Circular dichroism spectra of cytochrome c oxidase†
Artem V.
Dyuba
,
Alexander M.
Arutyunyan
,
Tatiana V.
Vygodina
,
Natalia V.
Azarkina
,
Anastasia V.
Kalinovich
,
Yuri A.
Sharonov
and
Alexander A.
Konstantinov
*
A. N. Belozersky Institute of Physico-Chemical Biology, Moscow State University, Moscow 119992, Russia. E-mail: konst@genebee.msu.su; Fax: +7(495)939-3181; Tel: +7(495)9495549
First published on 2nd February 2011
Abstract
Circular dichroism spectra of bovine heart aa3-type cytochrome c oxidase have been studied with a major focus on the Soret band π → π* transitions, B0(x,y), in the two iron porphyrin groups of the enzyme. The spectra of the fully reduced and fully oxidized enzyme as well as of its carbon monoxide and cyanide complexes have been explored. In addition, CD spectra of the reduced and oxidized ba3-type cytochrome c oxidase from Thermus thermophilus were recorded for comparison. An attempt is made to interpret the CD spectra of cytochrome c oxidase with the aid of a classical model of dipole–dipole coupled oscillators taking advantage of the known 3D crystal structure of the enzyme. Simultaneous modeling of the CD and absorption spectra shows that in the bovine oxidase, the dipole–dipole interactions between the hemes a and a3, although contributing significantly, cannot account either for the lineshape or the magnitude of the experimental spectra. However, adding the interactions of the hemes with 22 aromatic amino acid residues located within 12 Å from either of the two heme groups can be used to model the CD curves for the fully reduced and fully oxidized oxidase with reasonable accuracy. Interaction of the hemes with the peptide bond transition dipoles is found to be insignificant. The modeling indicates that the CD spectra of cytochrome oxidase in both the reduced and oxidized states are influenced significantly by interaction with Tyr244 in the oxygen-reducing center of the enzyme. Hence, CD spectroscopy may provide a useful tool for monitoring the redox/ionization state of this residue. The modeling confirms wide energy splitting of the orthogonal Bx and By transitions in the porphyrin ring of heme a.
Introduction
Cytochrome c oxidase (COX)‡ is the terminal enzyme in the respiratory chain of most aerobic organisms. The enzyme catalyzes 4-electron reduction of molecular oxygen to water by four molecules of cytochrome c.1 The reaction is highly exergonic (about 0.8 eV), and its free energy is utilized by COX to generate a transmembrane difference of hydrogen ion electrochemical potential (ΔμH+), mainly by pumping protons electrogenically (i.e., without charge compensation) across the membrane from the inside to the outside of mitochondria or bacteria. The overall reaction catalyzed by COX can be summarized by the scheme| 4 cyt c2+ + O2 + 8H+in = 4 cyt c3+ + 2H2O + 4H+out (+8q+↑) |
Crystal structures of cytochrome oxidases from bovine heart mitochondria2 and several bacteria3 have been solved and refined, which has given strong impetus to further studies of the enzyme. The catalytic redox core of cytochrome oxidase is comprised of two hemes (low-spin and high-spin, the latter reacting with oxygen) and two redox active copper centers (binuclear CuA and mononuclear CuB) (Fig. 1A). In COX from mitochondria and many bacteria, both hemes are represented by the same iron porphyrin species called heme A and are denoted as hemes a and a3. The high-spin iron of heme a3 and mononuclear CuB are located within 4–5 Å of each other and form an oxygen-reducing center of the enzyme, often called “the binuclear site”, whereas CuA and heme a serve to deliver electrons to the O2-reducing site. The electron transfer sequence is described by the scheme
| cyt c2+ → CuA → heme a → CuB/heme a3 → O2 |
 | ||
| Fig. 1 Structure of cytochrome c oxidase catalytic site. (A) Redox centers. Hemes a and a3 and nearby amino acid residues of highest importance (A) and (B). The structure shows the aromatic residues around the hemes (within ∼12 Å from either of the heme iron ions) included in the consideration in our model. The hemes are shown as red sticks, copper atoms marked as copper-colored sphere. Aromatic residues that are labeled on (A) are not labeled on (B). Carbon atoms are shown green for tyrosines, cyan for histidines, yellow for tryptophans, and purple for phenylalanines (PDB entry: 1V55). | ||
Deciphering the mechanism by which the electrons travel through the enzyme, propelling protons across the membrane, is one of the basic problems in studies of biological redox energy transduction on a molecular level. In particular, interactions between the two hemes of the enzyme are of great interest. The hemes are attached via the axial histidines to the same hydrophobic transmembrane α-helix of subunit I with a Fe-to-Fe distance of 13.4 Å and the closest edge-to-edge distance between the porphyrin groups of only 7.1 Å (cf.Fig. 1A). The two hemes have been amply studied by absorption, EPR, IR, Raman and magnetic circular dichroism spectroscopy (reviewed in ref. 1a,b,6). A number of important findings has been made with the use of ultrafast absorption spectroscopy (reviewed in ref. 7).
A method that is particularly sensitive to heme-heme interactions in multiheme proteins is circular dichroism (CD) spectroscopy. Detailed studies on the optical activity of myoglobin and hemoglobin for which crystal structures were available provided a solid basis for interpretation of the optical activity of hemoproteins in the absorption bands of the hemes and pointed out the feasibility of electronic interactions among even distant iron-porphyrin groups in multiheme proteins.8 It was predicted that heme-heme interactions should contribute significantly to the optical activity of many membrane-bound cytochrome complexes in their heme absorption bands.8b Indeed, excitonic interaction between the hemes was revealed, and heme-heme distances were evaluated for several membrane-bound multi-heme cytochromes, and in particular for the diheme cytochromes b in chromaffin granules,9 in several respiratory and photosynthetic cytochrome bc1 (b6f) complexes, as well as in succinate-quinone reductases (reviewed in ref. 10). More recently, a strong excitonic interaction was revealed between the high-spin hemes b595 and d in the oxygen-reducing center of bd-type quinol oxidase from Escherichia coli.11
Concerning cytochrome c oxidase, since the first publication in 196712 a number of studies on the optical activity of the enzyme have been carried out (reviewed in ref. 13). CD signals arising from the hemes were described for the reduced and oxidized COX from bovine heart in the free state and in the presence of exogenous ligands of the high-spin heme a3, such as cyanide, azide, CO and NO. While some evidence was found for interdependence of spectral characteristics of the two hemes,14 the major conclusion was that the two hemes are too far from each other for direct dipole–dipole interactions.13a Later CD spectra of bacterial ba3-type oxidase from Thermus thermophilus were described15 with the same inference of the absence of dipole–dipole interactions between the two hemes. Systematic studies ended around 1975, before a major body of biophysical/biochemical studies on the enzyme was obtained, and about 20 years before the first two crystal structures of COX were resolved.2a,3a,16 Thus the origin of the CD signals and their assignment were poorly clarified at that time.
Since early studies summarized in reviews,13 significant progress has been achieved in understanding the CD spectra of cytochromes. A great contribution to this progress is associated with the studies of Woody and coworkers.8,17 In particular, using a point monopole approximation, they showed that in myoglobin and hemoglobin it is heme interaction with the π → π* transitions of the adjacent aromatic residues that gives the major contribution to the optical activity.8a In addition, investigations into the optical activity of heme undecapeptide of cytochrome c (the so-called microperoxidase) revealed some intrinsic optical activity inherent in the polypeptide-bound iron porphyrin group due to heme ruffling, which breaks the symmetry of the heme;17b besides, the study pointed out interaction of the heme with the π → π* transitions of the peptide bonds (there are no aromatic groups in the undecapeptide other than a single histidyl residue). The classical theory of polarizability of DeVoe18 was used to calculate interactions among the hemes in the α,β- and α2,β2- forms of hemoglobin,8b and an extension of quantum-mechanical Kirkwood polarizability theory was applied to model the CD associated with the α-helices in ref. 17a.
Therefore, we feel it is timely to revisit the CD spectroscopy of cytochrome oxidase and, with the crystal structure of the enzyme at hand, try to understand the origin of the CD signals and evaluate potential usefulness of CD spectroscopy for gaining new insights into the mechanism of the enzyme. In this work we have measured CD spectra of aa3 bovine heart COX as well as of ba3-type oxidase from T. thermophilus in several redox and ligand bound states. A provisional model has been developed based on classical polarizability theory of optical rotation and circular dichroism by DeVoe18 that describes the experimental spectra with reasonable accuracy. The model indicates that optical activity in the COX heme absorption bands is determined by two major factors: (1) dipole–dipole interaction between the two hemes, and (2) interaction of the hemes with the dipoles of the aromatic groups of the protein. Interaction of the π → π* transitions of the hemes with the dipoles of the protein α-helices was also considered, but the contribution of this factor appears to be negligible.
Interestingly, the modeling predicts that the CD spectra of the oxidase in the Soret band are influenced significantly by interaction with Tyr244, a residue that forms a covalent bond with the His240 ligand of CuB (Fig. 1). This residue is supposed to participate directly in the catalytic function of the oxygen-reducing center, forming a radical state as well as deprotonated form in the course of catalysis.1b,19 The redox and/or protonation changes of this critically important and highly conserved tyrosine are expected to be reflected in the Soret CD spectra of COX, which may give a new spectroscopic tool to follow the state of the oxygen-reducing center.
