Phosphoglycerate mutase from Trypanosoma brucei is hyperactivated by cobaltin vitro, but not in vivo
Fazia Adyani Ahmad
Fuad
a,
Linda A.
Fothergill-Gilmore
a,
Matthew W.
Nowicki
a,
Lorna J.
Eades
b,
Hugh P.
Morgan
a,
Iain W.
McNae
a,
Paul A. M.
Michels
c and
Malcolm D.
Walkinshaw
*a
aStructural Biochemistry Group, Institute of Structural and Molecular Biology, University of Edinburgh, Michael Swann Building, The King's Buildings, Mayfield Road, Edinburgh, UK EH9 3JR
bSchool of Chemistry, University of Edinburgh, Joseph Black Building, The King's Buildings, West Mains Road, Edinburgh, UK EH9 3JJ
cResearch Unit for Tropical Diseases, de Duve Institute and Laboratory of Biochemistry, Université catholique de Louvain, Avenue Hippocrate 74, B-1200 Brussels, Belgium
First published on 13th October 2011
Abstract
Production of ATP by the glycolytic pathway in the mammalian pathogenic stage of protists from the genus Trypanosoma is required for the survival of the parasites. Cofactor-independent phosphoglycerate mutase (iPGAM) is particularly attractive as a drug target because it shows no similarity to the corresponding enzyme in humans, and has also been genetically validated as a target by RNAi experiments. It has previously been shown that trypanosomatid iPGAMs require Co2+ to reach maximal activity, but the biologically relevant metal has remained unclear. In this paper the metal content in the cytosol of procyclic and bloodstream-form T. brucei (analysed by inductively coupled plasma-optical emission spectroscopy) shows that Mg2+, Zn2+ and Fe2+ were the most abundant, whereas Co2+ was below the limit of detection (<0.035 μM). The low concentration indicates that Co2+ is unlikely to be the biologically relevant metal, but that instead, Mg2+ and/or Zn2+ may assume this role. Results from metal analysis of purified Leishmania mexicana iPGAM by inductively coupled plasma-mass spectrometry also show high concentrations of Mg2+ and Zn2+, and are consistent with this proposal. Our data suggest that in vivo cellular conditions lacking Co2+ are unable to support the maximal activity of iPGAM, but instead maintain its activity at a relatively low level by using Mg2+ and/or Zn2+. The physiological significance of these observations is being pursued by structural, biochemical and biophysical studies.
Introduction
Metals are abundant in biological macromolecules, and about one-third of all proteins contain metals that are essential for diverse functions such as enzyme activity, gene expression and cell signalling, as well as for the maintenance of structural integrity. The burgeoning discipline of metalloproteomics is providing insights into the wide range of functions of metalloproteins, and into the common structural signatures encountered in protein metal-binding sites.1–3 These studies are also sounding notes of caution concerning the possibilities for mis-identification of the native metal within a particular metalloprotein because of (i) lack of selectivity of metal-binding sites, (ii) variations in metal concentrations in different environments and (iii) artefactual problems encountered during protein purification. Replacement of a native metal by an alternative metal can have a substantial impact on the properties of the metalloprotein, and in the cases of metalloenzymes may even strikingly enhance the activity above that of the native enzyme. The replacement of Zn2+ by Co2+, Ni2+ or Cu2+ in aminopeptidase is an example in which the non-native metals enhance activity by up to 10-fold.4 Developments in atomic spectroscopy have enabled trace amounts of metals to be detected in scarce biological and purified protein samples,5,6 and therefore have encouraged studies to identify the biologically relevant native metal. Inductively coupled plasma-optical emission spectroscopy (ICP-OES) and inductively coupled plasma-mass spectrometry (ICP-MS) have been used in this study to measure the metal concentrations in subcellular samples derived from protist parasites and in purified protein samples, respectively.The glycolytic pathway of trypanosomatid parasites has been identified as a possible drug target because the production of ATP in the pathogenic bloodstream stage of trypanosomes is entirely dependent on glycolysis.7 In particular, the glycolytic enzyme cofactor-independent phosphoglycerate mutase (iPGAM) is an attractive target because it is completely different from the cofactor-dependent enzyme that catalyses the corresponding reaction in humans,8 and because it has also been genetically validated as a drug target.9Phosphoglycerate mutase (PGAM) catalyses the interconversion between 3-phosphoglycerate and 2-phosphoglycerate in the glycolytic and gluconeogenic pathways. Cofactor-independent PGAM (iPGAM) is active in the absence of the cofactor 2,3-bisphosphoglycerate, and is found in protists including Trypanosoma brucei and Leishmania mexicana, as well as in plants and in some nematodes, fungi, algae, bacteria and archaea. All experimentally characterised iPGAMs are monomeric with a mass of about 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da, and have been classified as members of the metal-dependent alkaline phosphatase superfamily.10 By contrast, cofactor-dependent PGAM (dPGAM) found in humans and other vertebrates, and in some yeasts and bacteria requires the bisphosphoglycerate cofactor for activity (but no metals) and is usually dimeric or tetrameric with subunits of about 27000 Da.11 The two types of PGAM are unrelated and share no sequence similarity.
