The puzzle posed by COMMD1, a newly discovered protein binding Cu(II)
Bibudhendra
Sarkar
*a and
Eve A.
Roberts
b
aMolecular Structure and Function Program, The Hospital for Sick Children Research Institute, Department of Biochemistry, University of Toronto, Toronto, Ontario, Canada. E-mail: bsarkar@sickkids.ca; Fax: +1 416-813-5022; Tel: +1 416-813-5921
bGenetics and Genome Biology Program, The Hospital for Sick Children Research Institute, Departments of Paediatrics, Medicine, and Pharmacology, University of Toronto, Toronto, Ontario, Canada
First published on 3rd November 2010
Abstract
Copper is critically important for cellular metabolism. It plays essential roles in developmental processes, including angiogenesis. The liver is central to mammalian copper homeostasis: biliary excretion is the major route of excretion for ingested copper and serves to regulate the total amount of copper in the organism. An extensive network of proteins manipulates copper disposition in hepatocytes, but comparatively little is known about this protein system. Copper exists in two oxidation states: most extracellular copper is Cu(II) and most, if not all, intracellular copper is Cu(I). Typical intracellular copper-binding proteins, such as the Cu-transporting P-type ATPases ATP7B (Wilson ATPase) and ATP7A (Menkes ATPase), bind copper as Cu(I). Accordingly, the recent discovery that the ubiquitous protein COMMD1 binds Cu(II) exclusively raises the question as to what role Cu(II) may play in intracellular processes. This issue is particularly important in the liver and brain. In humans, Wilson’s disease, due to mutations in ATP7B, exhibits progressive liver damage from copper accumulation; in some Bedlington terriers, mutations in COMMD1 are associated with chronic copper-overloaded liver disease, clinically distinct from Wilson’s disease. It seems unlikely that Cu(II), which generates reactive oxygen species through the Fenton reaction, has a physiological role intracellularly; however, Cu(II) might be the preferred state of copper for elimination from the cell, such as by biliary excretion. We argue that COMMD1 participates in the normal disposition of copper within the hepatocyte and we speculate about that role. COMMD1 may contribute to the mechanism of biliary excretion of copper by virtue of binding Cu(II). Additionally, or alternatively, COMMD1 may be an important component of an intracellular system for utilizing Cu(II), or for detecting and detoxifying it.
 Bibudhendra Sarkar | Bibudhendra Sarkar received his PhD in Biochemistry from the University of Southern California. He studied protein chemistry with Hal Dixon at the University of Cambridge and quantum biochemistry with Madame Alberte Pullman at the Université de Paris. At the University of Toronto and the Hospital for Sick Children, he established his research career on metal-caused genetic diseases. He discovered the Menkes disease treatment. He became full Professor in 1978, head of the department of Structural Biology and Biochemistry between 1990–2002 and director of the Advanced Protein Technology Center 1998–2002. Currently, he is Professor Emeritus and continues his research on molecular mechanisms of metal-caused diseases. |
 Eve A. Roberts | Dr Roberts was educated at Bryn Mawr College, the Johns Hopkins University School of Medicine, and Dalhousie University. She trained in hepatology with Dame Sheila Sherlock at the Royal Free Hospital in London. She joined the staff of the Hospital for Sick Children in 1984 as a translational clinician-scientist and developed an innovative paediatric hepatology programme. She was appointed Senior Scientist in the Research Institute in 1990 and full Professor in 1998. She was on the Toronto team which identified ATP7B. In 2008 she received the Gold Medal of the Canadian Liver Foundation/Canadian Association for the Study of the Liver. |
1. Introduction
Copper is very important in mammalian cellular metabolism. It plays a central role in developmental processes including cell growth and apoptosis.1 It functions as an essential cofactor to a variety of enzymes, mainly because it is redox active. It can exist as either Cu(I) or Cu(II) and cycle between these two oxidation states. Excess copper, particularly Cu(II), contributes to oxidative stress and thus is highly toxic to cells. Accordingly, almost all intracellular copper is bound to metallochaperones or other proteins. Most, if not all, intracellular copper is in Cu(I) form. The recent discovery2 that the ubiquitous protein COMMD1 binds only Cu(II) raises the interesting question as to what role Cu(II) may play in intracellular processes.2. Disposition of copper in humans
The total-body handling of copper in mammals is understood in some detail. Copper is absorbed in the proximal small intestine and then loosely bound to albumin or amino acids, principally histidine, for transport in the plasma compartment.3,4Copper-albumin and copper-histidine distribute copper to various tissues. The copper associated with these transporters is Cu(II).5 The portal circulation, a venous vascular bed between the intestinal tract and the liver, brings copper absorbed from the intestinal tract directly to the liver. The handling of copper in hepatocytes is particularly important because certain critical proteins, like ATP7B, also known as the Wilson ATPase, are expressed principally in the hepatocyte, which serves as the main route of copper excretion from the body, through secretion into bile (Fig. 1). Cu(II) brought to the liver via the portal vein must be reduced to Cu(I) before uptake into hepatocytes. It is not known how this reduction occurs. Dietary reductants or an unidentified reductase located on the hepatocellular plasma membrane may participate in this reaction. Copper is taken actively across the hepatocellular plasma membrane by the transporter CTR1, encoded by the gene SLC31A1.6 The structure of human CTR1 has been solved by solved by electron crystallography to an in plane resolution of 7 Å.7 The protein exists as a stable trimer spanning the membrane with a conducting pore for copper in the center of the trimer between the monomers. There is a conserved methionine cluster motif (MXXXM) at the pore entrance, in contrast to a cysteine cluster (CXXC) typical of the ATP7A, which is the Menkes ATPase, and the Wilson ATPase ATP7B, and various intracellular copper-binding molecules. A second copper transporter, CTR2, may mediate observed low-affinity copper uptake.8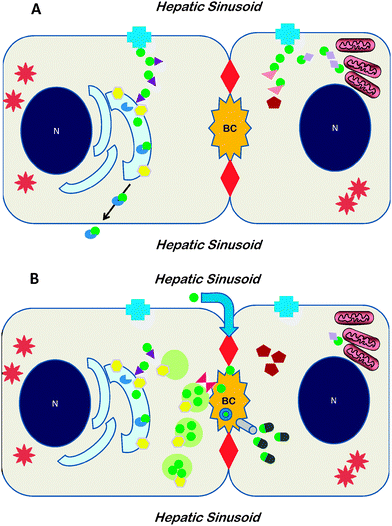 | ||
| Fig. 1 Cartoons depicting the network of proteins involved in copper disposition within the hepatocyte at low copper concentrations (A) or at high concentrations (B). A doublet of hepatocytes is illustrated, as part of a single-cell thick cord of hepatocytes, with sinusoids containing blood flowing from portal tract to hepatic venule on either side; the specialized bile canalicular membrane located between two tight junctions (large red diamonds); N: nucleus; BC: bile canaliculus. In A, when intracellularcopper is not in excess, copper (green circle) is taken up viaCTR1 (blue cross) in the plasma membrane and deployed within the hepatocytevia metallochaperones. ATOX1 (purple triangle) delivers copper to ATP7B (yellow hexagon) located in trans-Golgi network. Copper is transferred into the Golgi where it is inserted into apoceruloplasmin (incomplete blue disc) and subsequently the resulting holoceruloplasmin is secreted across the plasma membrane. As shown in the second hepatocyte, CCS1 (rose triangle) delivers copper to SOD1 (red pentagon) in cytoplasm, and Cox17 (lavender diamond) delivers copper to mitochondria. In B, when intracellularcopper is in excess, uptake by CTR1 is lessened. ATP7B is found in endosomes in the vicinity of the BC, and biliary excretion may be expedited by COMMD1 (scarlet hourglass). In the second hepatocyte some copper bound to glutathione (green-gray bullets) is excreted into by viaMRP (gray cylinder). A paracellular pathway (blue arrow through tight junction at top) may also function. Copper complexed in bile is shown as the green circle in a blue ring. Spiny stars represent metallothioneins in the cytoplasm of hepatocytes. Note that the complete network of proteins involved in hepatocellular copper disposition has not yet been identified. | ||
Once inside the hepatocyte, copper—now as Cu(I)—is not found as an ionic species. Intracellular copper can be conceptualized as constituting at least three functional compartments. The first functional compartment relates to processes carried out by the cell: mitochondrial energy production, protein synthesis, and local cytoprotection. With respect to this compartment, copper as Cu(I) is associated with any of a number of low molecular-weight intracellular proteins, known as metallochaperones, not all of which have yet been identified. Metallochaperones deliver copper to different specific target molecules within the hepatocyte (Fig. 1A). ATOX1, which has one CXXC copper-binding motif, delivers Cu(I) to the Wilson ATPase, in the trans-Golgi network. CCS, which has an MXCXXC motif in domain 1, delivers copper to superoxide dismutase 1 (SOD1, Cu/Zn superoxide dismutase) in the cytoplasm. Various proteins, notably Cox17, guide copper to components of complex IV (cytochrome c oxidase) of the oxidative-phosphorylation chain in the mitochondria, such as Sco1 and Sco2.9,10 The Wilson ATPase has a distinctive amino terminus tail containing six MXCXXC copper-binding motifs. Its N-terminal copper-binding region binds Cu with the stoichiometry of one Cu per metal-binding domain through a cooperative mechanism.11 Bound copper is Cu(I) coordinated by two cysteines in a distorted linear geometry, and the N-terminal region undergoes secondary and tertiary conformational changes in response to copper-binding.12 Recent characterization of the copper-binding units 4–6, as well as 1–6, of the Wilson ATPase by NMR spectroscopy shows that flexibility between these domains is critically important for transfer of copper from one domain to another and for interaction with ATOX1: this flexibility of linkers between is fundamental to the overall function of ATP7B in copper transport.13 When hepatocellular copper concentrations exceed what is required for physiological functioning, the Wilson ATPase relocates from the trans-Golgi network to vesicles in the region of the bile canalicular membrane of the hepatocyte (Fig. 1B), thus facilitating biliary excretion of copper.14 (Similarly the Menkes ATPase moves to the plasma membrane under conditions of intracellular copper excess, and movement toward one terminus or the other appears to be by amino acid signatures in the respective ATPase.15,16) A second functional pool is that copper associated with metallothionein, and the third, highly mobile pool of copper associated with glutathione.17−19Copper stored in lysosomes may be regarded as a fourth compartment: lysosomal membranes contain copper transporters,20 although the extent of bidirectional copper flux is unclear. This putative compartment attains importance in pathological states since copper-laden lysosomes account for the histochemically stainable copper in diseased hepatocytes which have a long-standing abnormal accumulation of copper.
The mechanisms by which copper gets into bile are complicated and the relative contribution of each possible mechanism to biliary excretion of copper is still poorly understood.21 A principal mechanism appears to be direct exocytosis into bile of copper from copper-containing cytoplasmic vesicles in the endosome-lysosome spectrum.22–25 Another possible mechanism involves the Wilson ATPase in direct transfer of copper across the bile canalicular membrane.16 Observations in the LEC rat, a model for Wilson’s disease, indicate that the Wilson ATPase is not required for at least some of the biliary excretion.26 One such alternative excretory pathway is via the transporter MRP2 (also known as cMOAT),18,27,28 which has been shown to operate in rats for rapid removal of copper administered intravenously.29 This transporter is typically down-regulated in chronic liver disease and specifically in Wilson’s disease. A paracellular pathway of biliary excretion of copper has recently been postulated.30 It seems to require the action of the Wilson ATPase, possibly as a regulator of tight junction ‘leakiness’ through binding the protein zona occludens 1 (ZO-1, also known as tight junction protein 1 or TJP1), a structural component of tight junctions. Whether biliary copper is exclusively Cu(II) or not remains uncertain. Evidence of copper excreted as Cu(II) from hepatocytesin vitro is unconvincing because of the dissimilarities between cells in culture and cells in an intact tissue. Abundant data show that copper in bile is complexed, mainly in aggregates containing lecithin and bile acids31; some copper secreted into bile may be bound to ceruloplasmin remnants.32Cu(II) appears to enhance binding of bile acids to the amino acid asparagine.33Copper in bile is unavailable for reabsorption in the gut.34
Among hereditary diseases of hepatic copper accumulation found in humans, Wilson’s disease is the most important. It develops because the Wilson ATPase is dysfunctional or absent, due to mutations in the gene ATP7B. Affected individuals develop hepatic copper overload, with copper bound as Cu(I) to metallothioneins accumulated in the cytoplasm of hepatocytes or aggregated in lysosomes. In Wilson’s disease, the serum concentration of enzymatically active ceruloplasmin is decreased and thus the serum copper concentration is low, except when the serum concentration of copper not incorporated in ceruloplasmin is elevated, usually as a result of progressive liver damage. Strictly speaking, this is not “free” copper: copper is present as copper-albumin and/or copper histidine and/or associated with other molecules in blood. Other hereditary disorders of copper handling in the liver include Indian childhood cirrhosis and Tyrolean childhood cirrhosis, which are eco-genetic disorders associated with progressive severe liver damage. Sporadic kindreds with other hereditary hepatic copper toxicosis disorders, as yet poorly characterized, may exist.35,36
3. Bedlington terrier copper toxicosis and COMMD1
In Bedlington terriers suffering from an autosomal recessive disorder, copper toxicosis (BT/CT, Bedlington terrier copper toxicosis), there is a massive copper accumulation in the liver, seen as dense granules in lysosomes, resulting in chronic hepatitis and cirrhosis.37,38 It is worth noting that dogs have a distinctive metabolism of copper compared to humans: in the plasma, non-ceruloplasmin-bound copper is mainly bound to amino acids because the canine albumin lacks a specific binding site for copper, due to a mutation of histidine to tyrosine in the third position39,40 and in the liver the copper concentration is elevated physiologically.41 The detailed pathogenesis of BT/CT remains unclear although defective biliary excretion of copper is typically specified as central to the disease mechanism.42 However, unlike Wilson’s disease, the plasma level of ceruloplasmin in BT/CT-affected animals is normal,43 indicating that copper uptake and transport to trans-Golgi network is normal. Proteins known to be involved in hepatocellular copper handling, such as the Wilson ATPase and ATOX1, are normal in this copper toxicosis.44–46In 2002, the MURR1 gene, which is present only in bile-producing organisms (vertebrates), was found to be mutated in most, but certainly not all,47,48 Bedlington terriers affected by BT/CT.47,49,50 The encoded protein is not produced. Subsequently, a family of proteins in diverse species, including flies and yeast, with structural and functional homology to MURR1 was discovered.51 A new nomenclature for this family of proteins has been assigned as COMMD (copper metabolism gene MURR1), which at present consists of 10 subgroups (COMMD1-COMMD10); MURR1 was renamed COMMD1. All these proteins are defined by the presence of a conserved and unique motif that is leucine-rich, 70–85 amino acids long, and near the carboxyl terminus. This domain functions as an interface for protein–protein interaction. COMMD1 is ubiquitously expressed in tissues and cell types.52 The protein is mainly located in cytoplasm but has also been found associated with cytoplasmic vesicular compartments of carcinoma cell lines. COMMD1 functions mainly as an intracellular regulatory protein with multiple functions; it shows no homology with other known proteins or motifs, outside of its protein family. Some evidence exists that COMMD1 interacts specifically with the Wilson ATPase.53COMMD1 was also found to interact with human epithelial sodium channel and to inhibit it;54 it also binds to creatine kinase and inhibits it.55COMMD1 appears to have an extensive role in regulating protein degradation. In the embryo COMMD1 affects degradation of the factor HIF-1α, a βHLH/PAS transcription factor which regulates angiogenesis and various cellular metabolic functions, by competing with HSP90β and HSP70 to regulate HIF-1αprotein degradation.56 Of note, both HSP70 and HSP90 were identified as members of the copper-metalloproteome in Hep G2 cells.57 Recent findings show that COMMD1 also competes with the ARNT protein (also known as HIF-1β) for binding with HIF-1α and thus influences HIF-1αbinding to DNA,58 a function possibly important for the growth behaviour of some cancers where COMMD1 expression is decreased.
COMMD1 may have a broader role in the control of cellular function relating to copper disposition. There is an inverse correlation between COMMD1 and the intracellular level of copper.59 An intracellular negative regulator of apoptosis, XIAP (X-linked inhibitor of apoptosis) can bind copper; after binding copper, XIAP is susceptible to more rapid degradation.60 This phenomenon is not common with metal-binding proteins, unless there is some conformational change causing the protein susceptible to enzymatic degradation. XIAP was identified as a negative regulator of COMMD1. The copper level in the liver of XIAP-deficient mice was lower than that in control animals. This protein was found to decrease the level of COMMD1 by promoting ubiquitination and degradation of COMMD1.61 Under the conditions of copper overload, such as in Wilson’s disease or other copper toxicosis disorders and cell cultures under high copper concentration, XIAP levels were markedly reduced. In untreated toxic milk mice (Jackson laboratory version, C3H background strain), a model for Wilson’s disease, COMMD1 expression rose over the first six months of life, compared to normal control, while immunodetectable XIAP progressively dropped over the same time period.62 It is not surprising that XIAP can bind copper. XIAP has five CXXC motifs in its primary structure. It has been identified as one of a family of regulatory proteins with baculoviral IAP repeat (BIR) motifs.63 Other proteins in the BIR-containing protein family also have several of these canonical CXXC-containing repeats in their primary structure. These proteins affect protein–protein interactions and have a role in modulating diverse proteins including inhibition of caspases, ubiquitination (notably by ubiquitin ligase E3 activity), and regulation of NKT cells and of BMP-associated developmental processes.
Relevant to Wilson’s disease, COMMD1 also influences mechanisms of apoptosis. It inhibits the degradation of IκB-α and thus keeps the transcription factor NF-κB in an inactive form, which is critical for making the resting CD4T cells non-permissive for HIV infection.64 Furthermore COMMD1 regulates the nuclear function of NF-κB by affecting the association of NF-κB with chromatin.
Thus far, no reports have indicated that COMMD1 is mutated in patients with either Indian Childhood Cirrhosis or Tyrolean childhood cirrhosis.65 In several series, mutations in human COMMD1 have not been identified in patients with Wilson’s disease or presumed Wilsonian liver disease.66–69 Thus, in general, human COMMD1 does not appear to act as a gene modifier of ATP7B, and it does not account for atypical presentations of Wilson’s disease.
4. Cu(II)-binding by COMMD1
In spite of various reports that suggested the role of COMMD1 in copper metabolism, it was shown only recently COMMD1 binds copper exclusively in Cu(II) form.2 By using various techniques including native-PAGE, EPR, UV-visible electronic absorption, intrinsic fluorescence spectroscopes as well as DEPC (diethylpyrocarbonate) modification of histidine, we demonstrated that COMMD1 binds copper specifically as Cu(II) in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry and does not bind other divalent metals. Moreover, the exon 2 product, COMMD(61–154), which is deleted in Bedlington terriers, alone was able to bind Cu(II) just like the wild type protein, with a stoichiometry of 1 mol of Cu(II) per protein monomer. The approximate binding was determined by DEPC modification and fluorescence spectroscopic studies. The protection of the DEPC modification of COMMD1 by Cu(II) involves histidine residues. Further investigation by DEPC modification of COMMD(61–154) and subsequent MALDI MS mapping and MS/MS sequencing identified the protection of His101 and His 134 residues in the presence of Cu(II). Fluorescence studies of single point mutants of the full length protein revealed involvement of Met110 in addition to His101 and His134 in direct Cu(II) binding.2 The postulated binding site is shown in Fig. 2. It is possible that one or more additional residues that are involved in the direct Cu(II) binding remain to be identified.
:1 stoichiometry and does not bind other divalent metals. Moreover, the exon 2 product, COMMD(61–154), which is deleted in Bedlington terriers, alone was able to bind Cu(II) just like the wild type protein, with a stoichiometry of 1 mol of Cu(II) per protein monomer. The approximate binding was determined by DEPC modification and fluorescence spectroscopic studies. The protection of the DEPC modification of COMMD1 by Cu(II) involves histidine residues. Further investigation by DEPC modification of COMMD(61–154) and subsequent MALDI MS mapping and MS/MS sequencing identified the protection of His101 and His 134 residues in the presence of Cu(II). Fluorescence studies of single point mutants of the full length protein revealed involvement of Met110 in addition to His101 and His134 in direct Cu(II) binding.2 The postulated binding site is shown in Fig. 2. It is possible that one or more additional residues that are involved in the direct Cu(II) binding remain to be identified.
 | ||
| Fig. 2 COMMD1 binds Cu(II) with a stoichiometry of 1 mol of Cu(II) per protein monomer. The protection of DEPC modification of COMMD1 by Cu(II), and subsequent MALDI MS mapping, and MS/MS sequencing identified the protection of His101 and His134 residues in the presence of Cu(II), indicating the involvement of these two His residues in Cu(II) binding. Fluorescence studies of single point mutants of the protein revealed the involvement of Met110, in addition to His101 and H134 in direct binding of Cu(II) to COMMD1 (see also ref. 2). | ||
Direct comparison of Cu(II)-binding sites in other proteins which are known to bind Cu(II) does not advance our assessment of Cu(II)-binding by COMMD1 to any great extent. The striking characteristic of the copper-binding units in Cu(II)-binding proteins is the diversity of their structures. Inherently Cu(II) can assume several different geometric arrangements of ligands arising from various parts of the protein. Potential for distortion in the ligand geometry, or some degree of instability in that architecture, may exist. In fact, the distorted structure is more of a rule than an exception. The cellular environment, the local redox environment, and the very proteins involved all influence the nature of the Cu(II)-binding site.
Sequence alignment of wild-type COMMD1 in dog, human, mouse and mutated COMMD1 in the Bedlington terrier with hepatic copper toxicosis (BT/CT dogs) is shown in Fig. 3. There are one casein kinase site, two PKC phosphorylation sites and two N-myristoylation sites. N-myristoylation promotes reversible protein-membrane and protein–protein interaction that regulate protein targeting.70 Most interestingly, the region deleted in BT/CT contains the PKC phosphorylation sites, myristoylation sites and the copper-binding site. COMMD1 might function as a specific cargo (substrate) selector in an AP-like complex, selecting ATP7B, the Wilson ATPase, and accompanying it to specific destination. The phosphorylation and myristoylation sites on COMMD1 could provide the signals for the trafficking of the Wilson ATPase from one location to the other, similar to the function of members of AP-3 family.
 | ||
| Fig. 3 Alignment of the deduced amino acid sequence of COMMD1 in dog, human, mouse and Bedlington terriers with copper toxicosis. The conserved residues of the protein kinase C phosphorylation sites (pink), the casein kinase II phosphorylation site (yellow) and the N-myristoylation sites (green) are indicated. The Cu(II)-binding residues are shown in blue, as deduced in ref. 2. | ||
5. Why do hepatocytes contain a Cu(II)-binding protein?
Since COMMD1 has been reported to bind to the Wilson ATPase but not to the Menkes ATPase, it seems possible that COMMD1 has a specific role in copper disposition in hepatocytes. The puzzle of COMMD1 is why such a protein would be present in hepatocytes to bind Cu(II). The predominant form of copper intracellularly is Cu(I). Several possibilities exist. On the assumption that the copper-binding site is functional, the least interesting possibility is that COMMD1 binds Cu(II) only in vitro but actually binds Cu(I) in the hepatocytein vivo. Although this possibility cannot be ruled out completely, from what we know about Cu(I)-binding motifs, the primary structure of the COMMD1 binding site, as shown in Fig. 3, strongly argues against this interpretation of the experimental data. Therefore it is entirely appropriate to speculate about the potential functional roles of COMMD1 within hepatocytes.The accumulation of copper in hepatocytes in BT/CT dogs with mutated COMMD1 suggests that COMMD1 participates in biliary copper excretion, although not merely as a carrier for Cu(II). This is currently the prevailing view. Down-regulation of functional COMMD1 by small interference RNA is associated with copper retention in cells.71,72 The apparent specificity of interaction between COMMD1 and ATP7B provides some general evidence for this role in biliary excretion.53 Such participation could be accomplished in various ways. It could be direct participation. One possibility is that the copper excretion process via a transporter in the bile canalicular membrane requires the formation of a complex between COMMD1 and ATP7B: COMMD1 and ATP7B, acting cooperatively, would then transfer copper to the transporter. This could be analogous to the interaction of COMMD1 and Re1A, which form a stable complex and migrate to the nucleus.73–75 Alternatively, a complex of COMMD1 and ATP7B might move to the bile canalicular membrane for exocytic excretion of copper. A recent report shows that COMMD1 binds to endosomal membranesvia specifically-phosphorylated phosphatidylinositols and forms oligomeric complexes which may regulate movement of intracellular membrane proteins.76 These observations may be relevant to how COMMD1 could participate in exocytosis of copper into bile. Any BT/CT dog which expressed mutant COMMD1 would not have this function because of the lack of the protein–protein interaction domain; however, it is a moot point because affected dogs typically express no COMMD1 at all. Another possibility is that the biliary excretion by whatever mechanism requires the Cu(II) bound form and COMMD1 mediates Cu(II) availability for excretion. This possibility requires definitive, preferably in vivo, evidence that Cu(II) is the copper species excreted. The role could be indirect. Binding of Cu(II) might serve a function related to sensing ambient concentrations of copper and mobilizing the excretory apparatus. At the present time few experimental data are available to evaluate this more complicated role for COMMD1 in the excretion of copper. Observations in a well-differentiated mouse hepatoma line indicate that COMMD1 regulates movement of the Wilson ATPase from the cytoplasmic vesicles near the cell periphery back to the trans-Golgi region when intracellular copper concentrations are low; unfortunately, this model did not permit assessment with respect to the bile canalicular membrane as such.72 In HEK293T cells (non-hepatic origin) no effect of COMMD1 on Wilson ATPase translocation, apparently in either direction, was found.77 Other proteins besides COMMD1 participate in the process of ATP7B relocation within the cell. The dynactin subunit p62 may mediate this intracellular movement of the Wilson ATPase toward bile canalicular membrane.78 Glutaredoxin1, which interacts with the amino-termini of both ATP7A and ATP7B, may play a key role in how the Wilson ATPase captures copper for utilization or export.79 Recent findings showed that deficiency in glutaredoxin1 impaired excretion of excess copper in HEK293T cells and intracellular glutathione depletion was associated with intracellularcopper retention and impaired ATP7B trafficking.80
Cu(II) is extremely toxic. In vitro studies show that Cu(II) increases oxidant stress in hepatocytes, with lysosomes as a main target.81 Additionally, Cu(II) adversely affects hepatocellular lipid metabolism and accelerates cell turnover.82 It may alter DNA.83 It is not known whether Cu(II) is produced spontaneously within liver cells, for example, through interaction with activated oxygen species in a Fenton reaction. If Cu(II) were produced intracellularly, COMMD1, functioning as a Cu(II)-binding protein, might bind and thus detoxify the Cu(II) produced. If it did not detoxify the Cu(II) directly, COMMD1 might serve as a sensor of intracellular Cu(II) and mobilize cytoprotective defences. This would be a sensing function for a purpose other than copper excretion. Are potential sources of hepatocellular Cu(II) within the hepatocyte itself currently overlooked? It is conceivable that binding of Cu(I) to metallochaperones or to glutathione is marginally unstable: such Cu(I) dislodged from a metallochaperone might convert to Cu(II). Another possible “intrinsic” source of Cu(II) in the hepatocyte is SOD1, whose breakdown might release Cu(II) into the cytoplasm. COMMD1 has recently been shown to regulate formation of SOD1.84
Thus there is no easy explanation for the intriguing discrepancy that ATP7B binds Cu(I) and COMMD1 binds Cu(II). Since biliary excretion of copper seems to be the process defective in BT/CT, cooperation between ATP7B and COMMD1 in delivering copper into bile may be most likely. However, an emerging concept is that COMMD1 plays a pivotal role in regulation of the Wilson ATPase, ATP7B, and the intracellular utilization of copper. Thus biliary excretion may not be the only function of COMMD1; indeed, a regulatory or quality-control function might be more important. A network of proteins regulates copper levels in hepatocytes and responds to elevated levels of copper by interacting with pathways which can mobilize apoptosis. Based only on the currently known, principal proteins involved in distribution of copper in hepatocytes, a possible configuration of the cellular pathophysiology of Wilson’s disease emerges.62,77 Decreased or absent function of the Wilson ATPase (ATP7B) leads to copper accumulation in the hepatocytes which in turn leads to decreased XIAP and consequent upregulation of COMMD1. This is a self-amplifying cycle. Secondary effects include increased apoptosis of hepatocytes, an important feature of the liver damage of Wilson’s disease.85,86 Additionally, the role of COMMD1 in regulating HIF-1α may be important in the overall pathophysiology of Wilson’s disease.
A few other proteins are known to bind Cu(II), apart from albumin. In hepatocytes the mitochondrial proteins Sco1 and Sco2 bind Cu(I) or Cu(II).9,87 Sco1 and Sco2 may serve regulatory roles in the intracellular disposition of copper,88,89 but emerging evidence indicates that they play a critical role in the biogenesis of cytochrome c oxidase in the mitochondrial oxidative-phosphorylation chain.87 Notably, these proteins function in the specialized (oxidizing) environment of the mitochondrion. The neuronal cytoplasmic protein α-synuclein, which accumulates in neurons in Parkinson’s disease, binds Cu(II). Studies of Cu(II)-binding by α-synuclein indicate that it is a highly dynamic process and suggest that the dynamic nature of Cu(II)-binding within the cell is critical to the physiological role of Cu(II)-binding proteins.90Copper may play an important role in determining localization and aggregation of α-synuclein.91 The interaction of hepatocyte growth factor (HGF) and its plasma membrane receptor Met is inhibited by Cu(II) resulting in decreased function, measured as cell scattering in culture, a quantifiable effect of HGF. A putative histidine-containing binding site for Cu(II) has been identified.92 Factor V in the coagulation cascade may also bind Cu(II),93 but this action would be expected to take place extracellularly.
6. Conclusion
In summary, various intracellular proteins which are capable of binding copper have recently been identified. Most of these proteins bind Cu(I), which is the predominant form of copper found in cells. COMMD1 binds Cu(II), the form of copper found in the plasma compartment. Whereas it is not surprising that proteins in the plasma compartment can bind Cu(II), COMMD1 is an intracellular protein. Very few other proteins within the cell bind Cu(II). Observations of liver disease in Bedlington terriers possessing a mutated COMMD1 which lacks the Cu(II) binding site suggest that in hepatocytes, COMMD1 might play a key role in the biliary excretion of copper. Since intracellular Cu(II) is very toxic as a pro-oxidant, diverse mechanisms to detect it and disarm it should exist. Thus COMMD1 might interact with Cu(II) produced within the cell. Although its role in copper disposition in hepatocytes remains unclear, COMMD1 appears either to participate directly in how Cu(II) is normally handled in hepatocytes or else it may serve to maintain the intracellular milieu whereby Cu(II) can be handled safely.References
- M. L. Turski and D. J. Thiele, J. Biol. Chem., 2009, 284, 717–21 CAS.
- S. Narindrasorasak, P. Kulkarni, P. Deschamps, Y. M. She and B. Sarkar, Biochemistry, 2007, 46, 3116–28 CrossRef CAS.
- B. Sarkar, Chem. Rev., 1999, 99, 2535–2544 CrossRef CAS.
- P. Deschamps, P. P. Kulkarni and B. Sarkar, Inorg. Chem., 2004, 43, 3338–40 CrossRef CAS.
- P. Deschamps, P. P. Kulkarni, M. Gautam-Basak and B. Sarkar, Coord. Chem. Rev., 2005, 249, 895–909 CrossRef CAS.
- M. J. Petris, Pfluegers Arch., 2004, 447, 752–5 CrossRef CAS.
- C. J. De Feo, S. G. Aller, G. S. Siluvai, N. J. Blackburn and V. M. Ungera, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4237–4242 CrossRef CAS.
- J. Lee, M. J. Petris and D. J. Thiele, J. Biol. Chem., 2002, 277, 40253–9 CrossRef CAS.
- P. A. Cobine, F. Pierrel and D. R. Winge, Biochim. Biophys. Acta, Mol. Cell Res., 2006, 1763, 759–72 CrossRef CAS.
- S. C. Leary, D. R. Winge and P. A. Cobine, Biochim. Biophys. Acta, Mol. Cell Res., 2009, 1793, 146–53 CrossRef CAS.
- M. DiDonato, S. Narindrasorasak, J. R. Forbes, D. W. Cox and B. Sarkar, J. Biol. Chem., 1997, 272, 33279–82 CrossRef CAS.
- M. DiDonato, H. F. Hsu, S. Narindrasorasak and L. Que, Jr. and B. Sarkar, Biochemistry, 2000, 39, 1890–1896 CrossRef CAS.
- N. Fatemi, D. M. Korzhnev, A. Velyvis, B. Sarkar and J. D. Forman-Kay, Biochemistry, 2010, 49, 8468–8477, DOI:10.1021/bi1008535.
- N. A. Veldhuis, A. P. Gaeth, R. B. Pearson, K. Gabriel and J. Camakaris, BioMetals, 2009, 22, 177–90 Search PubMed.
- M. J. Petris and J. F. Mercer, Hum. Mol. Genet., 1999, 8, 2107–15 CrossRef CAS.
- S. La Fontaine and J. F. Mercer, Arch. Biochem. Biophys., 2007, 463, 149–67 CrossRef.
- J. H. Freedman, M. R. Ciriolo and J. Peisach, J. Biol. Chem., 1989, 264, 5598–605 CAS.
- N. Ballatori, Drug Metab. Rev., 1991, 23, 83–132 CrossRef CAS.
- S. C. Luza and H. Speisky, Am. J. Clin. Nutr., 1996, 63, 812S–20S CAS.
- P. V. van den Berghe, D. E. Folmer, H. E. Malingre, E. van Beurden, A. E. Klomp, B. van de Sluis, M. Merkx, R. Berger and L. W. Klomp, Biochem. J., 2007, 407, 49–59 CrossRef CAS.
- M. Y. Bartee and S. Lutsenko, BioMetals, 2007, 20, 627–37 Search PubMed.
- J. B. Gross, Jr., B. M. Myers, L. J. Kost, S. M. Kuntz and N. F. LaRusso, J. Clin. Invest., 1989, 83, 30–9 CrossRef CAS.
- B. M. Myers, F. G. Prendergast, R. Holman, S. M. Kuntz and N. F. Larusso, Gastroenterology, 1993, 105, 1814–23 CAS.
- M. A. Cater, S. La Fontaine, K. Shield, Y. Deal and J. F. Mercer, Gastroenterology, 2005, 130, 493–506.
- L. Nyasae, R. Bustos, L. Braiterman, B. Eipper and A. Hubbard, Am. J. Physiol.: Gastrointest. Liver Physiol., 2007, 292, G1181–94 CAS.
- M. L. Schilsky, A. N. Irani, G. R. Gorla, I. Volenberg and S. Gupta, J. Biochem. Mol. Toxicol., 2000, 14, 210–4 CrossRef CAS.
- S. Itagaki, M. Sugawara, M. Kobayashi, K. Miyazaki, T. Hirano and K. Iseki, Drug Metab. Pharmacokinet., 2004, 19, 150–4 Search PubMed.
- P. Ferenci, G. Zollner and M. Trauner, J. Gastroenterol. Hepatol., 2002, 17, S105–12 CrossRef.
- M. Dijkstra, F. Kuipers, G. J. Van den Berg, R. Havinga and R. J. Vonk, Hepatology, 1997, 26, 962–6 CrossRef CAS.
- S. Hernandez, Y. Tsuchiya, J. Garcia-Ruiz, V. Lalioti, S. Nielsen, D. Cassio and I. V. Sandoval, Gastroenterology, 2008, 134, 1215–23 CrossRef CAS.
- K. O. Lewis, Gut, 1973, 14, 221–32 CrossRef CAS.
- G. F. Chowrimootoo, H. A. Ahmed and C. A. Seymour, Biochem. J., 1996, 315, 851–5 CAS.
- B. de Castro, J. L. Lima, I. H. Mayer and S. Reis, J. Pharm. Biomed. Anal., 1998, 16, 771–6 CrossRef CAS.
- T. Y. Tao and J. D. Gitlin, Hepatology, 2003, 37, 1241–7 CrossRef CAS.
- M. M. Fisher and S. Sherlock, Arch. Dis. Child., 1964, 39, 14–7 CrossRef CAS.
- J. H. Lefkowitch, C. L. Honig, M. E. King and J. W. Hagstrom, N. Engl. J. Med., 1982, 307, 271–7 CrossRef CAS.
- J. Ludwig, C. A. Owen, Jr., S. S. Barham, J. T. McCall and R. M. Hardy, Lab. Invest., 1980, 43, 82–7 Search PubMed.
- S. Haywood and E. J. Hall, Veterinary Record, 1992, 131, 272 Search PubMed.
- D. W. Appleton and B. Sarkar, J. Biol. Chem., 1971, 246, 5040–6 CAS.
- J. W. Dixon and B. Sarkar, J. Biol. Chem., 1974, 249, 5872–7 CAS.
- C. A. Goresky, T. H. Holmes and A. Sass-Kortsak, Can. J. Physiol. Pharmacol., 1968, 46, 771–84 CAS.
- L. C. Su, C. A. Owen, Jr, P. E. Zollman and R. M. Hardy, Am. J. Physiol., 1982, 243, G231–6 CAS.
- C. A. Owen, Jr. and J. Ludwig, Am. J. Pathol., 1982, 106, 432–4 CAS.
- S. L. Dagenais, M. Guevara-Fujita, R. Loechel, A. C. Burgess, D. E. Miller, V. Yuzbasiyan-Gurkan, G. J. Brewer and T. W. Glover, Mamm. Genome, 1999, 10, 753–6 CrossRef CAS.
- M. S. Nanji and D. W. Cox, Genomics, 1999, 62, 108–12 CrossRef CAS.
- M. Nanji, V. A. Coronado and D. W. Cox, Mamm. Genome, 2001, 12, 617–21 CrossRef CAS.
- V. A. Coronado, D. Damaraju, R. Kohijoki and D. W. Cox, Mamm. Genome, 2003, 14, 483–91 CrossRef CAS.
- C. Hyun, L. T. Lavulo and L. J. Filippich, Am. J. Vet. Res., 2004, 65, 1573–9 CrossRef CAS.
- B. van De Sluis, J. Rothuizen, P. L. Pearson, B. A. van Oost and C. Wijmenga, Hum. Mol. Genet., 2002, 11, 165–73 CrossRef CAS.
- O. P. Forman, M. E. Boursnell, B. J. Dunmore, N. Stendall, B. van den Sluis, N. Fretwell, C. Jones, C. Wijmenga, J. Rothuizen, B. A. van Oost, N. G. Holmes, M. M. Binns and P. Jones, Anim. Genet., 2005, 36, 497–501 CAS.
- E. Burstein, J. E. Hoberg, A. S. Wilkinson, J. M. Rumble, R. A. Csomos, C. M. Komarck, G. N. Maine, J. C. Wilkinson, M. W. Mayo and C. S. Duckett, J. Biol. Chem., 2005, 280, 22222–32 CrossRef CAS.
- A. E. Klomp, B. van de Sluis, L. W. Klomp and C. Wijmenga, J. Hepatol., 2003, 39, 703–9 CrossRef CAS.
- T. Y. Tao, F. Liu, L. Klomp, C. Wijmenga and J. D. Gitlin, J. Biol. Chem., 2003, 278, 41593–6 CrossRef CAS.
- W. Biasio, T. Chang, C. J. McIntosh and F. J. McDonald, J. Biol. Chem., 2004, 279, 5429–34 CAS.
- P. Li, S. Zhang and C. Fan, Int. J. Biochem. Cell Biol., 2009, 41, 2459–65 CrossRef CAS.
- B. van de Sluis, A. J. Groot, J. Vermeulen, E. van der Wall, P. J. van Diest, C. Wijmenga, L. W. Klomp and M. Vooijs, PLoS One, 2009, 4, e7332 CrossRef.
- S. D. Smith, Y. M. She, E. A. Roberts and B. Sarkar, J. Proteome Res., 2004, 3, 834–40 CrossRef CAS.
- B. van de Sluis, X.-C. Mao, Y.-L. Zhai, A. J. Groot, J. F. Vermeulen, E. van der Wall, P. J. van Diest, M. H. Hofker, C. Wijmenga, L. W. Klomp, K. R. Cho, E. R. Fearon, M. Vooijs and E. Burstein, J. Clin. Invest., 2010, 120, 2119–30 CrossRef CAS.
- A. R. Mufti, E. Burstein and C. S. Duckett, Arch. Biochem. Biophys., 2007, 463, 168–74 CrossRef CAS.
- A. R. Mufti, E. Burstein, R. A. Csomos, P. C. Graf, J. C. Wilkinson, R. D. Dick, M. Challa, J. K. Son, S. B. Bratton, G. L. Su, G. J. Brewer, U. Jakob and C. S. Duckett, Mol. Cell, 2006, 21, 775–85 CrossRef CAS.
- G. N. Maine, X. Mao, P. A. Muller, C. M. Komarck, L. W. Klomp and E. Burstein, Biochem. J., 2009, 417, 601–9 CrossRef CAS.
- E. A. Roberts, C. H. Lau, T. R. da Silveira and S. Yang, Biochem. Biophys. Res. Commun., 2007, 363, 921–5 CrossRef CAS.
- S. M. Srinivasula and J. D. Ashwell, Mol. Cell, 2008, 30, 123–35 CrossRef CAS.
- L. Ganesh, E. Burstein, A. Guha-Niyogi, M. K. Louder, J. R. Mascola, L. W. Klomp, C. Wijmenga, C. S. Duckett and G. J. Nabel, Nature, 2003, 426, 853–7 CrossRef CAS.
- T. Muller, B. van de Sluis, A. Zhernakova, E. van Binsbergen, A. R. Janecke, A. Bavdekar, A. Pandit, H. Weirich-Schwaiger, H. Witt, H. Ellemunter, J. Deutsch, H. Denk, W. Muller, I. Sternlieb, M. S. Tanner and C. Wijmenga, J. Hepatol., 2003, 38, 164–8 CrossRef CAS.
- B. Stuehler, J. Reichert, W. Stremmel and M. Schaefer, J. Mol. Med., 2004, 82, 629–34 CAS.
- V. A. Coronado, J. A. Bonneville, H. Nazer, E. A. Roberts and D. W. Cox, Clin. Genet., 2005, 68, 548–51 CrossRef CAS.
- K. H. Weiss, U. Merle, M. Schaefer, P. Ferenci, J. Fullekrug and W. Stremmel, World J. Gastroenterol., 2006, 12, 2239–42 Search PubMed.
- Z. Y. Wu, G. X. Zhao, W. J. Chen, N. Wang, B. Wan, M. T. Lin, S. X. Murong and L. Yu, J. Mol. Med., 2006, 84, 438–442 CrossRef CAS.
- T. A. Farazi, G. Waksman and J. I. Gordon, J. Biol. Chem., 2001, 276, 39501–4 CrossRef CAS.
- E. Burstein, L. Ganesh, R. D. Dick, B. van De Sluis, J. C. Wilkinson, L. W. Klomp, C. Wijmenga, G. J. Brewer, G. J. Nabel and C. S. Duckett, EMBO J., 2004, 23, 244–54 CrossRef CAS.
- T. Miyayama, D. HIraoka, F. Kawaji, E. Nakamura, N. Suzuki and Y. Ogra, Biochem. J., 2010, 429, 53–61 CrossRef CAS.
- P. de Bie, B. van de Sluis, E. Burstein, K. J. Duran, R. Berger, C. S. Duckett, C. Wijmenga and L. W. Klomp, Biochem. J., 2006, 398, 63–71 CrossRef CAS.
- M. Lian and X. Zheng, J. Biol. Chem., 2009, 284, 17998–8006 CrossRef CAS.
- X. Mao, N. Gluck, D. Li, G. N. Maine, H. Li, I. W. Zaidi, A. Repaka, M. W. Mayo and E. Burstein, Genes Dev., 2009, 23, 849–61 CrossRef CAS.
- J. L. Burkhead, C. T. Morgan, U. Shinde, G. Haddock and S. Lutsenko, J. Biol. Chem., 2009, 284, 696–707 CAS.
- P. de Bie, B. van de Sluis, E. Burstein, P. V. van de Berghe, P. Muller, R. Berger, J. D. Gitlin, C. Wijmenga and L. W. Klomp, Gastroenterology, 2007, 133, 1316–26 CrossRef CAS.
- C. M. Lim, M. A. Cater, J. F. Mercer and S. La Fontaine, J. Biol. Chem., 2006, 281, 14006–14 CrossRef CAS.
- C. M. Lim, M. A. Cater, J. F. Mercer and S. La Fontaine, Biochem. Biophys. Res. Commun., 2006, 348, 428–36 CrossRef CAS.
- W. C. Singleton, K. T. McInnes, M. A. Cater, W. R. Winnall, R. McKirdy, Y. Yu, P. E. Taylor, B. X. Ke, D. R. Richardson, J. F. Mercer and S. La Fontaine, J. Biol. Chem., 2010, 285, 27111–21 CrossRef CAS.
- R. Seth, S. Yang, S. Choi, M. Sabean and E. A. Roberts, Toxicol. in Vitro, 2004, 18, 501–9 CrossRef CAS.
- D. Kennedy, R. Lyn and J. Pezacki, J. Am. Chem. Soc., 2009, 131, 2444–5 CrossRef CAS.
- S. Oikawa and S. Kawanishi, Biochemistry, 1996, 35, 4584–4590 CrossRef CAS.
- W. I. Vonk, C. Wijmenga, R. Berger, B. van de Sluis and L. W. Klomp, J. Biol. Chem., 2010, 285, 28991–9000 CrossRef CAS.
- S. Strand, W. J. Hofmann, A. Grambihler, H. Hug, M. Volkmann, G. Otto, H. Wesch, S. M. Mariani, V. Hack, W. Stremmel, P. H. Krammer and P. R. Galle, Nat. Med., 1998, 4, 588–93 CrossRef CAS.
- E. A. Roberts, B. H. Robinson and S. Yang, Mol. Genet. Metab., 2008, 93, 54–65 CrossRef CAS.
- S. C. Leary, F. Sasarman, T. Nishimura and E. A. Shoubridge, Hum. Mol. Genet., 2009, 18, 2230–40 CrossRef CAS.
- S. C. Leary, P. A. Cobine, B. A. Kaufman, G. H. Guercin, A. Mattman, J. Palaty, G. Lockitch, D. R. Winge, P. Rustin, R. Horvath and E. A. Shoubridge, Cell Metab., 2007, 5, 9–20 CrossRef CAS.
- J. J. Briere and A. Tzagoloff, Mol. Cell., 2007, 25, 176–8 CrossRef CAS.
- D. R. Brown, Metallomics, 2010, 2, 186–194 RSC.
- X. Wang, D. Moualla, J. A. Wright and D. R. Brown, J. Neurochem., 2010, 113, 704–14 CrossRef CAS.
- T. G. Wright, J. Tsai, Z. Jia and B. E. Elliott, J. Biol. Chem., 2004, 279, 32499–32506 CrossRef CAS.
- E. T. Adman, Adv. Protein Chem., 1991, 42, 145–97 CAS.
| This journal is © The Royal Society of Chemistry 2011 |