Structure–reactivity relationships and substituent effect additivity in the aqueous oxidation of chlorophenols by cerium(IV)†
Adrienn
Simon
,
Csaba
Ballai
,
Gábor
Lente
* and
István
Fábián
Department of Inorganic and Analytical Chemistry, University of Debrecen, Debrecen, Hungary. E-mail: lenteg@delfin.unideb.hu; Fax: +36 52-518-660; Tel: +36 52-512-900 Ext. 22373
First published on 25th October 2010
Abstract
The oxidation of all 19 chlorophenols and unsubstituted phenol by cerium(IV) was studied in an acidic aqueous solution in order to carry out a systematic test of chlorine substituent effects on the reactivity. All reactions were found to show 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶1 cerium(IV) ∶phenol stoichiometry and a simple second-order rate equation. Rate constants did not correlate well with characteristic parameters such as pK values, carbon-13 NMR chemical shifts or Hammett substituent constants. Nevertheless, a strict additivity of chlorine substituent effects was found in both characteristic and reactivity parameters. The data suggest that a proton-coupled electron transfer mechanism could be operative. 2,4,6-Trichlorophenol was found to show exceptionally high reactivity towards cerium(IV).
∶1 cerium(IV) ∶phenol stoichiometry and a simple second-order rate equation. Rate constants did not correlate well with characteristic parameters such as pK values, carbon-13 NMR chemical shifts or Hammett substituent constants. Nevertheless, a strict additivity of chlorine substituent effects was found in both characteristic and reactivity parameters. The data suggest that a proton-coupled electron transfer mechanism could be operative. 2,4,6-Trichlorophenol was found to show exceptionally high reactivity towards cerium(IV).
Introduction
Chlorophenols constitute a group of widely used compounds that are currently listed as priority pollutants by European and American environmental protection agencies.1,2 Their removal from industrial wastewater usually requires special techniques as they are resistant to chemical oxidation or microbial degradation.3–16 From an environmental point of view, the stoichiometric oxidant in a chemical treatment method should be an abundant and cost-effective reagent, the by-product of which does not cause major environmental concerns. Hydrogen peroxide and peroxomonosulfate ion (oxone) seem ideal oxidants for this purpose and a number of catalytic systems have been reported using these compounds. The best results were obtained with iron complexes as catalysts.3,6–8,10–12 Mechanistic details of such catalyzed processes are not easy to explore. A useful but not very often used tool in those kinetic studies is a systematic change of the number of chlorine substituents on the oxidizable substrates.In one of our earlier studies, an interesting structure–reactivity relationship was revealed in the FeIIITPPS-catalyzed (FeIIITPPS: iron(III) meso-tetra(4-sulfonatophenyl)porphine) oxidation of chlorophenols by hydrogen peroxide.10 Using various substituted chlorophenols, the logarithm of the rate constant of the reaction between the active form of the catalyst and the chlorophenols was shown to correlate with the 13C NMR chemical shift of the carbon atom that is directly connected to the oxygen in the chlorophenol.10 No similar relationship with the pK values of various phenols was found in the same work.10 The correlation must have far-reaching mechanistic implications, but the lack of similar examples from the earlier literature was a serious obstacle to making a convincing point about the consequences. Based on some independent information on the homolytic bond dissociation energies of chlorophenols,17 it was suggested that the observed trend might be indicative of hydrogen atom transfer in the rate determining step.10 The interpretation of these results would be much easier in comparison with similar substituent effect studies in stoichiometric redox reactions of chlorophenols in non-catalytic systems.
The three major one-electron oxidation pathways of phenols are hydrogen atom transfer (HAT), proton-coupled electron transfer (PCET), and sequential proton-loss electron transfer (SPLET). In water, SPLET is equivalent to the acid dissociation of the phenol followed by electron transfer from the phenolate anion. This is easily identified by the pH dependence as this sort of mechanism should show inverse first-order dependence on the concentration of hydrogen ion in the acidic region. No such distinction is easily made between HAT and PCET and recent research has focused a lot of attention on this question.18–28
The aim of the present work was a thorough study of the effect of chlorine substituents on the one-electron oxidation of phenols using all possible chlorinated phenols. Cerium(IV) was found to be an ideal oxidant for this purpose as it is a strong one-electron oxidant which can only be used in highly acidic medium, ensuring that any rate contributions from pathways involving the phenolate ion (SPLET) could only be minor. This paper reports our detailed stoichiometric and kinetic results and correlation analysis between rate constants and selected characteristic parameters.
Experimental section
Materials
Chlorinated phenols were purchased from Aldrich, Lancaster Synthesis and Supelco. When necessary (i.e. when the purchased material was not completely white), they were purified by vacuum sublimation before use. The purity of all compounds has been tested in 1H and 13C NMR measurements. Throughout this paper, different chlorophenols will be abbreviated by giving only the positions of the chlorine substituents on the phenol (e.g. 236 means 2,3,6-trichlorophenol). Solutions of Ce(IV) were prepared by dissolving Ce(SO4)2 in sulfuric acid. The Ce(IV) concentrations of these solutions were standardized by iodometry. All other chemicals used in this study were of analytical reagent grade and purchased from commercial sources. Doubly deionized and ultrafiltered water from a Millipore Q system was used to prepare the stock solutions and samples.Instruments and softwares
Shimadzu UV-2501PC (scanning), Shimadzu UV-3101PC (scanning), Shimadzu MultiSpec-1500 (diode-array), Perkin Elmer Lambda 25 (scanning), Perkin Elmer Lambda 2S (scanning), and HP-8543 (diode-array) spectrophotometers were used in this study using standard quartz cells with 1.000 cm optical path length. All kinetic measurements were performed with an Applied Photophysics DX-17 MV sequential stopped-flow apparatus in the absorbance mode using 1.00 cm optical path length. The dead time of the instrument was measured to be 1.09 ± 0.02 ms by published methods.29,30 pH was measured by standard combination glass electrodes (Hanna Instruments or Metrohm) connected to either a precision Hanna Instruments pH-meter or a computer-controlled Metrohm Titrino system. The calibration of the electrodes was based on relevant IUPAC recommendations.311H and 13C NMR spectra were recorded on Varian VRX-300, Varian VRX-400, Bruker DRX-360 and Bruker DRX-400 instruments and were calibrated using the residual signal of the solvent CDCl3. In addition to the operating softwares of individual instruments, Scientist32 and Matlab33 were used in the fitting procedures.Results
Acid dissociation constants and chemical shifts
The acid dissociation constants of all studied chlorophenols were determined in this study in order to use strictly identical conditions and measuring methods. A combined pH-potentiometric and spectrophotometric method was used, whereby the UV-vis spectra of chlorophenols were recorded as a function of measured pH. Fig. S1 in the ESI† gives a series of spectra for 2356 as an example. The data were evaluated based on the following equation with the use of linear algebraic transformations detailed in the ESI†: | (1) |
![[small script l]](https://www.rsc.org/images/entities/char_e146.gif) is the path length, ελ,HA and ελ,A are the molar absorption coefficients for the protonated and deprotonated forms of the phenol at wavelength λ. All determined pK values are shown in Table 1 along with some relevant literature data for comparison.34–36 The standard error for the determined parameter pK was smaller than 0.01 when calculated by the usual statistical analysis. This is viewed as unreasonably small as the actual accuracy of the pH measurements was 0.01 in all cases. Therefore, the conservative estimate of 0.02 will be used for the standard errors for all pK values determined in this work. 1H NMR and 13C chemical shifts were determined for all chlorophenols in CDCl3 by routine measurements. The results are listed in Table 1 and Table S1 in the ESI.†
is the path length, ελ,HA and ελ,A are the molar absorption coefficients for the protonated and deprotonated forms of the phenol at wavelength λ. All determined pK values are shown in Table 1 along with some relevant literature data for comparison.34–36 The standard error for the determined parameter pK was smaller than 0.01 when calculated by the usual statistical analysis. This is viewed as unreasonably small as the actual accuracy of the pH measurements was 0.01 in all cases. Therefore, the conservative estimate of 0.02 will be used for the standard errors for all pK values determined in this work. 1H NMR and 13C chemical shifts were determined for all chlorophenols in CDCl3 by routine measurements. The results are listed in Table 1 and Table S1 in the ESI.†
| pKa | Literature pKb | δC1c/ppm | k/M−1s−1 | ||
|---|---|---|---|---|---|
| Ref: d,e | Ref: f | ||||
| a Measured in this work, standard error: ±0.02 (see text for detailed explanation). b Literature data typically measured at varying ionic strength and medium, sometimes with the addition of non-aqueous solvents up to 50%. c Precision: ±0.1 ppm, solvent: CDCl3. d NIST Database (ref. 34). e SC Database (ref. 35). f Ref. 36. | |||||
| Phenol | 9.86 | 9.79d | 10.02 | 155.3 | (3.72 ± 0.03) × 104 |
| 2-Chlorophenol | 8.33 | 8.53d | 8.48 | 151.5 | (9.07 ± 0.05) × 103 |
| 3-Chlorophenol | 8.85 | 8.78d | 9.02 | 155.9 | (3.48 ± 0.01) × 104 |
| 4-Chlorophenol | 9.34 | 9.20d | 9.38 | 153.8 | (4.43 ± 0.02) × 104 |
| 2,3-Dichlorophenol | 7.64 | 7.70d | 7.45 | 152.8 | (9.08 ± 0.11) × 103 |
| 2,4-Dichlorophenol | 7.83 | 7.89d | 7.75 | 150.3 | (5.72 ± 0.05) × 103 |
| 2,5-Dichlorophenol | 7.40 | 7.51d | 7.35 | 152.0 | (9.79 ± 0.03) × 103 |
| 2,6-Dichlorophenol | 6.77 | 6.79d | 6.79 | 148.1 | (1.58 ± 0.01) × 103 |
| 3,4-Dichlorophenol | 8.56 | 8.59d | 8.39 | 154.6 | (4.54 ± 0.05) × 104 |
| 3,5-Dichlorophenol | 7.99 | 8.18d | 7.93 | 156.5 | (3.48 ± 0.04) × 104 |
| 2,3,4-Trichlorophenol | 7.00 | — | 7.59 | 151.4 | (1.00 ± 0.05) × 104 |
| 2,3,5-Trichlorophenol | 6.68 | — | 7.23 | 153.0 | (1.06 ± 0.06) × 104 |
| 2,3,6-Trichlorophenol | 5.75 | — | 6.12 | 149.2 | (1.84 ± 0.01) × 103 |
| 2,4,5-Trichlorophenol | 6.97 | 6.71e | 7.33 | 150.8 | (1.63 ± 0.03) × 104 |
| 2,4,6-Trichlorophenol | 6.15 | 6.42e | 6.42 | 147.1 | (2.27 ± 0.08) × 104 |
| 3,4,5-Trichlorophenol | 7.82 | — | 7.74 | 154.2 | (5.6 ± 0.3) × 104 |
| 2,3,4,5-Tetrachlorophenol | 6.31 | — | 6.96 | 151.0 | (2.3 ± 0.2) × 104 |
| 2,3,4,6-Tetrachlorophenol | 5.32 | — | — | 147.9 | (2.21 ± 0.08) × 103 |
| 2,3,5,6-Tetrachlorophenol | 5.31 | — | 5.44 | 150.1 | (2.7 ± 0.1) × 103 |
| Pentachlorophenol | 4.66 | 5.25e | 5.26 | 148.3 | (6.0 ± 0.9) × 103 |
Reaction stoichiometry
The stoichiometry of the reaction between cerium(IV) and each chlorophenol was studied by spectrophotometric titration in 0.10 M H2SO4. A solution of the studied phenol was titrated in a cell and the spectra were recorded after the addition of each increment. As an example, the titration of 246 is shown in Fig. 1, which also displays the UV-vis spectra of Ce(IV) and Ce(III). In all cases, the reaction was shown to feature Ce(IV)∶phenol stoichiometry of 2∶1 corresponding to an overall two-electron oxidation of the phenols. This is valid for conditions under which the phenol is in excess and the product of this initial reaction was shown to be oxidized further by Ce(IV) on a time scale longer (from several minutes to hours) than that of the direct reaction between the original reagents. This clearly demonstrated the need for titrating a phenol solution with small increments of Ce(IV) solution in the spectrophotometric titrations, results from a reverse titration would have been difficult to interpret. The kinetics of the process was always studied at phenol excess, where the 2∶1 stoichiometry clearly prevails.