Experimental
Chemicals
Dodecyl maltoside (DM) of “SOL-GRADE” purity was from “Anatrace”. Bis-tris propane ([1,3-bis(tris(hydroxymethyl)methylamino)propane], BTP), ascorbate, and K3Fe(CN)6, were purchased from “Sigma”. Phenazine methosulfate (PMS) was from “Serva”, and sodium dithionite (“LAB GRADE”, >87%) was from “Merck”.Preparations
COX was prepared from beef heart mitochondria by a procedure based on a modification of the method of Fowler et al.20 The concentration of aa3 was determined using extinction coefficient Δε605–630 = 27 mM−1 cm−1 for the reduced–minus-oxidized difference absorption spectrum. The basic incubation medium contained 100 mM buffer (BTP), pH 8, 50 mM K2SO4, 0.5 mM Na-EDTA, and 0.05% dodecyl maltoside (DM). Cytochrome ba3 was purified from T. thermophilus according to ref. 21. The concentration of the enzyme was determined using extinction coefficient Δε560–590 = 26 mM−1 cm−1 for the absolute spectrum of the fully reduced oxidase.22Spectroscopy
CD spectra were recorded with a Chirascan dichrograph (Applied Photophysics) in standard quartz cells for fluorescence measurements with an optical pathlength of 10 mm and total volume of 4 ml. Recordings were performed with 0.5 nm steps (scan rate, 0.5 s/step), and an optical bandwidth of 1 nm. Absorption spectra were collected from the samples during the recordings of the CD spectra in the Chirascan, as allowed by the instrument. Alternatively, a CaryBio 300 double-beam spectrophotometer (Varian) was used. The CD and absorption spectra of the buffer were subtracted from the spectra of the samples. All spectra have been normalized to the concentration of the enzyme and are given as ε, mM−1 cm−1 for the absorption spectra, and ΔεL–R, M−1 cm−1 for the CD curves.Spectral modeling. Theoretical model
General description
In any medium, light intensity decreases with distance as| I = I0e−kl = I010−Dl; k = 2.303D; | (1) |
| θ = arctan(as/al) | (2) |
| θ(rad) = (kL − kR)/4 = Δk/4 | (3) |
| [θ] = 2.303·(4500/π)·(εL − εR) = 3300ΔεL−R | (4) |
In the approach used for calculation of ellipticity of complex systems, the molecule is broken down into small units, and its optical activity is attributed to interactions among the units. The first such model was considered by Kirkwood23 within the framework of polarizability theory with the calculation based on approximated perturbation treatment. Later DeVoe18 described a matrix method that avoids the perturbation approximation in calculations and is easily applicable to the polarizability method. The point dipole interaction model can be treated by this method exactly, and this is the basic approach used in our studies.
In DeVoe's model, a complex molecule is treated as a set of coupled dipole oscillators arising from the interaction with the light. Each oscillator is assigned one electronic transition and is defined by: (i) natural frequency (energy of the corresponding electronic transition), (ii) polarizability, which is wavelength-dependent, (iii) oscillator strength and (iv) orientation within the molecule. To a first approximation, the factors which could give rise to CD signal of COX and should be taken into consideration by our model are (1) interaction between the heme a and heme a3 (b and a3 in the case of ba3 oxidase), (2) interaction of the hemes with the aromatic amino acid residues and (3) interactions with peptide bonds.
The model requires the following input parameters.
1. 3D structure of the bovine heart enzyme (PDB:1V54 for the oxidized form and PDB:1V55 for the reduced enzyme24).
2. Characteristics of the oscillators including natural frequencies (transition wavenumbers/wavelengths), oscillator strengths and transition half-widths. For the aromatics the parameters were taken from ref. 25 for phenylalanine, tyrosine and tryptophan side chains, and from ref. 8a for imidazole mimicking the histidine residue. The transition half-width for all aromatic groups were set to 1100 cm−1. The parameters for the hemes were dealt with as follows.
Two mutually perpendicular π → π* electronic transitions in the plane of the porphyrin ring (Bx and By components of γ-absorption band) have been considered for each of the hemes. The half-widths for the Bx and By transitions in one heme were assumed to be equal. Accordingly, the oscillator characteristics of the four transitions in the two hemes are described by a string containing ten parameters: four natural transition frequencies, four oscillator strength values and two transition half-widths. The ten parameters were optimized using a genetic algorithm searching for minimal deviation of the CD and absorption curves calculated with these parameters according to DeVoe's method18 from the experimental spectra. The initial population for the genetic algorithm to start optimization included 20 “individuals” (i.e., 20 sets with ten parameter values each) derived from the primary set of the ten input values by random variation within the limits indicated below.
Bx and By transition wavelengths: oxidized heme a, 426 ± 2 nm; oxidized heme a3, 416.5 ± 2.5 nm; reduced heme a, 440 ± 10 nm; reduced heme a3, 445 ± 5 nm. These values correspond reasonably well to the absolute and difference absorption spectra of the individual heme a and heme a3 resolved experimentally.26
Oscillator strengths: values of 0.52 ± 0.26 were assigned to all heme transitions in either redox state. These values were chosen to provide for appropriate magnitudes of the calculated absorption spectra.
Transition half-widths: 900 ± 600 cm−1 values were assigned initially to all heme transitions in either redox state. The values correspond to a half-width of approximately 16 ± 10.5 nm in the Soret region, which covers the range of experimentally observed heme a and heme a3 bandwidths.26 For each redox state, each of the ten parameters was optimized independently.
3. Orientations of the transition dipole moments for the oscillators (hemes, aromatic residues and peptide bonds) within cytochrome oxidase were obtained from the intramolecular orientations of the transitions taken from the literature8a,25 and the position of the groups within the crystal structure of COX (PDB:1V54 for the oxidized form and PDB:1V55 for the reduced enzyme24). The two in-plane transition dipole moments in each of the hemes were assumed to be oriented along the lines connecting the opposite pyrrol nitrogens, so they are almost perpendicular (there is slight deviation from orthogonality in the actual structure of the hemes, which are not exactly planar).
A standard genetic algorithm was used to find an optimal correspondence between the Soret region of the calculated and experimental CD and absorption spectra simultaneously. The algorithm interpretes a set of ten parameters (see above) as an “individual”, and the parameter values belonging to the set as “genes”. A set of 20 “individuals” forms a “population”. For each “individual”, the algorithm calculates the absorption and CD curves according to DeVoe's model18 and obtains an “error” as a measure of discrepancy with the experimental spectra. Then the population is “improved” by choosing “individuals” with the minimal error, removing “individuals” with the maximal error, making “crossover” between “individuals” and “mutating” the “genes” in them. Finally, the parameters evolve into a state with the error minimized.
DeVoe's procedure allows the calculation of the spectral curves from the initial “intrinsic” input parameters for the oscillators (i.e., those prior to dipole–dipole interactions), but does not give the parameter values of the transitions as altered by the dipole–dipole interactions in the final state. The latter could be evaluated with the normal mode model.27 This model requires that oscillators in the system are described by Lorentzian curves with equal half-widths (see the next section “Specific description”). So these parameters have been calculated approximately from the optimized intrinsic parameters of the hemes found by the fitting according to DeVoe's procedure but assuming that all transition half-widths (including aromatics) are equal to 500 cm−1 or 1050 cm−1 for the reduced and oxidized enzyme, respectively. Despite this obvious oversimplification, the spectral curves constructed according to the Applequist method were very close to the spectra calculated according to DeVoe's model.
Specific description
1. In our model the dipole oscillators correspond to transitions in the hemes and aromatic amino acid residues. The total number of oscillators is N. Quantum theory describes light absorbance through electronic transition from the ground to the excited state. It defines electric transition dipole moment for each transition, which is a matrix element of the electric dipole moment operator:| μie = 〈Ψ0|μe|Ψi〉 | (5) |
While interacting with light, a molecule gets an induced dipole moment caused by all the electronic transitions that have occurred. Neglecting magnetic dipole moment for the hemes and aromatic amino acids, we can describe the contribution of each electronic transition to the induced dipole moment of the molecule as oscillating dipole moment μi proportional to the effective electrical field E′i at the dipole:
| μi = αi(E′i·ei)ei | (6) |
The electric field at the location of oscillator i consists of the applied field plus the fields of all other induced dipoles in the molecule. Using the Lorentz–Lorenz approach, we can get the following expression for the local field:
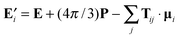 | (7) |
| Tij,αβ = |rij|−3δαβ − 3|rij|−5rij,αrij,β | (8) |
Now we can rearrange eqn (6) to:
| Aμ = E′, | (9) |
| Aij = α−1iδij + eiTijej | (10) |
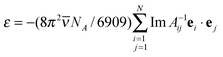 | (11) |
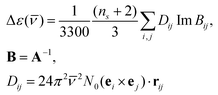 | (12) |
![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) is a wavenumber, ns is the refractive index of the solvent, and N0 is Avogadro's number. A−1 is the inverse of matrix A.
is a wavenumber, ns is the refractive index of the solvent, and N0 is Avogadro's number. A−1 is the inverse of matrix A.
2. To find the approximate parameters of the heme transitions after the interactions (Table 2), the normal mode model of Applequist27 was applied as follows:
(a) The frequency dependency of the polarizability is Lorentzian:
| αi = ci(2i − 2 + iΓi)−1 | (13) |
i is natural transition frequency, and Γi is the transition half-width, i = (−1)1/2.
(b) All transition half-widths are equal to Γ.
The dipole interaction is treated by the normal mode method. Each mode has contributions from each of the initial absorption bands, and the sum of these modes gives the calculated spectrum. So calculated spectra are sums of Lorentzians with the maxima shifted relative to the initial band maxima and half-widths equal to Γ.
With these assumptions, the frequency dependency of the elements of matrix B can be explicitly expressed by
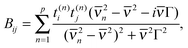 | (14) |
2n are eigenvectors and eigenvalues (n = 1,2,…,p) that satisfy the following equations:| A0t(n) = 2iCt(n) |
| A0 − |2nC| = 0, | (15) |
| t(m)TCt(n) = δmn, |
= 0: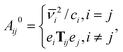 | (16) |
| Cij = δij/cij | (17) |
ε = −(8π2NA/2303)Im ![[small alpha, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0c2.gif) , , | (18) |
| Δε = −(64π32NA/2303)Im β, | (19) |
means average calculated polarizability of the molecule = (αxx + αyy)/3, where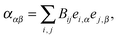 | (20) |
 | (21) |
Results
CD spectra of bovine cytochrome oxidase in various redox and ligand-bound states are shown in panels A of Fig. 2, 4 and 5. For comparison, we also include CD spectra of ba3-type oxidase from T. thermophilus(Fig. 3) in which the spectrum of the high-spin heme a3 is not overlayed so heavily by the absorption bands of the low-spin heme a. In each figure, panel B gives the absolute absorption spectra of the samples. All the spectra have been normalized to the concentration of the enzyme and are given in molar (CD) or millimolar (absorption) extinction coefficients. Positions of the extrema and the corresponding extinction coefficient values (in brackets) are indicated in the figures.In general, the CD spectra of the bovine oxidase observed in this work are in reasonable agreement with the data of the early studies13,14 (Table 1). The differences in absolute magnitude of the signals may arise from different procedures (different extinction coefficients) used to determine concentration of cytochrome oxidase from the absorption spectra of the samples. However, the magnitude ratios for different bands should be independent of this factor, which is not always the case.
| The enzyme state | Position of extremum | Magnitude of extremum | Source of the data |
|---|---|---|---|
| λ, nm | CD, M−1 cm−1 | ||
| a Deoxycholate preparation. | |||
| Oxidized (a3+a33+) | 431 | +97 | this work |
| 427.5 | +73 | 14a | |
| 426 | +73 | 30 | |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||
| Reduced (a2+a32+) | 431.5 | −15 | this work |
| 422 | −6 | 14a | |
| 430 | −11 | 30 | |
|
|||
| 446 | 163 | this work | |
| 446 | 146 | 14a | |
| 445 | 115 | 30 | |
| 444 | 140 | 30 | |
|
|||
| 609 | 16 | this work | |
| 607.5 | 14 | 14a | |
|
|||
| Oxidized-CN (a3+a33+-CN) | 431 | 78 | this work |
| 430 | 70 | 14a | |
| 428–430 | 71 | 30 | |
|
|||
| Reduced-CN (a2+a33+-CN) | 443 | 53 | this work |
| 445.5 | 91 | 14a | |
| 443.5 | 62 | 30 | |
|
|||
| 607.0 | 10 | this work | |
| 606.0 | 15 | 14a | |
|
|||
| Reduced-CO (a2+a32+-CO) | 420 | −29 | this work |
| 419 | −33 | 14a | |
|
|||
| 440 | 80 | this work | |
| 439.5 | 82 | 14a | |
| 435–439 | 74–85 | 30 | |
|
|||
| 606 | 16 | this work | |
| 604 | 21 | 14a |
CD spectra of ligand-free bovine COX
In Fig. 2A, CD spectra of the fully oxidized and fully reduced enzyme are shown, and the corresponding absorbance spectra are given in Fig. 2B. The Soret band CD spectrum of the oxidized cytochrome c oxidase (dotted line) shows a positive peak at 431 nm, which is close to the absorption maximum of the low-spin heme a and far from the peak of the free high-spin heme a3 at ∼414 nm.28 The CD peak is rather symmetric (see also Fig. 7 below) with a half-width of ∼14 nm, i.e. much narrower than the absorption bands (ca. 22 nm for heme a and 20 nm for heme a3, cf.ref. 26a). On the blue side of the peak the CD curve of the oxidized COX shows a weak negative rather structureless region with small negative ΔεL−R values. | ||
| Fig. 2 Circular dichroism (A) and absorption (B) spectra of bovine cytochrome c oxidase. Dotted lines, oxidized enzyme; solid lines, reduced enzyme. 5.4 μM COX in basic medium containing 100 mM BTP buffer, pH 8, 50 mM K2SO4, 0.5 mM Na-EDTA, and 0.05% dodecyl maltoside. The enzyme was reduced by addition of a small excess of dithionite with 5 μM PMS. | ||
In the fully reduced COX, the Soret absorption bands of hemes a and a3 become very close to each other, with the overall peak at 443–444 nm26,28 (Fig. 1B). A shoulder at about 420 nm is also seen in most preparations. Its nature has not been firmly established; it may represent the vibronic component of the B0 band or some other transition of the hemes. As compared to the oxidized enzyme, the CD spectrum of the fully reduced form (solid line) is characterized by a much more intense and narrow symmetrical signal in the Soret with maximum at 446 nm and half-width of ∼10 nm. In the visible a positive CD signal at 609 nm appears upon reduction, which is presumably associated with the α-band of heme a. Notably, like in the case of the oxidized COX, the CD peak at 446 nm is significantly narrower than the absorption bands of either heme a or heme a3. The linewidth of 10 nm is actually close to that of the individual Bx and By electronic transitions of the hemes.
CD spectra of ba3 oxidase
The ba3-type oxidase from T. thermophilus, for which the crystal structure is also available,3c,d has essentially the same structure of the redox centers as the mitochondrial aa3 oxidase, but the low spin iron-porphyrin group is a heme B instead of a heme A. Accordingly, the absorption bands of the two hemes in the reduced state are much better separated than in the aa3 oxidases15,29 (Fig. 3B, spectrum 1). There is a clear shoulder of the ferrous heme a3 at ∼440 nm in the Soret band, and the α-bands in the visible are fully separated (560 nm for heme b2+ and 613 nm for heme a32+). For the oxidized form of the enzyme the opposite is true: the Soret bands of the high-spin a3 at ∼414 nm and low-spin heme b at 410–412 nm merge, giving rise to an intense symmetric peak at 412 nm (spectrum 2). | ||
| Fig. 3 Circular dichroism (A) and absorption (B) spectra of ba3-type cytochrome c oxidase from T. thermophilus. Dotted lines, oxidized enzyme; solid lines, dithionite-reduced ba3. Enzyme concentration, 7.9 μM. Other conditions as in Fig. 2. | ||
The CD spectra of the ba3 oxidase (Fig. 3A) were briefly considered earlier15 and differ substantially from those of the aa3 oxidase. In both the reduced and oxidized states, the Soret band CD curves are much more conservative than in the bovine oxidase, with extensive negative lobes of the curves at 400 nm for the oxidized and at 419 nm for the reduced forms of the enzyme. The positive extrema of the ba3 CD curves are significantly blue-shifted relative to the aa3 oxidase (437 nm vs. 446 nm for the reduced, and 424 nm vs. 431 nm for the oxidized state). Also the peak-to-trough magnitudes of the CD signals are considerably less in the ba3 oxidase, particularly in the case of the fully reduced enzymes (∼100 M−1 cm−1 for ba3vs. ∼180 M−1 cm−1 for aa3). These spectral characteristics are similar to those in ref. 15 with respect to position of the extrema, but the magnitudes of the signals differ markedly.
In the visible, the reduced form of ba3 shows a conservative symmetric derivative-type CD signal with a zero-crossing point close to the α-band maximum of heme b at 560 nm with a peak-to-trough magnitude of ca. 11 M−1 cm−1 (curve 1), which is very close to the data in ref. 15, whereas the signal at 609 nm observed in the aa3 oxidase and associated tentatively with the α-band of heme a is absent. Notably, there is no discernible signal at the α-band of the ferrous heme a3 at 613 nm.
CD spectra of ligand-bound COX
Carbon monoxide and cyanide are classical inhibitors of cytochrome oxidase that bind to the high-spin heme a3 at the sixth axial coordination position and, hence, prevent reaction of the enzyme with oxygen at this site. CO binds exclusively with the ferrous heme iron, while CN− has a strong preference for binding with the ferric heme. Both ligands convert heme a3 to the low-spin hexacoordinated state and therefore affect significantly the spectral characteristics of the heme.CD spectra of the CO-bound fully reduced COX are given in Fig. 4A. In the Soret, CO binding to heme a32+ shifts the positive extemum of the CD signal from 446 nm to 440 nm and halves its magnitude (from 163 M−1 cm−1 to 80 M−1 cm−1). The negative extremum of the curve also shifts to the blue (from 431 nm to 420 nm) and grows ∼2-fold (from −15 to −29 M−1 cm−1). In the visible, the positive signal associated with the α-band of heme a shifts from 609 nm to 606 nm without changing significantly in magnitude or lineshape. These effects of CO agree fairly well with published data.14a,30
 | ||
| Fig. 4 Circular dichroism (A) and absorption (B) spectra of aa3-type bovine cytochrome c oxidase. 4.6 μM bovine COX in basic medium. The reduced CO-complex of the oxidase was obtained by addition of excess solid dithionite and 5 μM PMS to the enzyme in the optical cell and subsequent bubbling with CO for 1 min. After formation of the complex, the cell was tightly closed. | ||
Fig. 5 shows the spectra of the cyanide-ligated COX in the fully oxidized (a3+a33+-CN) and mixed-valence states (a2+a33+-CN). The absolute CD spectrum of the oxidized cyanide complex of COX (Fig. 5A, curve 2) shows a positive Cotton effect in the Soret with a maximum around 430–431 nm and, actually, is very similar both in the lineshape and magnitude to the spectrum of the free oxidized enzyme (Fig. 2). The Soret band CD signals for the two states are compared on an expanded scale in Fig. 6. The two spectra are very close to each other in lineshape and magnitude except for a small loss (ca. 10%) in signal intensity in the cyanide complex, noted also in ref. 14a. This is in contrast to the data in ref. 30, where cyanide was found to shift the Soret CD maximum of the oxidized COX by ca. 2–3 nm, removing a significant part of the CD signal on the blue side of the extremum without changing the magnitude of the peak (cf. Fig. 5 of ref. 30).
 | ||
| Fig. 5 Circular dichroism (A) and absorption (B) spectra of cyanide-complexed bovine cytochrome c oxidase. 7.9 μM COX in basic medium. Dotted lines, oxidized cyanide complex (a3+a33+-CN) obtained by 2.5 h incubation of the enzyme with 10 mM KCN in the presence of 100 μM ferricyanide and 5 μM PMS. Solid lines, mixed-valence cyanide complex (a2+a33+-CN) obtained by addition of 2 mM ascorbate to the oxidized cyanide adduct. | ||
 | ||
| Fig. 6 Effect of cyanide binding on CD (A) and absorption spectra (B) of oxidized cytochrome c oxidase in the Soret band. Solid lines, absolute spectra of oxidized enzyme. Dashed lines, absolute spectra of oxidized cyanide complex. Difference spectra (a3+a33+minus a3+a33+-CN) are marked with black circles. | ||
Note that all the experiments shown in this paper were performed at pH 8 to avoid enzyme conversion to the so-called “slow form” at more acidic pH,31 while the earlier works14a,30 were done at pH 7.2–7.4. At pH 8 a significant part of the pentacoordinate high-spin heme a3 with an absorption maximum at ∼414 nm converts to some hexacoordinate complex (hydroxide, or even peroxide at the sixth axial coordination bond have been proposed as the ligands2b,32), which absorbs maximally around 428 nm, close to low-spin heme a or CN-ligated low-spin heme a3.31,33 Indeed, the overall Soret absorption peak of the oxidized COX in our experiments (Fig. 2B) is narrower and more intense and is red-shifted relative to the spectra at pH 7–7.4 (γ-band at ∼422–424 nm). Accordingly, the CD signal of the oxidized enzyme in our experiments is shifted by ∼4 nm to the red relative to the spectra in ref. 14a and 30 (431 nm vs. 426–427.5 nm, Table 1). This circumstance may explain why the effect of cyanide on the absorption and CD spectra of the oxidized COX in our experiments is relatively small (Fig. 6).
Addition of a reductant to the cyanide adduct of ferric COX converts the enzyme to the so-called mixed-valence state (a2+a33+-CN) in which heme a becomes reduced, while heme a3 remains oxidized and bound with cyanide (Fig. 5A, solid line). Upon the reduction of heme a, the positive signal at 431 nm disappears and is replaced by a smaller peak at 443 nm. The peak is probably associated mainly with the ferrous heme a, as also evidenced by the appearance of a positive signal at 607 nm in the visible. The Soret band CD spectrum of the mixed-valence cyanide complex agrees well with the data in ref. 30, but differs markedly both in magnitude and peak position from the signal reported by the van Gelder's group14a (Table 1).
Reduced-minus-oxidized difference CD spectra of hemes a and a3
Selective reduction of heme a in the cyanide complex of COX can be used to formally obtain a difference CD spectrum (reduced-minus-oxidized) of heme a by subtraction:| a2+a33+-CN minus a3+a33+-CN ∼ a2+minus a3+ |
 | ||
| Fig. 7 “Individual” redox difference CD spectra of hemes a and a3 in the Soret band. (A) Redox difference CD spectrum of heme a. (B) Redox difference CD spectra of heme a3 obtained by two methods. The spectra were obtained as described in the text. | ||
As shown above (Fig. 5 and 6), cyanide binding to the ferric heme a3 does not significantly affect the CD spectrum of the oxidized COX at pH 8. Therefore, it is possible to obtain an approximate redox difference CD spectrum for the reduced heme a3 in a second way by subtracting the absolute spectrum of the mixed-valence cyanide complex (a2+a33+-CN ∼ a2+a33+) from the absolute spectrum of the fully reduced free enzyme (a2+a32+). Such a spectrum is shown in Fig. 7B by the solid line. It can be seen that the two redox difference spectra of heme a3 are very close to each other, virtually coinciding in the positive lobe and differing but slightly in the negative part. The reduced-minus-oxidized CD curve of heme a3 shows an asymmetric signal with an intense positive lobe at 446 nm (Δε of ∼115 M−1 cm−1), and a well defined trough at ∼434 nm (Δε of −40 to −50 M−1 cm−1). The spectrum can be compared to that of the fully reduced COX in Fig. 2. Apparently the reduction of heme a3 contributes very significantly to the Soret band CD of the reduced COX. Peculiarly, the CD spectrum of the ferrous heme a3 is very close in lineshape to the MCD spectrum.35
It must be emphasized that whereas the “ligand subtraction method” is essentially correct for obtaining the individual absorption spectra of hemes a and a3,26,28,34 it is no more than an empirical procedure in the case of the CD spectra, suitable mainly for the purpose of data comparison among different studies, because the two hemes strongly affect each other and the contributions of the individual hemes are not additive.
Calculations of the spectra of aa3 oxidase with the developed model (see above)
The CD and absorption spectra calculated according to the model described in the “Experimental” section are given in Fig. 8 and 9 by dotted lines and can be compared to the experimental spectra (solid lines). All calculations were done assuming the π → π* electronic transitions in both a and a3 hemes to be directed through the pyrrol nitrogens. The CD curves calculated for interaction between the two hemes only are also shown in Fig. 8A and C by the curves labeled with circles. Obviously, the conservative curves for the heme–heme excitonic interactions cannot reproduce the non-conservative lineshape of the experimental spectra, and under no conditions do they reach the magnitude of the experimental curves, whereas the magnitudes of the calculated absorption spectra tend to exceed the experimental values (not shown). Apparently, interaction between the hemes, while contributing significantly, is not sufficient to explain the observed CD signals, and it is necessary to invoke interaction of the hemes with other light absorbing components of the enzyme. To this end, 22 aromatic amino acid residues located within 12 Å from either of the two heme iron ions (see Fig. 1B) were included in the model. | ||
| Fig. 8 Modeling of CD (A,C) and absorption (B,D) spectra of bovine oxidase. (A,B), reduced COX. (C,D), oxidized COX. Experimental curves are given by thick solid lines. Curves labeled with circles in (A) and (C) show calculated CD spectra taking into consideration only interactions between the two hemes. Dotted curves correspond to the calculations according to the full model (i.e., including interactions of the hemes between themselves as well as with all aromatic residues within 12 Å from each of the heme irons). Calculated characteristics of heme transitions in absence of interactions (“intrinsic” parameters) and approximate parameters as affected by dipole–dipole interactions are given in Table 2. | ||
Results for the reduced aa3 are shown in Fig. 8A and B. The calculated CD curve (A, dotted line) reproduces fairly well the magnitude and lineshape of the experimental spectrum (solid line). The magnitude of the modeled curve is slightly different, and the negative extremum of the calculated CD curve is somewhat more pronounced and shifted to the red. The simultaneously modeled absorption curve (panel B, dotted curve) reproduces adequately the magnitude and peak position of the experimental spectrum as well as the lineshape on the red side of the peak. A strong deviation of the modeled absorption curve on the blue side of the maximum is due to the presence of an additional band in the absorption spectrum of the reduced COX at about 420 nm. The nature of this shoulder observed in most preparations of COX (e.g.ref. 26,30,36) is not established, and the band has not been included in our model, which takes into account only the two B00 transitions in each of the hemes. In any case, the ∼420 nm shoulder in the Soret absorption band of the reduced oxidase appears to be not associated with significant CD signals.
Modeling the oxidized enzyme spectra is complicated by the presence of multiple states of heme a33+ so that the Soret peak of the “as isolated” ferric enzyme is known to vary from ca. 419 nm to 427 nm in different preparations and at different pH values.31 At pH 8 part of the heme retains a state with a maximum at ∼414 nm, while a significant fraction is likely to convert to a form, probably still high-spin,37 absorbing close to the low-spin heme a at ∼428 nm. We were able to model the spectra assuming either of these states of ferric heme a3 in the initial set of parameters, but not for their mixture, since further increase in the number of parameters to be optimized makes the procedure unreliable. Therefore, the data for the free oxidized COX should be considered as provisional and are included mainly for illustrative purpose. A reasonably good fit could still be obtained for the CD spectra of the oxidized enzyme assuming either of the spectral forms of heme a3 in the initial set of parameters (one of the variants is shown in Fig. 8C and Table 2). However, approximation of the absorption band for the oxidized COX is significantly worse than for the reduced enzyme, although it reproduces roughly the magnitude and position of the Soret peak.
 | ||
| Fig. 9 Modeling of CD (A) and absorption (B) spectra of oxidized CN-complex of bovine oxidase. Experimental curves are given by thick solid lines. Dotted curves correspond to calculations according to the full model (i.e., including interactions of hemes between themselves as well as with all aromatic residues within 12 Å from each heme iron). Calculated characteristics of the heme transitions in the absence of interactions (“intrinsic” parameters) and approximate parameters as affected by dipole–dipole interactions are given in Table 2. | ||
| Heme | Optimized transition parameters before interaction | Transition half-width, cm−1 | Transition parameters after interaction | ||||
|---|---|---|---|---|---|---|---|
| Natural transition frequency | Oscillator strength | Wavenumber, cm−1 | Wavelength, nm | Oscillator strength | |||
| Wavenumber, cm−1 | Wavelength, nm | ||||||
| Values correspond to modeled spectra shown in Fig. 8. The four columns “Optimized transition parameters before interaction” give the “intrinsic” parameters of the four oscillators in the two hemes, i.e., the values expected if there were no interaction of the hemes with each other or with the aromatics. Parameters have been optimized from starting values by an iterative procedure based on a genetic algorithm searching for best fit of spectra calculated with the parameters according to DeVoe's method18 to the experimental curves (see “Modeling spectra. Theoretical model”). In the calculations the transition half-widths for all aromatic amino acid residues have been set to 1100 cm−1. The three columns “Transition parameters after interaction” give parameters of transitions altered by dipole–dipole interactions. These parameters were calculated approximately from optimized “intrinsic” values according to the Applequist normal mode model27 (see text). The Applequist model requires that the transition half-widths for all oscillators in the system are set equal and that all bands have Lorentzian shape. For the reduced COX spectrum all transition half-widths were set to 500 cm−1, for the oxidized spectrum, to 1050 cm−1, and for oxidized CN-complex, to 1107 cm−1. | |||||||
| Reduced | |||||||
| a 2+ | 22941 | 435.9 | 0.45 | 452 | 22889 | 436.9 | 0.38 |
| 22917 | 436.4 | 0.34 | 22238 | 449.7 | 0.13 | ||
| a 3 2+ | 22561 | 443.2 | 0.45 | 635 | 22453 | 445.4 | 0.52 |
| 22727 | 440 | 0.34 | 22584 | 442.8 | 0.37 | ||
|
|||||||
| Oxidized | |||||||
| a 3+ | 23456 | 426.3 | 0.56 | 991 | 24056 | 428.7 | 0.47 |
| 23481 | 425.9 | 0.38 | 23692 | 442.4 | 0.26 | ||
| a 3 3+ | 23926 | 418 | 0.75 | 1275 | 23329 | 415.7 | 0.41 |
| 24155 | 414 | 0.38 | 22603 | 422.1 | 0.67 | ||
|
|||||||
| Oxidized-CN | |||||||
| a 3+ | 23348 | 428.3 | 0.41 | 1096 | 22693 | 440.7 | 0.18 |
| 23425 | 426.9 | 0.33 | 23238 | 430.3 | 0.38 | ||
| a 3 3+-CN | 23381 | 427.7 | 0.47 | 1118 | 23451 | 426.4 | 0.35 |
| 23458 | 426.3 | 0.42 | 23126 | 432.4 | 0.51 | ||
A homogenous spectral form of COX in which both hemes are oxidized is provided by the cyanide complex of the ferric enzyme (Fig. 5 and 6). Cyanide binding converts heme a3 to low-spin state absorbing close to heme a. Hence, the cyanide complex is a clean model for the alkaline form of the oxidized COX, free of the a3 heterogenity problems and suitable for analysis. Modeling of the CD and absorption spectra of the cyanide complex of ferric COX is shown in Fig. 9. A fairly good fit is obtained for the CD spectrum, except for the part below 420 nm, where the calculated curve does not account for the negative broad feature seen in the experimental curve. As to the absorption spectrum, a strong deviation is observed on the blue side of the maximum, similarly to the case of the reduced enzyme (cf.Fig. 8B). It is likely that in the cyanide complex, as in the reduced COX, the Soret absorption contains a significant contribution from a band located on the high-energy side of the electronic Bx,y transition peak at ∼430 nm. The band appears to be centered around 410 nm and may correspond to the 420 nm shoulder observed in the Soret band of the reduced enzyme (e.g., a vibronic component of the B0 band). As all the absorption bands in the ferric enzyme are broader than in the reduced form, this high-energy satellite band is not resolved as a distinct shoulder in the spectrum of the cyanide complex of the oxidized form, but nevertheless is manifested clearly by an extended decay of the Soret peak on the blue side (Fig. 9B). It may well be that this band is responsible for the broad negative part of the CD signal below 420 nm in Fig. 9A.
The parameters of the electronic transitions of the hemes corresponding to the modeled spectra shown in Fig. 8 and 9 are given in Table 2. In our model the Bx,y-transitions in the reduced heme a per se are found not to be split intrinsically, but diverge to the ∼437 and ∼450 nm transitions as a result of interaction with heme a3 and aromatic amino acid residues. A strong splitting of the Bx and By transition in the reduced heme a corroborates the data of Copeland and coworkers,38 who reported the Soret absorption of the reduced heme a in COX to contain two bands with maxima at ∼442 and 450 nm, while no such splitting was observed for isolated heme a or for bis-histidine-coordinated heme a incorporated into an artificial protein scaffold. Accordingly, strong separation of the x and y bands of heme a in the Q0 band of COX was demonstrated by MCD studies.39
Transition parameters found for the oxidized enzyme are likely to be biased on heterogeneity of heme a3, as discussed above, and their discussion should await detailed studies of the pH-dependence of the spectra. It is more interesting to consider the parameters found for the cyanide complex of the oxidized enzyme, which is spectrally homogenous. The half-widths found for the B0 transitions of the ferric hemes are about twice broader than for the reduced hemes. As in the case of the reduced COX, the Bx and By transitions in each of the two hemes are found to be very close to each other intrinsically (i.e., in the absence of interactions); however, dipole–dipole interactions tend to split widely the x and y transitions of heme a, whereas only a moderate divergence of the x and y transitions is induced in heme a33+-CN.
It is noted that splitting of the Bx and By bands found for the oxidized heme a3+ may be exaggerated, since the location of the By band at ∼440 nm is in obvious (Fig. 8D) or at least mild (Fig. 9B) disagreement with the experimental spectra. The reason for such a strong splitting of the heme a absorption band found by the program remains to be established. Varying the model parameters, we found that the lineshape of the absorption curve is very sensitive to interaction of heme a with the two axial histidines, whereas this interaction is not modeled correctly with the point dipole approximation (see “Discussion”). For instance, manual increase in the Fe3+-to-imidazole distance by 10% eliminates completely the 440 nm shoulder from the calculated absorption spectrum of the oxidized enzyme (Fig. 8D), whereas the lineshape of the CD spectrum is not so much affected, but just loses some magnitude.
Effect of specific residues
The model used allows to remove any of the interacting species, either a heme or an aromatic residue, from the calculations of the spectra. Such a “model experiment” was performed for several specific aromatic groups, and some of the results are shown in Fig. 10. Interestingly, the calculations predict that CD spectra of COX, in both the reduced and oxidized states, should be affected markedly by Tyr244. The effects of Tyr244 “removal” on the calculated absolute CD spectra of COX are shown in panels A and B of Fig. 10 for the reduced and oxidized enzyme, respectively. As compared to the full system, the magnitude of the CD spectra in the systems lacking Tyr244 decreases (Fig. 8A and C, dotted lines), whereas the absorption spectra in the Soret remain essentially unchanged (not shown). The effect can be better seen in the CD difference spectra (panel C). The magnitude of the difference spectrum attains ∼55 M−1cm−1 for the reduced and ∼30 M−1 cm−1 for the oxidized state of COX. The results suggest that changes in the state of the functionally important Tyr244 would be observable by CD spectroscopy in the Soret region. For instance, assuming a red shift of the main optical transitions of tyrosine by ca. 2500 cm−1 upon the phenolic group ionization40 (and cf.ref. 41 for the ionization-induced absorptions changes of the covalent Tyr-His model compound), the model predicts a difference CD spectrum induced by Tyr244 ionization in the oxidized COX shown in panel D of Fig. 10. The CD peak-to-trough difference of ∼5 M−1 cm−1 is well within the reach of modern dichrograph sensitivity. Another aromatic residue that may affect noticeably the CD spectrum of COX is Trp126 (cf.Fig. 1). The difference CD spectra arising from “removal” of Trp126 from the calculations are shown in Fig. 10D by triangle labeled curves. | ||
| Fig. 10 Specific contribution of interactions with Tyr244 and Trp126 to calculated CD spectra of bovine oxidase. (A) and (B), effect of Tyr244 on calculated absolute CD spectra for fully reduced and fully oxidized oxidase, respectively. Solid lines, spectra calculated according to the full model (same as shown in Fig. 8A and C by dotted lines). Dotted lines (ΔTyr244) show the calculated spectra omitting interactions with Tyr244 from the model. Panel (C) shows calculated difference CD spectra for omitting interactions with Tyr244 (ΔTyr244) and Trp126 (ΔTrp126, curves labeled with triangles) from the model. Solid lines, reduced enzyme. Dotted lines, oxidized enzyme. (D) Calculated difference CD spectrum induced by ionization of Tyr244 in the oxidized enzyme. Numeric experiments assumed that upon deprotonation the energies of the main Tyr244 optical transitions decrease by 2498 cm−1.40 | ||
Effect of interactions with peptide bond tansition dipoles
Finally, using the simplified version of the model (see eqn (20)–(39) in ref. 27), we found that interaction of the hemes and aromatics with the peptide bond oscillators does not affect the calculated spectra significantly (not shown). This finding confirms that the optical activity of bovine oxidase originates mainly from interaction of the two hemes with each other and with the nearby aromatic amino acid residues.Discussion
Experimental data
In general, the experimental CD spectra of COX obtained in this work are in reasonable agreement with the results of the early studies, except for several details mentioned above. Two types of CD signals in the Soret band of the fully reduced oxidase have been described in the past, some showing a sharp negative extremum at ca. 430 nm on the blue side of the major 445–447 nm peak,12,14a some not.30 This controversy was assigned tentatively to the difference in preparations and, more specifically, to different detergents used.13a,30 The preparations in ionic detergent (deoxycholate) showed the negative extremum at ca. 430 nm, while in non-ionic detergents like Emasol or Tween-20 only a positive CD signal was observed.30 In our experiments the reaction medium contained a non-ionic detergent (DM), but the negative extremum on the blue side of the positive peak is still observed. Interestingly, it has been shown recently42 that deoxycholate binds to COX at a specific site, tentatively proposed to be responsible for the regulatory effect of the steroid hormones on the enzyme. Experiments are in progress to test whether the specific binding of deoxycholate can affect the CD spectrum of COX. Another possible reason for different lineshapes of the CD spectra of bovine COX in different detergents13a,30 might be different aggregation state of the enzyme. For example, formation of dimers or higher aggregates might bring about additional weak inter-monomer heme-heme interactions.Attribution of CD signals to individual hemes
Dipole–dipole interactions between the two hemes are likely to be responsible for a significant part of the ellipticity revealed by the CD spectra of COX in both the reduced and oxidized enzyme. Therefore, it may not be correct to assign the CD signals to the individual hemes. It is possible, however, to make some remarks concerning the sensitivity of the CD spectra to changes in the state of the individual hemes. Two points may be noted. First, it is likely that the ferrous heme a3per se contributes but little to the CD signal of the reduced enzyme in the visible (Q00 band). This is evidenced most clearly by the CD spectra of cytochrome ba3, in which the low-spin heme a no longer overlays with the absorption of heme a3 in the Q00 band (Fig. 3 of this work, ref. 15). No significant signals around the Q-band of heme a3 at 612 nm can be seen in the ba3 oxidase, while a rather strong signal in the 605–609 range is observed for the aa3 oxidase states in which heme a is reduced (Fig. 2, 4 and 5).Second, at pH 8, when the major part of heme a3 converts to a state absorbing near 430 nm, the Soret CD signal of the oxidized COX is changed only slightly by cyanide binding to the ferric heme a3 in accordance with only minor changes in the absorption spectrum of the enzyme (Fig. 6). Quoting the data at pH 7.4, review43 states that “cyanide shifts the absorption spectrum of the oxidized oxidase to the red, but does not affect position or magnitude of the CD signal”. However, inspection of the original data (Fig. 5 in ref. 30) shows that cyanide binding to ferric COX at pH 7.4, (i.e., when a large part of heme a3 is in the high-spin state absorbing at ∼414 nm33,37), results in a significant red shift of the Soret band CD signal, narrowing of the peak and loss of the area around 414 nm. Taken together, the data imply contrary to the earlier conclusions, that ferric heme a3 does contribute to the CD signal of COX, at least when in the 414 nm state.
Third, the intense CD peak at ∼446 nm characteristic of the reduced aa3 COX requires both ferrous hemes to be in the state absorbing around 444 nm. Modulation of the heme a3 Soret band by CO-binding in bovine oxidase, or replacement of the ferrous low-spin heme a by low-spin heme b in the ba3 oxidase (with a concomitant shift of the Soret absorption band of the low-spin heme) result in loss of the unique narrow positive CD band observed in the ligand-free fully reduced bovine oxidase.
Modeling of spectra
This work is the first pilot step of an attempt to interpret the CD spectra of cytochrome c oxidase, and we are aware of the many shortcomings and oversimplifications inherent in the crude model used. Nevertheless, we believe it may serve as a good starting point for further refinement of the approach.The most general inference from our modeling is that the CD curves are modeled much better than the absorption spectra. This has been observed for all forms of the oxidase tested—reduced, oxidized and cyanide-liganded oxidized forms. The circumstance may indicate that the Soret band CD curves originate largely from the electronic Bx,y transitions in the porphyrin rings of hemes a and a3, which have been incorporated in our modeling, whereas the absorption spectra include significant contribution from additional transitions not considered by the model. These transitions are located on the high-energy side of the Soret peaks and contribute but little to the CD spectra.
Origin of optical activity of COX
The hemes A per se are achiral and will not show optical activity. Hence, if the CD signal of cytochrome c oxidase in the heme absorption bands is non-zero, the following principal factors may be invoked to explain the ellipticity.(1) The planar symmetric structure of the iron porphyrin rings groups in the enzyme can be distorted by asymmetric steric/bonding interactions with the protein. This factor cannot be checked rigorously at the moment. To evaluate the intrinsic ellipticity of the hemes in situ (i.e., within the cytochrome c oxidase protein), quantum chemical calculations of the electronic structure of the protein-bound hemes are required. However, the contribution of such heme structure distortion is not expected to be large (cf.ref. 17b). Deviation of the hemes from planarity in the crystal structure of COX is rather small and is comparable to distortion of the heme in the crystal structure of cytochrome c heme undecapeptide (microperoxidase).17b For such a state of the heme, rotational strength of ∼0.01 DBM is predicted by investigations of the optical activity of microperoxidase in solution by molecular dynamics simulation,17b whereas the observed rotatory strength for the CD spectra of the reduced and oxidized COX in our experiments is about 0.78 and 1.02 DBM, respectively. Hence, the intrinsic optical activity of the hemes in COX is not likely to be a significant factor.
(2) The CD signal can arise from asymmetric electronic interaction of the hemes with the nearby transition dipoles, and this is likely to be the major factor for the optical activity of COX in the absorption bands of the hemes. There are several possibilities for such interactions.
(i) First, interaction of the two hemes with each other is to be considered; such an interaction is expected to give rise to a conservative CD signal (equal areas for the curve above and below zero) in the heme absorption bands.
(ii) There are a number of aromatic residues around each of the hemes absorbing in the near UV range. Interaction of the hemes with the π → π* transition dipoles of these residues could contribute to the observed CD signals in the Soret and visible regions.
(iii) The hemes can interact asymmetrically with the transition dipoles of the protein peptide bonds.
The last possibility was evaluated according to a simplified variant of Applequist`s model (cf. eqn (20)–(39) in ref. 27); apparently, dipole–dipole interactions of the hemes with the peptide bonds are insignificant and can be neglected. The possibilities (i) and (ii) are discussed below.
The dipole–dipole interactions between the two hemes are revealed more clearly in the rather conservative lineshape of the CD spectra of the ba3-type oxidase. A pronounced negative lobe in the Soret band CD spectra of ba3 oxidase is observed for both the reduced and oxidized states (Fig. 3). Comparison of the 3D structures of bovine aa3 and T. thermophilus ba3 oxidases shows that the number and spatial distribution of the aromatic groups around the hemes in ba3 oxidase are rather similar to those in the aa3 enzyme. Actually, there are more aromatic groups in the ba3 oxidase (27 residues vs. 22 in the aa3 oxidase within the 12 Å distance from the hemes). Hence, the more conservative type of the CD curves observed for ba3 oxidase is unlikely to be explained by weaker interaction of the hemes with the aromatic residues. Preliminary results of the modeling indicate that it is the higher oscillator strength of the π → π* optical transitions in the low-spin heme b, as compared to the low-spin heme a, that is at least partly responsible for the stronger heme-heme interactions in ba3 oxidase.
Our modeling has been made assuming that the orthogonally polarized B0x and B0y transitions in the planes of the porphyrin rings are directed through the pyrrol nitrogens in both hemes a and a3. However, this is not necessarily the case. If the Bx and By transitions in each of the hemes were equivalent and strictly perpendicular to each other, the CD spectra of COX are not expected to be sensitive to the specific orientation of the transitions within the heme planes. However, the genetic algorithm used for fitting the experimental spectra finds that the Bx and By transitions in each of the individual hemes are not equivalent, and they may differ in oscillator strength and transition energies (Table 2). In this case the CD spectra should depend somewhat on the orientation of the transitions within the heme plane in each of the two iron porphyrin groups. Indeed, if we retain the optimized parameters found by the fitting procedure for the four individual B transitions (oscillator strengths, natural frequencies and half-widths) and then vary orientation of the transitions in hemes a and a3, either in a correlated way or independently, the calculated CD and absorption spectra are affected significantly. Interestingly, the spectra are much more sensitive to rotation of the heme a3 transitions (data not shown). Understandably, such rotation, while retaining the parameters determined for the best fit orientation, impairs the fit.
Unfortunately, actual directions of the orthogonally polarized π → π* transitions in the hemes a and a3 are not known. Elucidation of the factors that determine preferential orientation of these transitions in the porphyrin planes of the two hemes is of great importance. To this end, polarization optical spectroscopy studies on cytochrome oxidase crystals as well as relevant quantum mechanical calculations are most desirable.
At this stage, our model for the interactions is rather crude. The following shortcomings are to be mentioned. First, it is not quite correct to use the point dipole approximation for evaluation of the heme interactions with the axial histidines, as the distances are too small. The magnitudes of the calculated transition dipoles in the hemes are in the order of 5 Debye, which corresponds to dipole length of ∼1 Å. Let us assume that there are only two such oscillators in the system and that a distance between them is equal to the distance between the iron atom and the center of the imidazole ring of the axial histidine residue in the heme (ca. 4 Å). The coupling between dipole oscillators brings about an energy shift in their natural frequencies that is proportional to the inverse cube of the distance between the point dipoles. Then, because of an uncertainty in the dipole center location, the relative error in the energy shift for the parameters specified above can be up to 50% depending on relative orientation of the dipoles. The maximal error corresponds to the case when the dipoles are oriented along the same line so that the coupling between them is minimal. However, according to the actual orientation of the axial histidines in 3D structure of COX, this is not the case, and we can expect the relative error to be no more than 10%. Calculations of the absorption spectra are particularly sensitive to this factor, which tends to overestimate the splitting of the x,y transitions within the B00 band.
Second, the transition moments and energies for the aromatic amino acid residues given in ref. 8a and 25 have been calculated for the molecules in the gas phase. Accordingly, the oscillator strengths for tyrosines, tryptophans and phenylalanines taken from the experimental data25 were determined for the molecules in the gas phase and may be subject to certain changes inside the protein molecule.
Third, in our model we consider His240 and Tyr244 as individual residues, whereas they make a covalent bond, which has to change significantly the oscillator characteristics of both Tyr and His. For a rigorous approach, quantum mechanical calculations for the entire system of closely interacting oscillators including CuB together with its ligands (His290, His291 and His240-Tyr244 dimer) are desirable; unfortunately, there are no data of this kind available in the literature. Some reasonable empirical corrections as to the natural frequencies and oscillator strengths of the optical transitions in covalently bound His240-Tyr244 may be inferred in the future from analysis of the data on various synthetic models of the Tyr-His dimer (e.g., ref. 41). As soon as the parameters of such models necessary for our calculations become available, appropriate refinement of the model will be easy to do.
An interesting prediction of the model is that interaction of the hemes with Tyr244 should make a strong contribution to the CD spectra of COX (Fig. 10). The strong interaction is explained by the close proximity of the residue to heme a3 (Fig. 1) and, presumably, by favorable orientation of the transition dipoles in Tyr244 and in the porphyrin ring of heme a3. Conceivably, quantitative description of the effect is likely to be biased by treatment of Tyr244 as an individual residue, while it makes a covalent dimer with His240. Nevertheless, strong interaction of Tyr244 with heme a3 in the covalent Tyr244-His240 dimer is very likely to take place. The interaction found by the model is so significant (ΔΔεL–R of ∼55 M−1 cm−1 for the reduced and ∼30 M−1 cm−1 for the oxidized COX in the CD difference spectrum, Fig. 10) that changes in the ionization or redox state of Tyr244 may be well within the sensitivity of modern CD spectrometers. Ionization of Tyr244 in oxidized COX is suggested by recent studies.44 Such ionization has been calculated to give a CD difference spectrum in the Soret band of ca. 5 M−1 cm−1 (Fig. 10D), which is an easily measurable effect.
Acknowledgements
This work was supported in part by a Howard Hughes Medical Institute International Scholar Award 55005615 to AAK. Thanks are due to Dr Claudio Soares from ITQB in Lisbon New University for discussion of the electronic structure of the Tyr-His dimer and to Dr Petra Hellwig (University of Strasbourg) for kindly sending us her data on the characteristics of Tyr-His model compounds prior to publication.Notes and references
- (a) S. Ferguson-Miller and G. T. Babcock, Heme/copper terminal oxidases, Chem. Rev., 1996, 7, 2889–2907 CrossRef; (b) I. Belevich and M. I. Verkhovsky, Molecular mechanism of proton translocation by cytochrome c oxidase, Antioxid. Redox Signaling, 2008, 10, 1–29 Search PubMed; (c) P. Brzezinski and R. B. Gennis, Cytochrome c oxidase: exciting progress and remaining mysteries, J. Bioenerg. Biomembr., 2008, 40, 521–531 CrossRef CAS.
- (a) T. Tsukihara, H. Aoyama, E. Yamashita, T. Takashi, H. Yamaguichi, K. Shinzawa-Itoh, R. Nakashima, R. Yaono and S. Yoshikawa, The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8, Science, 1996, 272, 1136–1144 CAS; (b) S. Yoshikawa, K. Shinzawa-Itoh, R. Nakashima, R. Yaono, N. Inoue, M. Yao, M. J. Fei, C. P. Libeu, T. Mizushima, H. Yamaguchi, T. Tomizaki and T. Tsukihara, Redox-coupled crystal structural changes in Bovine heart cytochrome c oxidase, Science, 1998, 280, 1723–1729 CrossRef CAS.
- (a) S. Iwata, C. Ostermeier, B. Ludwig and H. Michel, Structure at 2.8 resolution of cytochrome c oxidase from paracoccus denitrificans, Nature, 1995, 376, 660–669 CrossRef CAS; (b) C. Ostermeier, A. Harrenga, U. Ermler and H. Michel, Structure at 2.7 A resolution of the paracoccus denitrificans two-subunit cytochrome c oxidase complexed with an antibody Fv fragment, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 10547–10553 CrossRef CAS; (c) T. Soulimane, G. Buse, G. B. Bourenkov, H. D. Bartunik, R. Huber and M. E. Than, Structure and mechanism of the aberrant ba3-cytochrome c oxidase from thermus thermophilus, EMBO J., 2000, 19, 1766–1776 CrossRef CAS; (d) L. M. Hunsicker-Wang, R. L. Pacoma, Y. Chen, J. A. Fee and C. D. Stout, A novel cryoprotection scheme for enhancing the diffraction of crystals of recombinant cytochrome ba3 oxidase from Thermus thermophilus, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2005, D61, 340–343 CrossRef CAS; (e) L. Qin, C. Hiser, A. Mulichak, R. M. Garavito and S. Ferguson-Miller, Identification of conserved lipid/detergent-binding sites in a high-resolution structure of the membrane protein cytochrome c oxidase, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 16117–16122 CrossRef CAS.
- O. Einarsdottir and I. Szundi, Time-resolved optical absorption studies of cytochrome oxidase dynamics, Biochim. Biophys. Acta, Bioenerg., 2004, 1655, 263–73 CrossRef CAS.
- (a) D. Zaslavsky, A. Kaulen, I. A. Smirnova, T. V. Vygodina and A. A. Konstantinov, Flash-induced membrane potential generation by cytochrome c oxidase, FEBS Lett., 1993, 336, 389–393 CrossRef CAS; (b) A. A. Konstantinov, S. Siletsky, D. Mitchell, A. Kaulen and R. B. Gennis, The roles of the two proton input channels in cytochrome c oxidase from Rhodobacter sphaeroides probed by the effects of site-directed mutations on time resolved electrogenic intraprotein proton transfer, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 9085–9090 CrossRef CAS; (c) A. Jasaitis, M. I. Verkhovsky, J. E. Morgan, M. L. Verkhovskaya and M. Wikstrom, Assignment and charge translocation stoichiometries of the electrogenic phases in the reaction of cytochrome c with dioxygen, Biochemistry, 1999, 38, 2697–2706 CrossRef CAS.
- W. H. Woodruff and R. B. Dyer, in Biological Spectroscopy, Part B, ed. R. J. H. Clark and R. E. Hester, John Wiley Press, Chichester, PA, 1993, pp. 189–233 Search PubMed.
- M. H. Vos, A. Battistoni, C. Lechauve, M. C. Marden, L. Kiger, A. Desbois, E. Pilet, E. de Rosni and U. Liebl, Ultrafast heme-residue bond formation in six-coordinate heme proteins: implications for functional ligand exchange, Biochemistry, 2008, 47, 5718–5723 CrossRef CAS.
- (a) M.-C. Hsu and R. W. Woody, The origin of the heme cotton effects in myoglobin and hemoglobin, J. Am. Chem. Soc., 1971, 93, 3515–3525 CrossRef CAS; (b) R. W. Woody, Heme-heme Interactions in tetramers and dimers of hemoglobin subunits: DeVoe theory calculations, Chirality, 2005, 17, 1–6 CrossRef CAS.
- M. Degli Esposti, Y. A. Kamensky, A. M. Arutjunjan and A. A. Konstantinov, A model for molecular organization of cytochrome b-561 in chromaffin granule membranes, FEBS Lett., 1989, 254, 74–78 CrossRef CAS.
- G. Palmer and M. D. Esposti, Application of exciton coupling theory to the structure of mitochondrial cytochrome b, Biochemistry, 1994, 33, 176–185 CrossRef CAS.
- A. M. Arutyunyan, V. B. Borisov, V. I. Novoderezhkin, J. Ghaim, J. Zhang, R. B. Gennis and A. A. Konstantinov, Strong excitonic interactions in the oxygen-reducing site of bd-type oxidase: The Fe-to-Fe distance between hemes d and b595 is 10 A, Biochemistry, 2008, 47, 1752–1759 CrossRef CAS.
- D. W. Urry, W. W. Wainio and D. Grebner, Evidence for conformational differences between oxidized and reduced cytochrome oxidase, Biochem. Biophys. Res. Commun., 1967, 27, 625–631 CrossRef CAS.
- (a) Y. P. Myer and A. Pande, in The Porphyrins, ed. D. Dolphin, Academic Press, San Francisco, 1978, vol. IIIA, pp. 271–322 Search PubMed; (b) Y. P. Myer, Academic Press, 1985, vol. 14, pp. 149–188.
- (a) R. H. Tiesjema and B. F. van Gelder, Biochemical and biophysical studies on cytochrome c oxidase XVI. Circular dichroic study of cytochrome c oxidase its ligand complexes, Biochim. Biophys. Acta, Bioenerg., 1974, 347, 202–214 CrossRef CAS; (b) R. H. Tiesjema, G. P. M. A. Hardyg and B. F. van Gelder, Biochemical and biophysical studies on cytochrome c oxidase. XVIII. Potentiometric titrations of cytochrome c oxidase followed by circular dichroism, Biochim. Biophys. Acta, Bioenerg., 1974, 357, 24–33 CrossRef CAS.
- R. A. Goldbeck, O. Einarsdottir, T. D. Dawes, D. B. O'Connor, K. K. Surerus, J. Fee and D. S. Kliger, Magnetic circular dichroism study of cytochrome ba3 from thermus thermophilus: spectral contributions from cytochromes b and a3 and nanosecond spectroscopy of CO photodissociation intermediates, Biochemistry, 1992, 31, 9376–9387 CrossRef CAS.
- T. Tsukihara, H. Aoyama, E. Yamashita, T. Tomizaki, H. Yamaguchi, K. Shinzawa-Itoh, T. Nakashima, R. Yaono and S. Yoshikawa, Structures of metal sites of oxidized bovine heart cytochrome c oxidase at 2.8, Science, 1995, 269, 1069–1074 CrossRef CAS.
- (a) R. W. Woody and I. Tinoco, Optical rotation of oriented helices. III. Calculation of the rotatory dispersion and circular dichroism of the alpha- and 310-helix, J. Chem. Phys., 1967, 46, 4927–4945 CrossRef CAS; (b) G. Blauer, N. Sreerama and R. W. Woody, Optical activity of hemoproteins in the Soret region. Circular dichroism of the heme undecapeptide of cytochrome c in aqueous solution, Biochemistry, 1993, 32, 6674–6679 CrossRef CAS.
- H. DeVoe, Optical properties of molecular aggregates. II. Classical theory of the refraction, absorbtion, and optical activity of solutions and crystals, J. Chem. Phys., 1965, 43, 3199–3208 CrossRef CAS.
- (a) D. A. Proshlyakov, M. A. Pressler, C. DeMaso, J. F. Leykam, D. L. DeWitt and G. L. Babcock, Oxygen activation and reduction in respiration: involvement of redox-active tyrosine 244, Science, 2000, 290, 1588–1591 CrossRef CAS; (b) D. A. Proshlyakov, UV optical absorption by protein radicals in cytochrome c oxidase, Biochim. Biophys. Acta, Bioenerg., 2004, 1655, 282–289 CrossRef CAS.
- L. R. Fowler, S. H. Richardson and Y. Hatefi, A rapid method for the preparation of highly purified cytochrome oxidase, Biochim. Biophys. Acta, 1962, 64, 170–173 CrossRef CAS.
- Y. Chen, L. Hunsicker-Wang, R. L. Pacoma, E. Luna and J. A. Fee, A homologous expression system for obtaining engineered cytochrome ba3 from Thermus thermophilus HB8, Protein Expression Purif., 2005, 40, 299–318 CrossRef CAS.
- O. Farver, Y. Chenc, J. A. Feec and I. Pechtb, Electron transfer among the CuA-, heme b- and a3-centers of Thermus thermophilus cytochrome ba3, FEBS Lett., 2006, 580, 3417–3421 CrossRef CAS.
- J. G. Kirkwood, On the theory of optical rotatory power, J. Chem. Phys., 1937, 5, 479 CAS.
- T. Tsukihara, K. Shimokata, Y. Katayama, H. Shimada, K. Muramoto, H. Aoyama, M. Mochizuki, K. Shinzawa-Itoh, E. Yamashita, M. Yao, Y. Ishimura and S. Yoshikawa, The low-spin heme of cytochrome c oxidase as the driving element of the proton-pumping process, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 15304–15309 CrossRef CAS.
- D. M. Rogers and J. D. Hirst, Ab Initio Study of Aromatic Side Chains of Amino Acids in Gas Phase and Solution. J. Phys. Chem. A, 2003, 107, 11191–11200 Search PubMed.
- (a) W. H. Vanneste, The stoichiometry and absorption spectra of components a and a3 in cytochrome c oxidase, Biochemistry, 1966, 65, 838–848 CrossRef; (b) G.-L. Liao and G. Palmer, The reduced minus oxidized spectra of cytochromes a and a3, Biochim. Biophys. Acta, Bioenerg., 1996, 1274, 109–111 CrossRef.
- J. Applequist, K. R. Sundberg, M. L. Olson and L. C. Weiss, A normal mode treatment of optical properties of a classical coupled dipole oscillator system with Lorentzian band shapes, J. Chem. Phys., 1979, 70, 1240–1246 CrossRef CAS.
- M. Wikstrom, K. Krab and M. Saraste, Cytochrome Oxidase—A Synthesis, Academic Press, New York, 1981 Search PubMed.
- B. H. Zimmermann, C. I. Nitsche, J. A. Fee, F. Rusnak and E. Mnck, Properties of a copper-containing cytochrome ba3: A second terminal oxidase from the extreme thermophile thermus thermophilus, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 5779–5783 CrossRef CAS.
- Y. P. Myer, Conformation of cytochromes. V. Cytochrome c oxidase, J. Biol. Chem., 1971, 246, 1241–1248 CAS.
- A. J. Moody, “As prepared” forms of fully oxidised haem/Cu terminal oxidases, Biochim. Biophys. Acta, Bioenerg., 1996, 1276, 6–20 CrossRef.
- (a) H. Michel, Cytochrome c oxidase: catalytic cycle and mechanism of proton pumping—A discussion, Biochemistry, 1999, 38, 15129–15140 CrossRef CAS; (b) H. Aoyamaa, K. Muramotoc, K. Shinzawa-Itoh, K. Hiratad, E. Yamashita, T. Tsukihara, T. Ogurac and S. Yoshikawa, A peroxide bridge between Fe and Cu ions in the O2 reduction site of fully oxidized cytochrome c oxidase could suppress the proton pump, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 2165–2169 CrossRef CAS.
- P. G. Papadopoulos, S. A. Walter, J. Li and G. M. Baker, Proton interactions in the resting form of cytochrome oxidase, Biochemistry, 1991, 30, 840–850 CrossRef CAS.
- S. Horie and M. Morrison, Cytochrome c oxidase components. I: purification and properties, The Journal Of Biological Chemistry, 1963, 238 Search PubMed.
- L. Vickery, T. Nozawa and K. Sauer, Magnetic circular dichroism studies of myoglobin complexes. Correlation with heme spin state and axial ligation, J. Am. Chem. Soc., 1976, 98, 343–350 CrossRef CAS.
- Y. Orii, Determination and novel features of the absolute absorption spectra of the heme a moieties in cytochrome c oxidase, Journal of Bioenergetics and Biomembranes, 1998, 30, 47–53 Search PubMed.
- G. M. Baker and G. Palmer, Effect of high pH on the spectral and catalytic properties of beef heart cytochrome oxidase, Biochemistry, 1987, 26, 3038–3044 CrossRef CAS.
- (a) R. A. Copeland, Long-distance cofactor interactions in terminal oxidases studied by second-derivative absorption spectroscopy, J. Bioenerg. Biomembr., 1993, 25, 93–102 CrossRef CAS; (b) J. S. Felsch, M. P. Horvath, S. Gursky, M. R. Hobaugh, P. N. Goudreau, J. A. Fee, W. T. Morgan, S. J. Admiraal, M. Ikeda-Saito, T. Fujiwara, Y. Fukumori, T. Yamanaka and R. A. Copeland, Protein Sci., 1994, 3, 2097–2103 CrossRef CAS.
- K. Carter and G. Palmer, Models of the two heme centers in cytochrome oxidase, J. Biol. Chem., 1982, 257, 13507–13514 CAS.
- E. R. Holiday, Spectrophotometry of proteins: Absorption spectra of tyrosine, tryptophan and their mixtures. II. Estimation of tyrosine and tryptophan in proteins, Biochem. J., 1936, 30, 1795–1803 CAS.
- M. Voicescu, Y. E. Khoury, D. Martel, M. Heinrich and P. Hellwig, Spectroscopic analysis of tyrosine derivatives: on the role of the tyrosine-histidine covalent linkage in cytochrome c oxidase, J. Phys. Chem., 2009, 113, 13429–13436 Search PubMed.
- L. Qin, D. A. Mills, L. Buhrow, C. Hiser and S. Ferguson-Miller, A conserved steroid binding site in cytochrome c oxidase, Biochemistry, 2008, 47, 9931–9933 CrossRef CAS.
- Y. P. Myer and P. A. Bullock, Cytochrome b562 from Escherichia coli: conformational, configurational, and spin-state characterization, Biochemistry, 1978, 17, 3723–3729 CrossRef CAS.
- E. A. Gorbikova, M. Wikstrom and M. I. Verkhovsky, The protonation state of the cross-linked tyrosine during the catalytic cycle of cytochrome c oxidase, J. Biol. Chem., 2008, 283, 34907–34912 CrossRef CAS.
Footnotes |
| † This article is published as part of a themed issue on Cytochromes, Guest Edited by Norbert Jakubowski and Peter Roos. |
| ‡ Abbreviations: BTP, [1,3-bis(tris(hydroxymethyl)methylamino)propane; CD, circular dichroism; COX, cytochrome c oxidase; DM, dodecyl maltoside; PMS, phenazine methosulfate. |
| This journal is © The Royal Society of Chemistry 2011 |