000 Da, and have been classified as members of the metal-dependent alkaline phosphatase superfamily.10 By contrast, cofactor-dependent PGAM (dPGAM) found in humans and other vertebrates, and in some yeasts and bacteria requires the bisphosphoglycerate cofactor for activity (but no metals) and is usually dimeric or tetrameric with subunits of about 27000 Da.11 The two types of PGAM are unrelated and share no sequence similarity.
There is an absolute requirement for divalent metals by iPGAM, and the enzyme is inactive after treatment with metal chelators.12–14 The crystal structures of iPGAMs from Bacillus stearothermophilus (Bs iPGAM)15 and from L. mexicana (Lm iPGAM)16 share 33% sequence identity, and in their closed conformations possess essentially identical topologies with all 16 active-site residues being the same (Fig. 1). The additional availability of the crystal structure of iPGAM from Bacillus anthracis (Ba iPGAM, 78% sequence identity to Bs iPGAM) in a strikingly different open conformation has enabled detailed structural and molecular dynamic comparisons between the two Bacillus iPGAMs.17 The two metal binding sites illustrated in Fig. 1 have different coordination geometries in these open and closed iPGAM structures, indicating the essential roles played by the metals not only for catalysis, but also for enzyme conformation. Interestingly, the specific metal required by iPGAMs varies from one organism to another, despite overall conservation of the structure. Bs and Ba iPGAMs require Mn2+,18 but T. brucei iPGAM (Tb iPGAM) and Lm iPGAM prefer Co2+,19,20 although other divalent metals may support low activity of trypanosomatid iPGAMs. In addition, Mg2+ is favoured by nematode iPGAMs,21 and Zn2+ is required by alkaline phosphatase, a more-distant member of the metal-dependent phosphatase family.22 Plant iPGAMs are also known to depend on divalent metals, but the identities of the physiologically relevant metals remain controversial.
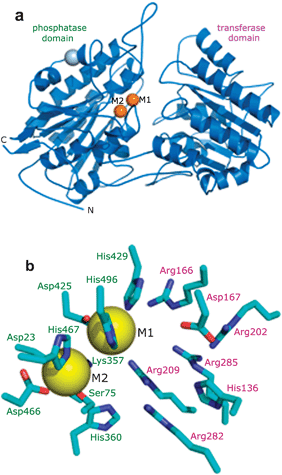 | ||
| Fig. 1 Structure of iPGAM as illustrated by the enzyme from L. mexicana.16 (a) A monomer of iPGAM is folded into two distinct domains designated phosphatase and transferase. The two divalent metal ions at the active site are shown by gold spheres. A Na+ ion (grey sphere) is distant from the active site, and probably plays a structural role. (b) The active site of iPGAM crystallised in the presence of 4 mM Co2+. The 16 amino acid residues shown (Asp23 and Lys357 are mostly hidden by the sphere of M2) are highly conserved in all iPGAMs. Those labelled in green are from the phosphatase domain, and those in purple from the transferase domain. Ser75 becomes phosphorylated during the catalytic mechanism. Metal site 1 (M1) shows high occupancy even at low concentrations of Co2+ (10 μM), but metal site 2 (M2) is only partially occupied under these conditions. Site M1 adopts octahedral geometry, whereas site M2 is tetrahedral. | ||
Co2+ is rare in nature, and it has been suggested that it, like Ni2+, may in fact, represent a relic of early life before the advent of dioxygen.23 In the reducing and sulphide-rich conditions of early life, the availability of Zn2+ was low because of its precipitation as a sulphide, but Co2+ and Ni2+ could have acted as valuable metal biocatalysts. The change to a dioxygen environment encouraged a switch to metal catalysts such as Mn2+ and Zn2+. In these circumstances, it is possible to envisage that an enzyme active site that had originally evolved to use Co2+ may also be able to bind Zn2+, but would function with lower catalytic efficiency. It is relevant to note in this context that epr, nmr and X-ray crystallographic studies of metal binding sites such as those in carbonic anhydrase have shown that it is particularly easy to substitute Zn2+ by other metals such as Co2+.24–27 In these circumstances it is likely that the ‘native’ metal used by a particular enzyme may be more a function of the concentration of acceptable divalent metals in its surroundings than of the selectivity of the enzyme metal binding site.
A main goal of our research is to identify new drugs for the treatment of neglected diseases such as human African trypanosomiasis, Chagas disease and a range of leishmaniases which are all caused by protists belonging to the family Trypanosomatidae. Together, these parasites cause severe public health and economic burdens on populations that are often already caught in a tragic cycle of poverty, poor nutrition and disease. As indicated previously, iPGAM is an attractive drug target, but for high-throughput and virtual screening, as well as for subsequent medicinal chemistry strategies it is essential to be able to understand and manipulate the conformation of the enzyme. This is particularly true for iPGAM with its small active site, so that conditions may be identified that favour the open conformation. Metals are likely to be essential players in this work.
We now report the metal analyses of cytosolic fractions from T. brucei, and of purified bacterially expressed Lm iPGAM samples by ICP-OES and by ICP-MS, respectively. The results indicate that Zn2+ and Mg2+ instead of Co2+ are the biologically relevant metals, and that the cellular activity of iPGAM is only a small fraction of its potential activity.
Experimental
Preparation of T. brucei cytosolic fractions
T. brucei cytosolic fractions were prepared by differential centrifugation as described previously, in isotonic buffer (250 mM sucrose (UCB S.A., Brussels, Belgium), 25 mM Tris-HCl (Fisher Scientific), pH 7.4, 1 mM EDTA (Fluka Biochemika)).28EDTA was included primarily as a proteolytic inhibitor, but is also a useful form of metal sequestration. It was not removed from the fractions, and would thus still be present with its bound metals during the ICP-OES measurements. Soluble proteins in the cytosolic fractions were obtained in the supernatant after centrifugation at 50000×g. Seven samples (3 mL each) were obtained that had been prepared on different occasions: three cytosolic samples from in vitro cultured procyclic insect-stage parasites (S1, S2, S3), and four cytosolic samples from pathogenic bloodstream-form parasites (S4, S5, S6 and S7). Samples S5 and S6 gave metal analysis results with relatively high standard deviations, and have been omitted.
Protein determination
Concentrations of proteins in cytosolic samples were measured by Bradford assay,29 with Coomassie Plus Protein Assay Reagent supplied by Thermo Scientific, and with purified Lm iPGAM as a standard. Concentrations of purified Lm iPGAM were determined by UV absorbance at A280 nm using a Jasco V-550 UV-VIS spectrophotometer using the standard Beer–Lambert law, with known extinction coefficient (42080 M−1 cm−1) and molecular mass (60723 Da).
Enzyme assays
iPGAM activity was measured by monitoring the conversion of 3-phosphoglycerate (3-PGA) to 2-phosphoglycerate (2-PGA) coupled with the oxidation of NADH to NAD+ by lactate dehydrogenase (LDH) viaenolase and pyruvate kinase. Assays of each cytosolic fraction were done using a Multimode Plate Reader-Molecular Devices M5 instrument, by the addition of a 10 μL sample of the cytosolic fraction to give a final reaction mixture of 100 μL containing 100 mM triethanolamine (TEA)-HCl buffer pH 7.6 (Sigma Life Sciences), 1.5 mM 3-PGA (Sigma-Aldrich), 5 mM MgCl2 (Fisher Scientific), 50 mM KCl (Fisher Scientific), 0.8 mM NADH (Roche), 1 mM ADP (Sigma-Aldrich), 2 units of enolase (Sigma-Aldrich), 4 units of pyruvate kinase (Sigma-Aldrich) and 6 units of LDH (Fluka Analytical). The decreased absorbance of NADH at A340 nm was used to obtain the rate of reaction for specific activity measurements (one unit corresponds to the conversion of 1 μmol of substrate min−1 mg−1protein under standard conditions). Assays done during the purification of Lm iPGAM used 10 μL samples containing ∼0.2 mg mL−1 iPGAM which were added into a cuvette to give a final reaction mixture of 1000 μL containing 100 mM triethanolamine (TEA)-HCl buffer, pH 7.6, 1.5 mM 3-PGA, 5 mM MgCl2, 50 mM KCl, 0.2 mM NADH, 1 mM ADP, 2 units of enolase, 4 units of pyruvate kinase and 6 units of LDH. The decrease in A340 nm was monitored using a Jasco V-550 UV-VIS spectrophotometer.iPGAM expression and purification
Lm iPGAM with a C-terminal His6 tag was expressed in Escherichia coliBL21(DE3) cells, and cell pellets were collected by centrifugation, with storage at -80 °C as described previously.30Cell lysis was done with a Constant Systems TS Cell Disruptor set to a maximum pressure of 22000 psi in buffer A (50 mM NaH2PO4 (AnalaR BDH), 300 mM NaCl (Fisher Scientific) pH 8) with the addition of EDTA-free Protease Inhibitor Mixture (Roche) tablets (1 tablet per 25 mL of extract). The extract was centrifuged for 45 min at 4 °C at 43400×g, and the supernatant injected onto an immobilised metal affinity chromatography column (FastFlow IMAC, GE Healthcare) pre-charged with 0.1 M Ni2+ (nickel(II) sulphate hexahydrate, Sigma-Aldrich), and washed with water, then buffer A in an ÄKTA HPLC system maintained at 10 °C. Following sample injection, the column was washed with buffer A, followed by 20 column volumes of buffer A containing 25 mM imidazole (Fisher Scientific). The Lm iPGAM was then eluted with a step of buffer A containing 500 mM imidazole, and desalted and buffer exchanged on a HiPrep 26/10 column (GE Healthcare) eluted with buffer B (20 mM HEPES (Sigma Life Sciences) pH 7.6). The protein was then buffer exchanged into 20 mM TEA-HCl, pH 7.6 and 50 mM NaCl using a PD10 column (GE Healthcare), and concentrated using a Vivaspin concentrator (10000 MW cut off) (GE Healthcare) to ∼2 mg mL−1. The protein was stored in the cold room (4 °C) for further biochemical and biophysical assays or at −20 °C with the presence of 10% glycerol.
ICP-OES
ICP-OES was used to analyse cytosolic fractions with a Perkin Elmer Optima 5300 DV instrument, using a radio frequency (RF) forward power of 1400 W, with argon gas flows of 15, 0.2 and 0.75 L min−1 for plasma, auxiliary, and nebuliser flows, respectively. Using a peristaltic pump, the samples were taken up into a Gem Tip cross-Flow nebuliser and Scotts spray chamber at a rate of 1.50 mL min−1. The instrument was operated in axial mode, and the selected wavelengths for each element were analysed in fully quantitative mode (three points per unit wavelength). Three replicate runs per sample were carried out. Initially two wavelengths were selected for each element, but after the analysis was completed the following wavelengths were chosen for reporting results: Co 228.616 nm, Cu 327.393 nm, Fe 238.204 nm, Mg 285.213 nm, Mn 257.610 nm, Ni 231.604 nm and Zn 206.200 nm.To assess the limit of detection (LOD) for each metal and the matrix effects of the buffers, a range of calibration standards was prepared both in deionised water (18Ω, Elga USF) and in EDTA-free cytosol buffer (250 mM sucrose (Fisher Scientific), 25 mM Tris-HCl (Sigma Life Sciences), pH 7.4). Standards of 0, 0.1, 10 and 100 mg L−1 were prepared by sequential dilution from single element 1000 mg L−1 stock solutions (Fisher Scientific). With all of the calibration lines the correlation coefficients for the linear regression were 0.9992 or better. Ten blank samples of each buffer were analysed after the instrument was calibrated to calculate the limits of detection for the analysis. LOD = 2.26*SDb/Slope (where SDb is the standard deviation of the 10 blanks and 2.26 is the student t-test at the 95% confidence limit). The limits of detection are indicated in Fig. 3.
For analysis of the samples, a range of calibration standards from 0–500 mg L−1 was prepared in cytosol buffer. The majority of the samples fell in the range 200–500 mg L−1, and results were calculated against a calibration from 10–500 mg L−1. However where values were lower than 20 mg L−1, the samples were reintegrated against a calibration only extending to 10 mg L−1. Where values fell below the limit of detection they were quoted as a < value.
It is relevant to mention that attempts were initially made to analyse the cytosolic fractions by ICP-MS, but were found to be unsuitable because of technical problems.
ICP-MS
Purified protein samples were analysed using an Agilent 7500ce (with octopole reaction system) instrument, employing an RF forward power of 1540 W and reflected power of 1 W, with argon gas flows of 0.82 L min−1 and 0.2 L min−1 for carrier and makeup flows, respectively. For the collision cell the helium flow was 0.6 L min−1. Sample solutions were taken up into the Micro mist using a peristaltic pump at a rate of approximately 1.2 mL min−1. Skimmer and sample cones were made of nickel. The instrument was operated in spectrum multi-tune acquisition mode and five replicate runs per sample were employed. Each mass was analysed in fully quantitiative mode (three points per unit mass). The following masses were selected for analysis: 55Mn, 59Co and 63Cu which were analysed in ‘standard gas mode’, and 26Mg, 56Fe, 60Ni and 66Zn which were analysed in ‘helium gas mode’ using the collision cell to remove any polyatomic interferences.Calibration standards with a range of metal concentrations (0, 1, 10 and 100 μg L−1) were prepared by sequential dilution from single element 1000 mg L−1 stock solutions (Fisher Scientific) in 20 mM TEA buffer pH 7.6 and 50 mM NaCl, as well as in water to assess matrix effects. Samples were analysed using the standards prepared in 20 mM TEA buffer pH 7.6. Ten blank samples of the 20 mM TEA buffer pH 7.6 buffer were analysed to calculate the LOD for each metal, and are indicated in Fig. 5. Where values fell below the limit of detection they were quoted as a < value.
Bioinformatics
Protein structure diagrams were generated using the program PyMOL.31 The calculated extinction coefficient and molecular mass of Lm iPGAM were obtained from the Expasy ProtParam website (http://web.expasy.org/protparam/).Results and discussion
Preparation and characterisation of Tb iPGAM cytosolic fractions
Trypanosomatids possess a unique subcellular organelle, the glycosome, with multiple copies in each cell which sequester the first seven of the ten glycolytic enzymes. iPGAM however, is located in the cytosol8,20,32 along with enolase and pyruvate kinase. Cytosolic samples were prepared at the Université catholique de Louvain, Brussels, Belgium from both procyclic and bloodstream stages of T. brucei. The former stage occurs in the tsetse fly vector, whereas the latter stage corresponds to the pathogenic form found in mammalian blood. The cytosolic fractions were analysed by SDS-PAGE (Fig. 2), and gave sharp bands indicative of the absence of proteolytic degradation. The patterns of bands are broadly similar across all the fractions, with the exception of a prominent band with a mass of ∼50 kDa in sample S4. The identity of this protein is not known. | ||
| Fig. 2 Electrophoretic analysis of cytosolic fractions. A 12% SDS-PAGE gel was used with 5 μL samples of each cytosolic fraction. The masses (in kDa) of standard marker proteins are indicated in lane M. Abbreviations: M, marker proteins; S1–S3, cytosolic fractions from procyclic T. brucei; S4 and S7, cytosolic fractions from bloodstream-form T. brucei. | ||
The protein concentrations of the fractions showed variation (i.e. from 2.8 to 8.1 mg mL−1), so the results for specific activity and for metal contents were all normalised to 1 mg of protein. The protein concentrations, iPGAM specific activities and estimated iPGAM concentrations of the fractions are given in Table 1. The specific activities were measured for the cytosolic fractions with no additional Co2+. It can be seen that the specific activities of iPGAM in the procyclic cytosolic fractions were higher than in the bloodstream-form cytosolic fractions. As anticipated, the addition of Co2+ gave a substantial enhancement of activity (about 10-fold, data not shown).
| S1a | S2 | S3 | S4 | S7 | |
|---|---|---|---|---|---|
| a Samples: S1, S2 and S3 are procyclic fractions; S4 and S7 are from bloodstream parasites. b The calculation was made using the assumption that pure iPGAM has a specific activity of 21 μmole min−1 mg−1protein (under the same assay conditions as those used to measure the activity of iPGAM in the cytosol). | |||||
| Protein concentration (mg mL−1) | 4.0 | 3.9 | 2.8 | 8.1 | 5.4 |
| Specific activity (nmol min−1 mg−1protein) | 16.0 | 19.7 | 6.29 | 1.12 | 5.80 |
| iPGAM concentration in cytosolic fractions (μmoles L−1)b | 50.1 | 60.3 | 13.8 | 7.12 | 24.5 |
ICP-OES analysis of T. bruceicytosol shows the presence of Mg2+, Zn2+ and Fe2+, and absence of Co2+
The concentrations of seven different metals (Co2+, Cu2+, Fe2+, Mg2+, Mn2+, Ni2+ and Zn2+) were measured by ICP-OES analysis of cytosolic fractions from procyclic- and bloodstream-form T. brucei. This type of analysis gives the total metal concentrations, and does not distinguish between metals bound to macromolecules or to EDTA, and those unbound. Elemental standards were prepared as described in the Experimental section, and gave linear concentration-response plots in both water and in cytosol buffer. Any background readings or noise signals collected from the control data were deducted from the readings of the samples. Mg2+, Zn2+ and Fe2+ were found to be the most abundant metals (∼200, 30 and 15 μM, respectively; Fig. 3), with Cu2+, Mn2+ and Ni2+ below 1 μM, and Co2+ below the limit of detection (<0.035 μM). It can be seen that the samples from the two different life stages of T. brucei gave similar results. | ||
| Fig. 3 ICP-OES of cytosolic fractions from procyclic and bloodstream-form T. brucei. The concentrations of metals in the cytosolic fractions are shown. The blue bars correspond to the procyclic cytosolic fractions S1, S2 and S3, whereas the pink bars indicate bloodstream cytosolic fractions S4 and S7. (a) Results for the relatively abundant metals, Fe2+, Mg2+ and Zn2+, with the y axis scale 0–200 μM. (b) Results for the less abundant metals, Co2+, Cu2+, Mn2+ and Ni2+, with the y axis scale 0–1.0 μM. The values for Co (0.0130–0.0296 μM) are below the limit of detection (<0.0352 μM). The following limit of detection values (all μM) were obtained (see Experimental section): Fe, 0.0819; Mg, 0.495; Zn, 0.122; Co, 0.0352; Cu, 0.116; Mn, 0.0920; Ni, 0.104. | ||
The specific activity of bacterially expressed Tb iPGAM purified in the absence of Co2+ is ∼25 μmol min−1 mg−1protein,8,19 very similar to the value of 21 μmol min−1 mg−1protein determined for pure bacterially expressed Lm iPGAM in this study (see next section). The cytosolic concentrations of Tb iPGAM may be estimated from the specific activity of the pure enzyme compared to the specific activities within the cytosolic fractions, and give a value of approximately 10-50 μM (Table 1). It is clear from the ICP-OES results that the Co2+ concentrations in the cytosolic fractions are <0.035 μM, and are therefore insufficient to satisfy even a small proportion of the iPGAM present.
Purification and characterisation of Lm iPGAM
The bacterial expression system for Lm iPGAM was the same as described previously.20 In the present study however, purified Lm iPGAM was obtained in the absence of added Co2+ from a two-step purification procedure: IMAC chromatography and desalting with buffer exchange (Fig. 4). The Ni2+-containing IMAC column in this procedure replaced the previously used Co2+-containing Talon (Clontech) column,30 and the 10 μM CoCl2 used throughout previous purification procedures was omitted. The yield of protein obtained from 1 L of cell culture was found to be ∼20 mg. SDS-PAGE analysis showed that the purified Lm iPGAM gave a single prominent band with mass ∼60700 Da, corresponding to the expected mass (Fig. 4). The specific activity was calculated to be 21 μmol min−1 mg−1protein for the conversion of 3-PGA to 2-PGA, similar to the value of 26 μmol min−1 mg−1protein obtained for pure bacterially expressed Tb iPGAM that had also been purified on a Ni2+ IMAC column without the addition of any divalent metals.19Lm iPGAM purified in the Edinburgh laboratory from a GE Healthcare IMAC column pre-charged with 0.1 M Co2+ or from a Talon column had specific activities of 370 ± 2 μmol min−1 mg−1 or ∼400 μmol min−1 mg−1protein,33 respectively. These values correspond closely with Lm iPGAM purified from a Talon column in the Brussels laboratory with a specific activity of 419 ± 4 μmol min−1 mg−1.20 The activity assays for all these enzymes were carried out in the presence of Co2+, and clearly all gave an approximately 20-fold enhancement of activity over Lm iPGAM purified and assayed in the absence of Co2+.
 | ||
| Fig. 4
Purification of Lm iPGAM. The two-step procedure involved (a) IMACvia the enzyme's C-terminal His tag on a Ni2+-charged column, followed by (b) desalting and buffer exchange with a HiPrep 26/10 column. SDS-PAGE gels of peak fractions from both steps are shown. The standard protein markers are as in Fig. 2. The purified enzyme was homogenous with the expected mass of 60700 Da, as shown by the SDS-PAGE gel in panel b. Blue traces correspond to absorbance at 280 nm, and the green trace in panel a indicates the concentration of imidazole. | ||
ICP-MS analysis of purified Lm iPGAM fails to detect Co2+
The metal content of the essentially homogenous purified bacterially expressed Lm iPGAM was analysed by the more sensitive ICP-MS technique. As for ICP-OES, the concentrations of seven different metals (Co2+, Cu2+, Fe2+, Mg2+, Mn2+, Ni2+ and Zn2+) were measured. Elemental standards were prepared as described in the Experimental section, and gave linear concentration-response curves in both water and in enzyme storage buffer (20 mM TEA-HCl, pH 7.6 and 50 mM NaCl). In the case of the purified enzyme, Mg2+ was the most abundant metal (as it was in the cytosolic fractions), but the amount of Zn2+ was substantially enhanced, and approached the concentration of Mg2+ (Fig. 5). The concentration of Co2+ was 0.01 μM, above the limit of detection (<0.0002 μM), but very low compared to the concentration of iPGAM (0.99 μM). Taken together, these results show clearly that despite Co2+ being the metal that enables Tb iPGAM and Lm iPGAM to be maximally active, it is not present in the enzyme in cytosolic fractions or in enzyme purified in the absence of added Co2+. The molar ratios of Mg2+ and Zn2+ to Lm iPGAM in the purified enzyme were ∼8 and ∼6 μmoles of metal to 1 μmole of Lm iPGAM, respectively, indicating that either or both of these metals may be the preferred metal(s) in vivo. By contrast, Mn2+ that is known to be the preferred metal for bacterial iPGAMs15,18 was present in the purified Lm iPGAM at a molar ratio of less than 0.3. It is noteworthy that the concentration of Ni2+ was low, despite the purification being done on an IMAC column charged with Ni2+. | ||
| Fig. 5 ICP-MS analysis of purified Lm iPGAM. The relative molar concentrations of metals and purified Lm iPGAM are shown. (a) Abundant metals, Fe, Mg and Zn, with the y axis scale 0–8. (b) Low abundance metals, with the y axis scale 0–0.25. The following limit of detection values (all μM) were obtained (see Experimental section): Fe, 0.0313; Mg, 0.195; Zn, 0.00652; Co, 0.000238; Cu, 0.000936; Mn, 0.00128; Ni, 0.000803. | ||
Mg2+ and Zn2+ are likely to be the biologically relevant metals of trypanosomatid iPGAMs
It is generally accepted that enzymes are evolving by natural selection to attain a state of optimal catalytic activity, often with adaptation to a particular biological environment. As an example, the activity of the glycolytic enzyme triose phosphate isomerase is limited only by diffusion of substrate onto, and product off, the enzyme, and is therefore thought to have evolved to almost catalytic perfection.34 There is thus an understandable tendency to correlate the highest specific activity of an enzymein vitro with its ‘native’ state in vivo. This is clearly not the case for trypanosomatid iPGAMs where the optimal activity (by a factor of ten or more) is obtained with Co2+, but the concentration of this metal is essentially undetectable in the parasite cytosol. In this respect, iPGAM is not alone, and carboxypeptidase A,35aminopeptidase,4alcohol dehydrogenase,365′-nucleotidase37 and catechol dioxygenase38 are just some among many examples of Zn2+-containing enzymes in vivo that are hyperactive when Co2+ replaces the Zn2+in vitro.The results of ICP-OES indicate that only Mg2+ and Zn2+ are sufficiently abundant in the parasite cytosol to support the activity of a significant proportion of the iPGAM present (Fig. 3 and Table 1), whereas Cu2+, Mn2+ and Ni2+ were all well below the concentration of iPGAM. A similar trend is observed with the ICP-MS analysis of purified Lm iPGAM in which only Mg2+ and Zn2+ are sufficiently abundant to support the activity of the enzyme (Fig. 5). It thus seems that the biologically relevant metal for this enzyme is likely to be either Mg2+ or Zn2+, or both. As mentioned previously, iPGAM belongs to the metal-dependent alkaline phosphatase superfamily10 most of whose members require two divalent metals at the active site. Four enzymes are particularly well characterised structurally: E. colialkaline phosphatase,22Bs iPGAM (closed conformation),15Ba iPGAM (open conformation)17 and Lm iPGAM.16Alkaline phosphatase uses two Zn2+ ions, Bs iPGAM two Mn2+ ions, but Lm iPGAM by contrast, appears to function with a one-metal mechanism. Of the two metal sites of Lm iPGAM (Fig. 1), only site M1 shows high occupancy, with site M2 having only low occupancy (6%) even at 4 mM Co2+. Moreover, when site M2 is occupied, the side chain of Ser75 that is phosphorylated during catalysis adopts a position unfavourable for phospho transfer.16 Site M1 has octahedral geometry, indicating that it has a preference for Mg2+ and Co2+ (MESPEUS database, http://mespeus.bch.ed.ac.uk/MESPEUS/_1.jsp). The poorly occupied site M2 has tetrahedral geometry with a likely preference for Zn2+. It is not possible at this stage to decide definitively which metal is the biologically relevant one, and indeed it may be that both metals can support trypanosomatid iPGAM activity.
On a note of caution, it can be mentioned that the discovery that Tb iPGAM and Lm iPGAM are optimally active with Co2+ was an accident, and a direct consequence of two experimental developments: (i) the use of His tags as a very convenient purification method for bacterially expressed proteins, and (ii) the availability of the Co-containing Talon IMAC resin with enhanced affinity for His tags. Both innovations were adopted by the Brussels and Edinburgh research groups, and have been very efficient for the purification of large quantities of several trypanosomatid glycolytic enzymes, including iPGAMs. Furthermore, routine enzyme stability studies were carried out on Tb iPGAM and Lm iPGAM as a strategy to improve the outcome of crystallisation trials.33 These studies showed that Co2+ was an excellent stabilisation agent for the trypanosomatid iPGAMs, and 10 μM CoCl2 was thereafter frequently included in buffers.
An important corollary of the findings in this paper is that the specific activity of trypanosomatid iPGAMin vivo is relatively low compared with the specific activities of other glycolytic enzymes.9 This is thus another reason why it is an attractive drug target because it exerts a higher flux control than most of the other glycolytic enzymes under typical metabolic conditions. The only exception is glyceraldehyde 3-phosphate dehydrogenase, which has a similar flux control coefficient9 when measured under the same metabolic conditions. Moreover, as previously mentioned, there is essentially no problem about species specificity of inhibitors because human and parasite PGAMs share no similarity in structure or mechanism.
Conclusion
ICP-OES was used to determine the divalent metal contents of cytosolic fractions from procyclic and bloodstream-form T. brucei, and now provides for the first time essential information relevant to understanding the in vivo properties of trypanosomatid metal-dependent enzymes. Of particular interest is the metal dependency of iPGAM, which has previously been shown to be maximally active in the presence of Co2+. The ICP-OES results show clearly that the biologically relevant metal for this enzyme cannot be Co2+ which was undetectable in the cytosolic fractions. It is likely that iPGAM is in fact a relatively inefficient Mg2+ and/or Zn2+-dependent enzyme. The significance of these observations is being further explored with the development of a new discontinuous plate-reader activity assay coupled to enolase, pyruvate kinase and lactate dehydrogenase to be carried out with minimal metal interference of the latter enzymes. An understanding of the in vivo metal requirements of iPGAM will be important for the design and implementation of a drug discovery programme currently in progress.Acknowledgements
We thank Melisa Gualdron-Lopez and Muriel Mazet, de Duve Institute, Brussels, Belgium for providing the cytosolic fractions, and Dr Buabarn Kuaprasert (Siam Synchrotron Light Research Institute) for helpful discussions. This project has been funded by the Ministry of Higher Education Malaysia and the University of Science Malaysia. The Centre for Translational and Chemical Biology and the Edinburgh Protein Production Facility were funded by the Wellcome Trust and the BBSRC. We also acknowledge the use of the Joint Chemistry/Geosciences ICP Facility, University of Edinburgh.References
- M. M. Harding, M. W. Nowicki and M. D. Walkinshaw, Crystallogr. Rev., 2010, 16, 1–56 CrossRef.
- W. Maret, Metallomics, 2010, 2, 117–125 RSC.
- A. Cvetkovic, A. L. Menon, M. P. Thorgersen, J. W. Scott, F. L. Poole, F. E. Jenney, W. A. Lancaster, J. L. Praissman, S. Shanmukh, B. J. Vaccaro, S. A. Trauger, E. Kalisiak, J. V. Apon, G. Siuzdak, S. M. Yannone, L. A. Tainer and M. W. W. Adams, Nature, 2010, 779–782 CrossRef CAS.
- J. M. Prescott, F. W. Wagner, B. Holmquist and B. L. Vallee, Biochemistry, 1985, 24, 5350–5356 CrossRef CAS.
- J. R. Dean, S. Munro, L. Ebdon, H. M. Crews and R. C. Massey, J. Anal. At. Spectrom., 1987, 2, 607–610 RSC.
- D. J. Gray, R. Burns, P. Brunswick, J. Le Huray, W. Chan, M. Mast, K. Allamneni and S. Sreedharan, in Plasma Source Mass Spectrometry: Current Trends and Future Developments, ed. G. Holland and D. R. Bandura, Royal Society of Chemistry, 2005, pp. 43–58 Search PubMed.
- C. L. M. J. Verlinde, V. Hannaert, C. Blonski, M. Willson, J. Périé, L. A. Fothergill-Gilmore, F. R. Opperdoes, M. H. Gelb, W. G. J. Hol and P. A. M. Michels, Drug Resist. Updates, 2001, 4, 1–14 CrossRef.
- N. Chevalier, D. J. Rigden, J. Van Roy, F. Opperdoes and P. A. M. Michels, Eur. J. Biochem., 2000, 267, 1464–1472 CrossRef CAS.
- M.-A. Albert, J. R. Haanstra, V. Hannaert, J. Van Roy, F. R. Opperdoes, B. M. Bakker and P. A. M. Michels, J. Biol. Chem., 2005, 280, 28306–28315 CrossRef CAS.
- M. Y. Galperin, A. Bairoch and E. V. Koonin, Protein Sci., 1998, 7, 1829–1835 CrossRef CAS.
- L. A. Fothergill-Gilmore and P. A. M. Michels, Prog. Biophys. Mol. Biol., 1993, 59, 105–236 CrossRef CAS.
- G. C. Smith and L. F. Hass, Biochem. Biophys. Res. Commun., 1985, 131, 743–749 CrossRef CAS.
- G. C. Smith, A. D. McWilliams and L. F. Hass, Biochem. Biophys. Res. Commun., 1986, 136, 336–340 CrossRef CAS.
- C. M. Johnson, M. J. Gore and N. C. Price, Biochem. Soc. Trans., 1987, 15, 878 CAS.
- M. J. Jedrzejas, M. Chander, P. Setlow and G. Krishnasamy, EMBO J., 2000, 19, 1419–1431 CrossRef CAS.
- M. W. Nowicki, B. Kuaprasert, I. W. McNae, H. P. Morgan, M. M. Harding, P. A. M. Michels, L. A. Fothergill-Gilmore and M. D. Walkinshaw, J. Mol. Biol., 2009, 394, 535–543 CrossRef CAS.
- M. Nukui, L. V. Mello, J. E. Littlejohn, B. Setlow, P. Setlow, K. Kim, T. Leighton and M. J. Jedrzejas, Biophys. J., 2007, 92, 977–988 CrossRef CAS.
- N. J. Kuhn, B. Setlow, P. Setlow, R. Cammack and R. Williams, Arch. Biochem. Biophys., 1995, 320, 35–42 CrossRef CAS.
- J.-F. Collet, V. Stroobant and E. Van Schaftinger, FEMS Microbiol. Lett., 2001, 204, 535–543 CrossRef.
- D. G. Guerra, D. Vertommen, L. A. Fothergill-Gilmore, F. R. Opperdoes and P. A. M. Michels, Eur. J. Biochem., 2004, 271, 1798–1810 CrossRef CAS.
- S. Raverdy, Y. Zhang, J. Foster and C. K. Carlow, Mol. Biochem. Parasitol., 2007, 156, 210–216 CrossRef CAS.
- E. E. Kim and H. W. Wyckoff, J. Mol. Biol., 1991, 218, 449–464 CrossRef CAS.
- J. J. R. Fraústo da Silva and R. J. P. Williams, in The Biological Chemistry of the Elements, Oxford University Press, 2nd edn, 2001 Search PubMed.
- K. Håkansson and A. Wehnert, J. Mol. Biol., 1992, 228, 1212–1218 CrossRef.
- I. Bertini, C. Luchinat, R. Pierattelli and A. J. Vila, Eur. J. Biochem., 1992, 208, 607–615 CrossRef CAS.
- S. Lindskog, Pharmacol. Ther., 1997, 74, 1–20 CrossRef CAS.
- B. Bennett, in Metals in Biology: Applications of High-Resolution EPR to Metalloenzymes, ed. G. Hanson and L. Berliner, Springer Science, 2010, ch. 10 Search PubMed.
- O. Misset and F. R. Opperdoes, Eur. J. Biochem., 1984, 144, 475–483 CrossRef CAS.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS.
- B. Poonperm, D. G. Guerra, I. W. McNae, L. A. Fothergill-Gilmore and M. D. Walkinshaw, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2003, 59, 1313–1316 CrossRef.
- W. L. DeLano, DeLano Scientific, San Carlos, CA, 2002.
- K. K. Oduro, I. W. Flynn and I. B. R. Bowman, Exp. Parasitol., 1980, 50, 123–135 CrossRef CAS.
- B. Poonperm, PhD Thesis, University of Edinburgh, 2005 Search PubMed.
- W. J. Albery and J. R. Knowles, Biochemistry, 1976, 15, 5631–5640 CrossRef CAS.
- W. D. Behnke and B. L. Vallee, Proc. Natl. Acad. Sci. U. S. A., 1972, 69, 2442–2445 CrossRef CAS.
- M. Cavaletto, E. Pessione, A. Vanni and C. Giunta, J. Biotechnol., 2000, 84, 87–91 CrossRef CAS.
- L. McMillen, I. R. Beacham and D. M. Burns, Biochem. J., 2003, 372, 625–630 CrossRef CAS.
- A. J. Fielding, E. G. Kovaleva, E. R. Farquhar, J. D. Lipscomb and L. Que, J. Biol. Inorg. Chem., 2010 Search PubMed.
| This journal is © The Royal Society of Chemistry 2011 |