![Spectrophotometric titration of an aqueous solution of 2,4,6-trichlorophenol with cerium(iv) sulfate. Initial solution: V = 2.00 cm3, [246] = 0.690 mM. Titrant: [Ce(iv)] = 4.75 mM. Increments: 0.100 cm3. Optical path length: 1.000 cm, T = 25.0 °C. Inset: molar absorbance values of Ce(iv) and Ce(iii).](/image/article/2011/NJ/c0nj00529k/c0nj00529k-f1.gif) | ||
| Fig. 1 Spectrophotometric titration of an aqueous solution of 2,4,6-trichlorophenol with cerium(IV) sulfate. Initial solution: V = 2.00 cm3, [246] = 0.690 mM. Titrant: [Ce(IV)] = 4.75 mM. Increments: 0.100 cm3. Optical path length: 1.000 cm, T = 25.0 °C. Inset: molar absorbance values of Ce(IV) and Ce(III). | ||
No efforts were made to positively identify the two-electron oxidation products formed from the phenols beyond some observations in the reactions of 246 and 26. In the case of 246, the confirmed 2∶1 stoichiometry corresponds to a two-electron oxidation of the organic molecule, which usually leads to 2,6-dichloro-1,4-benzoquione according to well established literature information.8,10,12,37–40 Further experimental confirmation of this product was made possible by the fact that neither 246 nor the product Ce(III) has significant absorption above 320 nm. The UV-vis spectrum of the product of the reaction between 246 and Ce(IV) was indistinguishable from the independently known spectrum of an authentic sample of 2,6-dichloro-1,4-benzoquione.37 Furthermore, two characteristic reactions (a photoreaction37 and oxidation by H2O2 solution10) of this quinone were also confirmed to occur in test experiments after masking cerium(III) with EDTA (Fig. S3 in the ESI†). In the case of 26 (Fig. S4 in the ESI†), a similar analysis showed that the product was different from 2,6-dichloro-1,4-benzoquinone, which was formed in the FeTPPS-catalyzed oxidation of 26.10 The confirmed stoichiometry also rules out the formation of 2,6-dichloro-1,4-benzoquinone as it would require a four-electron oxidation of 26. The most logical two-electron oxidation product of 26 would be 3-chloro-1,2-benzoquinone. This is in agreement with the intense peak observed at 434 nm in the product spectrum, which is usually characteristic of ortho-quinones, and is also in agreement with the literature information.41–44 However, these observations by no means imply a positive product identification similar to the case of 246.
Reaction kinetics
The kinetics of the reactions was studied at chlorophenol excess by the stopped-flow method. In all cases, the following simple second-order rate equation was found to be valid: | (2) |
 | ||
| Fig. 2 Pseudo first-order rate constants as a function of chlorophenol concentration in the oxidation of various chlorophenols by cerium(IV) ion. Medium: 0.10 M H2SO4. T = 25.0 °C. | ||
245, 345, 2345, 2346, 2356 and 23456 were not soluble enough in water to cover a sufficiently large concentration range in kinetic experiments while still using a large excess of the chlorophenol over Ce(IV). In these cases, most measurements were carried out under second-order conditions. The absorbance-time traces were fitted to the following equation:45
 | (3) |
| Δ = [Cl–Ph]0 − 0.5[Ce(IV)]0, γ = [Ce(IV)]0/[Cl–Ph]0, kapp = kΔ | (4) |
All the determined rate constants are listed in Table 1. The values refer to measurements done in 0.10 M H2SO4 without any additional salts. Some detailed kinetic information for chlorophenols not covered in Fig. 2 are shown in Fig. S5–S8 in the ESI.† The rate did not depend on the acidity in the H2SO4 concentration range between 0.10 M (pH = 0.86) and 0.50 M (pH = 0.25).
Structure–reactivity correlations
Correlations were sought between two characteristic parameters, pK and the 13C NMR chemical shift of the carbon atom connected directly to the oxygen in each chlorinated phenol (δ13C1) and the logarithm of the second-order rate constant of the oxidation of the phenol by Ce(IV). Fig. 3 shows that the two characteristic parameters are significantly different as they do not correlate with each other. On closer inspection, the graph reveals that the chlorophenols can be divided into three groups depending on the number of chlorine substituents in the meta position. There is strong correlation between the two characteristic parameters within each group.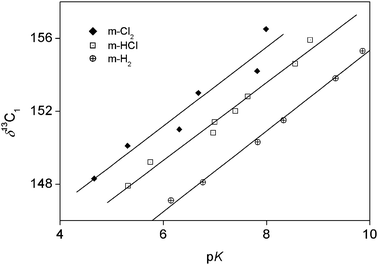 | ||
| Fig. 3 13C chemical shifts of the phenolic carbon as a function of the pK values for the studied chlorophenols. | ||
Fig. 4 shows that the correlation between the logarithm of the rate constants and δ13C1 is weak. The figure also displays similar data from an earlier work obtained in the FeIIITPPS-catalyzed oxidation of chlorophenols where a strong correlation was observed for comparison.10 There is no spectacular correlation between pK values and rate constants, either (Fig. S9 in the ESI†). Average chemical shifts of aromatic carbon atoms were also used as site-independent molecular descriptors, but failed to give any reasonable correlation with the rate constants (Fig. S10 in the ESI†).
 | ||
| Fig. 4 Rate constants of the oxidation chlorophenols by cerium(IV) as a function of the 13C chemical shift of the phenolic carbon. Grey triangles: similar plot obtained in the reaction of chlorophenols with hydrogen peroxide catalyzed by FeIII(TPPS).10 | ||
Additivity of substituent effects
The additivity of substituent effects was tested for both characteristic parameters and for the kinetic data as well by seeking substituent constants that best adhere to Hammett-type relationships.46 The first equation (3σ equation) assumed total additivity of substituent effects:| Y = Y0 + noσo + nmσm + npσp | (5) |
An approach containing more parameters has also been tried. In this approach, it was not assumed that two ortho substituents, for example, have the same effect on the property together as two independent ortho substituents would have. The corresponding equation (5σ equation) is given as:
 | (6) |
| Σ = noσo + nmσm + npσp | (7) |
 | (8) |
 | ||
| Fig. 5 pK values for the studied chlorophenols as a function of the sum of substituent constants. 3σ: with the use of eqn (5). 5σ: with the use of eqn (6). | ||
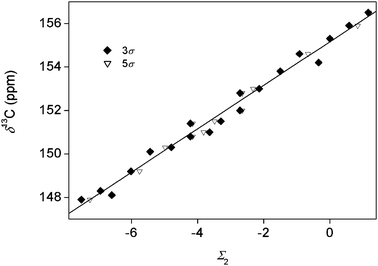 | ||
| Fig. 6 13C NMR chemical shifts for the studied chlorophenols as a function of the sum of substituent constants. 3σ: with the use of eqn (5). 5σ: with the use of eqn (6). | ||
 | ||
| Fig. 7 Rate constant measured in the oxidation of the studied chlorophenols by cerium(IV) as a function of the sum of substituent constants excluding 2,4,6-trichlorophenol. 3σ: with the use of eqn (5). 5σ: with the use of eqn (6). | ||
| σ o | σ m | σ p | r 3σ 2 | r 5σ 2 | |
|---|---|---|---|---|---|
| pK | −1.54 ± 0.03 | −0.79 ± 0.03 | −0.45 ± 0.06 | 0.9938 | 0.9949 |
| δ 13C | −3.3 ± 0.1 | 0.58 ± 0.10 | −1.50 ± 0.16 | 0.9865 | 0.9893 |
| log k | −0.53 ± 0.07 | 0.03 ± 0.07 | 0.24 ± 0.11 | 0.9519 | 0.9543 |
Discussion
The rate equation of the oxidation of chlorophenols by cerium(IV) shows that the phenolate anion cannot be a reactive species in these processes. Very clearly, inverse first-order acidity-dependence would be expected in this case under the highly acidic conditions used here. In addition, the true second order rate constant of the process would be higher than the diffusion controlled limit of 1010 M−1 s−1 for some of the studied compounds (phenol, 2, 3, 4, 23, 24, 25, 34, 35, 234, 245, 345) if electron transfer was initiated form the phenolate ion. This observation rules out the SPLET mechanism of one-electron transfer. It is also notable that none of the kinetic studies showed saturation of rate with increasing concentration of chlorophenol, which indicates that there is no substantial association (complex formation) between the reactants. The most notable deviation from the expected straight lines in the pseudo first-order rate constant-chlorophenol concentration plots occurs for 246 (shown in Fig. 2), where a careful look at the plot might hint of some curvature. However, this is statistically insignificant. Actually, it would have been very useful to do experiments at higher concentrations of 246 because any saturation character would have been more pronounced in that region. Unfortunately, this was not possible because the experiments already approached the solubility limit of 246. Therefore, there is no experimental reason to believe that there is a significant saturation character for this plot.Fig. 3 shows that the two characteristic parameters (pK and δ13C1) analyzed here are under different influences. A closer look at this figure reveals that the points actually give linear correlation in three distinct series depending on the number of metachloro substituents. The fact that the correlation is quite good within each of the three series (m-H2, m-HCl, and m-Cl2) indicates that the para and the ortho substituents have very similar effects on the pK and on δ13C1. This will be analyzed further at the discussion of the additivity tests.
Fig. 4 shows very poor correlation between the logarithm of rate constants and the δ13C1 values. An even worse correlation is found between log k and pK values (Fig. S9 in the ESI†). The points in Fig. 4 can be broken down to three clusters depending on the number of orthochloro substituents. Basically, o-Cl2 phenols (except 246) are oxidized relatively slowly and o-H2 phenols relatively fast, with o-HCl phenols occupying a middle position in this respect. This is quite different from the earlier case when we studied the FeIIITPPS/H2O2 system as shown by the grey points in Fig. 4.10 Even if one assumes a weak correlation for the rate constants of the cerium(IV) oxidation, this correlation is in the opposite direction (log k is smaller with larger δ13C1) than in the case of FeIIITPPS/H2O2 system.10 In addition, the product in one of the reactions was proved to be quite different: 26 is oxidized to 2,6-dichloro-1,4-benzoquinone in the case of FeIIITPPS/H2O2,10 whereas this is clearly not the case in the present study of cerium(IV) oxidation. On these grounds, it is very safe to conclude that these two reactions have quite different mechanisms. Since there is some reason to argue that the FeIIITPPS/H2O2 system oxidizes phenols by HAT,10 the cerium(IV) oxidation could follow the PCET pathway.
Fig. 5–7 show that the additivity of substituent effects is excellent for pK values, but the δ13C1 and log k data (excluding 246 in this last case) also show fits that are better than in most published examples of substituent effect relationships. It should be noted that the correlation coefficients only show a very modest improvement when eqn (6) is used. This, again, proves that the additivity of substituent effects is quite good for each of the studied properties and the fit cannot be improved by including further parameters.
The determined σ values summarized in Table 2 deserve more attention. The σ values obtained from different properties are not directly comparable as the absolute changes in pK, δ13C1 and log k are different. From the σ values obtained based on pK values, it is clear that distance is the primary factor in determining the acidity of the phenolic OH groups. All three σ constants are negative (any substitution with chlorine decreases the pK), which is probably due to the electron withdrawing effect of a chlorine substituent. The σ values decrease in the σo > σm > σp order. The two other properties show different behavior. The δ13C1 values reflect a behavior similar to the textbook direction rules in aromatic electrophilic substitution, which is based on a balance between electron withdrawing and inductive effects. Ortho and (to a smaller degree) para substituents decrease the chemical shift, which means that they increase the electron density on the phenolic carbon. The σo/σp ratio is 2.2 for δ13C1, which is not very far away from the similar ratio calculated based on pK values (3.4). This is the primary origin of the good correlation between pK and δ13C1 within each of the three series shown in Fig. 3. The σm value obtained from δ13C1 is positive, which means that meta substitution removes some electron density from the phenolic carbon. The magnitude of the effect, however, is much smaller than para or ortho effects. This is, again, in excellent agreement with the classic interpretation of the inductive effect.
Sigma values obtained from rate constants show a third trend. First of all, the rate constant of 246 did not follow the pattern set by the remaining 19 compounds. Therefore, the statistical analysis was done and Fig. 6 is shown based on 19 points. A version of this graph with all 20 points is shown as Fig. S11 in the ESI†, and illustrates that the effect of even a single point can hide an otherwise significant correlation in the statistical analysis. The largest substituent effect, similarly to the previous two cases, is exerted by the orthogroup, and the negative value of σo indicates that ortho substitution decreases the reactivity in the cerium(IV) oxidation. Earlier, there have been numerous attempts to characterize ortho substituents with Hammett constants.47–52 These efforts did not yield widely accepted σo values, which is classically interpreted as a consequence of steric effects. However, this interpretation would not be without contradictions in our special case. First of all, chlorine is a single atom, which is much closer in steric bulk to a hydrogen atom than any other chemical groups with several connected atoms. In addition, the effect compared to the effect of the para substituents is actually weaker than similar ratios for characteristic parameters (pK and δ13C1). Therefore, there is not much experimental ground for implying steric effects in this process.
Meta substitution has no significant effect on the reactivity as shown by the σm value, which is not significantly different from 0. Para substitution, on the other hand, clearly increases the reactivity: σp is positive and its value is about a half of −σo. Based on these observations, 4, 34 and 345 are expected to have the largest rate constants, which is in full agreement with the data shown in Table 1. It is also observed that the effects are strongly position-dependent: there is a 50-fold difference in rate constants and a pK change of more than 2.0 determined just within the series of six trichlorophenols. The chlorine substituents are not directly involved either in acid dissociation or in the rate determining step of the cerium(IV) reaction. Therefore, their effect must be exerted solely by modifying the electronic structure of the aromatic system. As pointed out previously, this can be often understood in terms of electron withdrawing and inductive effects.
As mentioned above, 246 did not fit the trend of rate constants defined by the rest of the chlorophenols. This is a quite unexpected finding and a warning for the future design of experiments because 246 has been used as a typical chlorophenol in many earlier studies.3,7,11 246 is not exceptional in its characteristic parameters, but the fact that it is the chlorophenol with the lowest δ13C1 should not go unnoticed. Therefore, it is the chlorophenol with the most electron rich phenolic carbon atom and it is not unfeasible that the oxidation mechanism for 246 is different from the remaining group of 19 compounds. It is also to be recalled that 246 is the case when some hints of rate saturation are seen in the rate equation which might be attributable to the complexation of reactants. No similar phenomena were observed experimentally for any of the 19 other compounds.
The generally accepted Hammett substituent constants45,46,48,52 for chlorine are σp = 0.23 and σm = 0.37. This fits the values obtained from the pK data in this work (σp/σm = 1.61 from the accepted values and 1.75 from the pK data of Table 2), which is perhaps not surprising as the Hammett substituent constants were originally defined based on the pK values of substituted benzoic acids.46 The Hammett constants are primarily used to interpret kinetic data. In the series of reactions studied in this work, they are unsuitable for this purpose, which is illustrated by a Hammett plot for the six chlorophenols not containing any ortho substituents (Fig. S12, ESI†). This plot does not show any correlation.
Conclusion
In this work, we have shown that the oxidation of chlorophenols by cerium(IV) follows 1∶2 stoichiometry and a simple second-order rate equation in the acidic region. pK values and 13C NMR chemical shifts have been determined as characteristic parameters. The rate constants do not correlate well with these characteristic parameters or with the conventional Hammett substituent constants. However, all three sets of parameters follow well defined additivity rules. 2,4,6-Trichlorophenol shows a substantially different behavior than other chlorophenols do, which should be taken into account when selecting typical chlorophenols as model compounds in future studies.
Acknowledgements
The authors thank the Tempus Foundation and Hungarian Science Foundation (OTKA) for financial support under grant No. K68668. The research was supported by the EU and co-financed by the European Social Fund through the Social Renewal Operational Programme under the project CHEMIKUT (TÁMOP-4.2.2-08/1-2008-0012). The financial support of TEVA Hungary Ltd. is also highly appreciated. Some experiments were carried out using the facilities of Iowa Sate University and Ames Laboratory in Ames, Iowa, US.References
- L. H. Keith and W. A. Telliard, Environ. Sci. Technol., 1979, 13, 416.
- Decision No 2455/2001/EC of the European Parliament and of the Council of 20 November 2001 establishing the list of priority substances in the field of water policy and amending Directive 2000/60/EC.
- A. Sorokin, J. L. Séris and B. Meunier, Science, 1995, 268, 1163 CrossRef CAS.
- F. J. Benitez, J. Beltran-Hereida, J. L. Acero and F. J. Rubio, Ind. Eng. Chem. Res., 1999, 38, 1341 CrossRef CAS.
- L. Padilla, V. Matus, P. Zenteno and B. Gonzalez, J. Basic Microbiol., 2000, 40, 243 CrossRef CAS.
- B. Meunier, Science, 2002, 296, 270 CrossRef CAS.
- S. Sen Gupta, M. Stadler, C. A. Noser, A. Ghosh, B. Steinhoff, D. Lenoir, C. P. Horwitz, K. W. Schramm and T. J. Collins, Science, 2002, 296, 326 CrossRef.
- G. Lente and J. H. Espenson, Chem. Commun., 2003, 1162 RSC.
- M. Fukushima, A. Sawada, M. Kawasaki, H. Ichikawa, K. Morimoto and K. Tatsumi, Environ. Sci. Technol., 2003, 37, 1031 CrossRef CAS.
- G. Lente and J. H. Espenson, New J. Chem., 2004, 28, 847 RSC.
- G. Lente and J. H. Espenson, Int. J. Chem. Kinet., 2004, 36, 449 CrossRef CAS.
- G. Lente and J. H. Espenson, Green Chem., 2005, 7, 28 RSC.
- G. Díaz-Díaz, M. Celis-García, M. C. Blanco-López, M. J. Lobo-Castanon, A. J. Miranda-Ordieres and P. Tunón-Blanco, Appl. Catal., B, 2010, 96, 51 CrossRef CAS.
- K. C. Christoforidis, M. Louloudi, E. R. Milaeva and Y. Deligiannakis, J. Catal., 2010, 270, 153 CrossRef CAS.
- K. C. Christoforidis, M. Louloudi and Y. Deligiannakis, Appl. Catal., B, 2010, 95, 297 CrossRef CAS.
- Q. Raffy, R. Ricoux, E. Sansiaume, S. Pethe and J. P. Mahy, J. Mol. Catal. A: Chem., 2010, 317, 19 CrossRef CAS.
- S. I. Ahonkhai, I. Wiater and R. Louw, Organohalogen Compd., 2000, 46, 74 CAS.
- J. M. Mayer, D. A. Hrovat, J. L. Thomas and W. T. Borden, J. Am. Chem. Soc., 2002, 124, 11142 CrossRef CAS.
- I. J. Rhile and J. M. Mayer, J. Am. Chem. Soc., 2004, 126, 12718 CrossRef CAS.
- C. Isborn, D. A. Hrovat, W. S. Borden, J. M. Mayer and B. K. Carpenter, J. Am. Chem. Soc., 2005, 127, 5794 CrossRef CAS.
- J. P. Aguer, G. Mailhot and M. Bolte, New J. Chem., 2006, 30, 191 RSC.
- F. d'Acunzo, C. Galli, P. Gentili and F. Sergi, New J. Chem., 2006, 30, 583 RSC.
- C. Costentin, M. Robert and J. M. Saveant, J. Am. Chem. Soc., 2006, 128, 4552 CrossRef CAS.
- O. V. Zalomaeva and A. B. Sorokin, New J. Chem., 2006, 30, 1768 RSC.
- G. Litwinienko and K. U. Ingold, Acc. Chem. Res., 2007, 40, 222 CrossRef CAS.
- C. Galli, P. Gentili, A. S. Nunes Pontes, J. A. F. Gamelas and D. V. Evtuguin, New J. Chem., 2007, 31, 1461 RSC.
- N. Song and D. M. Stanbury, Inorg. Chem., 2008, 47, 11458 CrossRef CAS.
- O. V. Zalomaeva, I. D. Ivanchikova, O. A. Kholdeeva and A. B. Sorokin, New J. Chem., 2009, 33, 1031 RSC.
- B. Tonomura, H. Nakatani, M. Ohnishi, J. Yamaguchi-Ito and K. Hiromi, Anal. Biochem., 1978, 84, 370 CrossRef CAS.
- G. Peintler, A. Nagy, A. K. Horváth, T. Körtvélyesi and I. Nagypál, Phys. Chem. Chem. Phys., 2000, 2, 2575 RSC.
- A. K. Covington, R. G. Bates and R. A. Durst, Pure Appl. Chem., 1985, 57, 531.
- SCIENTIST, Version 2.0, Micromath Software, Salt Lake City, UT, USA, 1995 Search PubMed.
- MatLab for Windows, Version 4.2c1, The Mathworks, Inc., Natick, MA, USA, 1994 Search PubMed.
- NIST Standard Reference Database 46 Version 8: Critically Selected Stability Constants of Metal Complexes (http://www.nist.gov/ts/msd/srd/nist46.cfm).
- The IUPAC Stability Constants Database, SC-Database (http://www.acadsoft.co.uk/).
- L. Lepri, P. G. Desideri and D. Heimler, J. Chromatogr., 1980, 195, 339 CrossRef CAS.
- G. Lente and J. H. Espenson, J. Photochem. Photobiol., A, 2004, 163, 249 CrossRef CAS.
- W. H. Hunter and M. Morse, J. Am. Chem. Soc., 1926, 48, 1615 CrossRef CAS.
- H. Lübbecke and P. Boldt, Angew. Chem., Int. Ed. Engl., 1976, 15, 608 CrossRef.
- R. S. Shukla, A. Robert and B. Meunier, J. Mol. Catal. A: Chem., 1996, 113, 45 CrossRef CAS.
- R. Willstätter and H. E. Muller, Ber. Dtsch. Chem. Ges., 1911, 44, 2182.
- R. D. Kaushik, R. P. Singh and V. Shashi, Asian J. Chem., 2003, 15, 1485 CAS.
- M. P. Shurygina, Y. A. Kurskii, N. O. Druzhkov, S. A. Chesnokov, L. G. Abakumova, G. K. Fukin and G. A. Abakumov, Tetrahedron, 2008, 64, 9784 CrossRef CAS.
- G. Albarran, W. Boggess, V. Rassolov and R. H. Schuler, J. Phys. Chem. A, 2010, 114, 7470 CrossRef CAS.
- J. H. Espenson, Chemical Kinetics and Reaction Mechanisms, 2nd edn, McGraw-Hill, New York, 1995, p. 25 and p. 226 Search PubMed.
- L. P. Hammett, Chem. Rev., 1935, 17, 125 CrossRef CAS.
- M. Charton, J. Am. Chem. Soc., 1969, 91, 6649 CrossRef CAS.
- C. H. Yoder, R. H. Tuck and R. E. Hess, J. Am. Chem. Soc., 1969, 91, 539 CrossRef CAS.
- M. Charton, Prog. Phys. Org. Chem., 1971, 8, 235 CAS.
- C. D. Schaeffer, Jr. and C. H. Yoder, J. Org. Chem., 1971, 36, 2201 CrossRef.
- T. Fujita and T. Nishioka, Prog. Phys. Org. Chem., 1976, 12, 49 CAS.
- C. Hansch and A. Leo, Substituent Constants for Correlation Analysis in Chemistry and Biology, Wiley, New York, 1979 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures, tables and mathematical derivations referred to in the text of the article. See DOI: 10.1039/c0nj00529k |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2011 |