govitrapong, P. Supavilai and Y. Sanvarinda, Neurosci. Lett., 2009, 462, 171–175 CrossRef CAS.
- M. H. Kossuga, A. M. Nascimento, J. Q. Reimao, A. G. Tempone, N. N. Taniwaki, K. Veloso, A. G. Ferreira, B. C. Cavalcanti, C. Pessoa, M. O. Moraes, A. M. Mayer, E. Hajdu and R. G. Berlinck, J. Nat. Prod., 2008, 71, 334–339 CrossRef CAS.
- P. Lee, J. Hur, J. Lee, J. Kim, J. Jeong, I. Kang, S. Y. Kim and H. Kim, Neurochem. Int., 2006, 48, 60–66 CrossRef CAS.
- D. Yuan, B. Ma, C. Wu, J. Yang, L. Zhang, S. Liu, L. Wu and Y. Kano, J. Nat. Prod., 2008, 71, 1271–1274 CrossRef CAS.
- Y. T. Shih, Y. Y. Hsu, F. R. Chang, Y. C. Wu and Y. C. Lo, Planta Med., 2010, 76, 120–127 CrossRef CAS.
- Y. Choi, M. K. Lee, S. Y. Lim, S. H. Sung and Y. C. Kim, Br. J. Pharmacol., 2009, 156, 933–940 CrossRef CAS.
- A. M. Mayer, M. L. Hall, S. M. Lynch, S. P. Gunasekera, S. H. Sennett and S. A. Pomponi, BMC Pharmacol., 2005, 5, 6 Search PubMed.
- H. W. Jung, Y. S. Chung, Y. S. Kim and Y. K. Park, Exp. Mol. Med., 2007, 39, 715–721 Search PubMed.
- M. Naoi, W. Maruyama, H. Yi, K. Inaba, Y. Akao and M. Shamoto-Nagai, J. Neural Transm., 2009, 116, 1371–1381 CrossRef CAS.
- T. Herraiz and C. Chaparro, Life Sci., 2006, 78, 795–802 CrossRef CAS.
- D. J. McKenna, G. H. N. Towers and F. Abbott, J. Ethnopharmacol., 1984, 10, 195–223 CrossRef CAS.
- A. Urbain, A. Marston, L. S. Grilo, J. Bravo, O. Purev, B. Purevsuren, D. Batsuren, M. Reist, P. A. Carrupt and K. Hostettmann, J. Nat. Prod., 2008, 71, 895–897 CrossRef CAS.
- M. K. Lee, B. Y. Hwang, S. A. Lee, G. J. Oh, W. H. Choi, S. S. Hong, K. S. Lee and J. S. Ro, Chem. Pharm. Bull., 2003, 51, 409–411 CrossRef CAS.
- H. Fujimoto, Y. Satoh, K. Yamaguchi and M. Yamazaki, Chem. Pharm. Bull., 1998, 46, 1506–1510 CAS.
- E. Okuyama, M. Homma, Y. Satoh, H. Fujimoto, M. Ishibashi, M. Yamazaki, M. Satake and A. B. Ghazali, Chem. Pharm. Bull., 1999, 47, 1473–1476 CAS.
- D. T. Huong, H. C. Choi, T. C. Rho, H. S. Lee, M. K. Lee and Y. H. Kim, Arch. Pharmacal Res., 1999, 22, 324–326 Search PubMed.
- T. Hatano, T. Fukuda, T. Miyase, T. Noro and T. Okuda, Chem. Pharm. Bull., 1991, 39, 1238–1243 CAS.
- J. S. Ro, S. S. Lee, K. S. Lee and M. K. Lee, Life Sci., 2001, 70, 639–645 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |