Molecular obesity, potency and other addictions in drug discovery
Michael M.
Hann
*
Computational and Structural Chemistry, GSK Medicines Research Centre, Gunnels Wood Rd., Stevenage, SG1 2NY, UK. E-mail: Mike.M.Hann@gsk.com
First published on 7th March 2011
Abstract
Despite the increase in global biology and chemistry knowledge the discovery of effective and safe new drugs seems to become harder rather than easier. Some of this challenge is due to increasing demands for safety and novelty, but some of the risk involved in this should be controllable if we had more effectively learnt from our failures. This perspective reflects on some of the learnings of recent years in relation to the causes of attrition. The term Molecular Obesity is introduced to describe our tendency to build potency into molecules by the inappropriate use of lipophilicity which leads to the premature demise of drug candidates.
Introduction
The road to the successful discovery of new medicines continues to frustrate all those involved. Patients are still in need of effective drugs for many diseases. Payers are increasingly only prepared to pay for innovative rather than derivative drugs. Legislators are demanding that only the very safest possible drugs are licensed. Researchers are equally frustrated in that, despite all the accumulated knowledge from the new technologies, the challenge of successfully navigating through it to find novel drugs seems to get harder rather than easier. In a recent study of the factors contributing to the estimated $1.8bn that it now takes to get a new drug to market, Paul et al. analysed the value and leverage of different components (risk and costs) from target ID, to Lead ID, to Candidate Selection and finally through clinical trial to a new drug.1 It is clear from their sensitivity analysis that the costs of later stage development and, in particular, that of clinical trials, have the highest cost impact of failure for a particular chemical entity. What is also highlighted is that taking the time to get the properties right in a molecular series at the outset does not add hugely to the overall cost, but it does of course, greatly improve the chance of a compound surviving and thus de-risking the propensity to failure in later stages. With current success in the clinical phases being only about 5% (i.e. 95% attrition), if we could only improve our success rate by just 5% then we would actually double the output of pharma and biotech R&D. An understanding of what drives these levels of attrition is therefore fundamental to achieving any reduction in its consequences.As noted by Philip Ball in a Nature News article he wrote under the title of “Economists need their own uncertainty principle” it is important to distinguish the genuinely predictable scientific activities (“which need maths”) from the more chaotic activities (“which need experience and intuition”).2 There is a striking parallel between his commentary on predictability, and lack of it, in economics and the issues we face in drug discovery.
In both cases we need to focus the understood and predictable aspect of our sciences on those parts of the surface that are controllable, while also being prepared for (and perhaps even enjoying) the chaotic nature of the rest. Ball also reminds us of Donald Rumsfeld's much quoted remarks about ‘known knowns, known unknowns and unknown unknowns’. What Rumsfeld neglected to include is the final part of the matrix which is for ‘unknown knowns’; these are those things that are known but have become unknown, either because we have never learnt them, or forgotten about them, or more dangerously chosen to ignore. The aim of this perspective is to look at some of the aspects of medicinal chemistry that have become increasingly known in recent years but often have a tendency to become unknown again!
The fundamental challenge in drug discovery
Medicinal Chemistry is at the heart of the drug discovery process and the creativity of medicinal chemists in identifying the best leads and then optimising them on a complex landscape of constraints is often the defining characteristic of successful drug discovery campaigns. This process more often resembles an unpredictable journey on a chaotic surface rather than a quantitative and predictive science.In addition, despite increased knowledge there is no doubt that our ability to know how to most effectively apply it to navigate what can be a chaotic journey on the multidimensional landscape idealised in Fig. 1 is not getting easier. When such a bounded chaotic system does have some kind of long term pattern but which is clearly not a simple periodic oscillation or orbit the term Strange Attractor is used.3 A strange attractor has non-integer dimensions and is often defined by its apparent chaotic behaviour. Such characteristics are typically sensitivity to initial conditions and the fact that many different end points can result from the same starting point. Experienced medicinal chemists can readily identify with these observations, whereby, for example, different teams in different companies will end up with different drugs (e.g. H2 antagonists), by taking a slightly different perspective on data or ideas generated from the same starting points (Histamine) which are then influenced by emerging observations as each programme evolves. This can, of course, also be exploited by a project team by taking parallel approaches, in the form of different leads, through the optimisation process. However the danger of just adding mass to the previous perceived best compound rather than continually reappraising it, is a recurring pitfall in the pursuit of the pressure to make rapid progress.
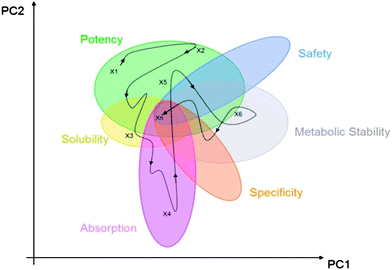 | ||
| Fig. 1 Schematic projection of medchem variables into a 2D principal component space representation of the high dimensional property space that drug discovery takes place in. | ||
Our mission as medicinal chemists is to maximise the use of what is known, and yet to understand the risks of what is not fully known and then develop strategies to integrate them, whilst always recognising that we are ultimately under the influence of strange attractor-influenced landscapes of drug discovery.
What are the leading causes of failure in drug discovery?
It is clear from data compiled by Kola and Landis (Fig. 2) that comparison of cited attrition reasons in 1991 to those in 2000 show a shift to increase in failure due to toxicological reasons and commercial reasons (the latter is not discussed in this perspective).4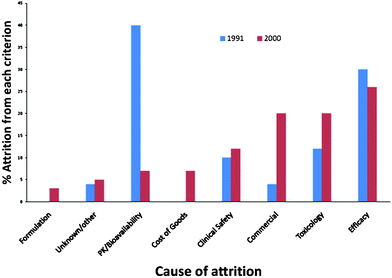 | ||
| Fig. 2 Leading courses of attrition 1991 and 2000 (redrawn with data estimated from Kola and Landis ref. 4). | ||
The issues of PK and biovailability that were the leading cause of attrition in the 1991 data, appear to have been largely controlled. It seems likely that this is due to both a better understanding of pharmacokinetic issues and also in the improved formulation of compounds that results in more chemical entities overcoming this hurdle. Some of this improvement in getting compounds into the body (and keeping them there) is likely related to the rise in attrition due to toxicological issues as the body responds to chemical entities that are “forced” into the body. Thus by using formulation technologies to deliver inappropriate molecules we may have only delayed failure (i.e. the realisation that a molecular series is inappropriate) to a more expensive part of the drug discovery activity.
So what defines inappropriate molecules? Clearly some of the toxicity of a molecule or series will be due to activity at the intended intervention target (or its pathway) which may only become apparent during development. However it is becoming increasingly clear that there are more generic influences behind a significant proportion of toxicology-related attrition. Much of this realisation has come from recently published analyses of internal data from large pharma companies and this has led to the emergence of new guidelines aimed at hopefully controlling this aspect of attrition in the future.5
The earliest of the rules of thumb that have become prevalent in contemporary drug discovery parlance is the Lipinski “rule of five” which has been adopted, and often erroneously used, as defining the limits of drug like space.6 Lipinski's rule actually refers to the likelihood of a compound having oral bioavailability, based on a set of compounds that made it to Phase IIa and were therefore assumed to be a good indicator of oral absorption. Thus compounds that have properties with one or more of either ClogP greater than 5, Molecular Weight greater than 500, Number of H-bond acceptors greater than 10 or Number of H-bond donors greater than 5 are less likely to be orally absorbed. Lipinski's rule of five is often misused as a definition of drug like space rather than oral absorption. It is now becoming increasingly clear that a much more tightly defined set of rules are appropriate if we are considering drug space from the viewpoint of toxicological risk rather than the risk of a compound not being orally absorbed.
One such publication is that from Hughes et al. at Pfizer and considers in vivo tolerability data on 245 compounds that were looked at in multiple assays – see Table 1.7 Compounds that have a ClogP < 3 and TPSA > 75 gave a 6-fold reduction in prevalence of in vivo toxicity versus compounds with ClogP > 3 and TPSA < 75. This reduction rose to a 24-fold reduction for basic compounds. This is often referred to as the Pfizer 3/75 rule and is a clear global sign post to avoiding likely problem compounds.
Another example of an analysis that has led to the emergence of a further set of guidelines is that of the GSK 4/400 rule which relates to compounds with a ClogP less than 4 and a MW less than 400 and how on average they have a more favourable ADMET profile.8 The analysis looked at ca. 30,000 neutral, basic, acidic and zwitterionic molecules that have been profiled in multiple physical chemistry and ADMET assays at GSK – see Fig. 3.
 | ||
| Fig. 3 GSK 4/400 rule for neutral molecules for ADMET issues (reproduced with permission from ref. 8). | ||
Another type of analysis which has been widely publicised is that of Leeson and Springthorpe in their seminal 2007 review.5 Among the many interesting observations in this review, they addressed the issue of receptor promiscuity by analysing the data from 2133 compounds that have been profiled in Cerep Bioprint assays – see Fig. 4. Using a promiscuity scale based on the number of Cerep targets that a compound showed activity at >30% when assayed at 10uM, they showed that compounds with a ClogP < 3 showed decreased promiscuity while compounds with ClogP > 4 had increased risks. The risks are also dependent on the charge state of the molecules with bases and quaternary salts having the most risk and acids the least. They have further codified this in terms of a Lipophilic Ligand Efficiency LLE index, defined as pIC50 − LogP. A value of greater than 5 is suggested as leading to significant reduction in the risk of toxicity for any given compound. The reason for this can be readily understood when it is remembered that the lipophilicity scales (such as clogP) are logarithmic and therefore an increase of just one unit in clogP means that there is now ten times more compound present in the highly lipophilic cellular membranes. These membranes are the home to many of the critical signalling systems and inappropriate triggering by local high concentrations of not very intrinsically potent compounds, can easily lead to unwanted effects leading to potentially toxicological events. While promiscuity of this non-specific type will be detrimental, there may be situations (e.g. for polypharmacology) where some degree of promiscuity is desirable.
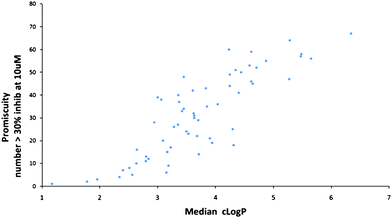 | ||
| Fig. 4 Promiscuity as function of ClogP (drawn using data supplied by Paul Leeson5). | ||
A further perspective on attrition related parameters is given by a count of the number of aromatic rings. While clearly related in a non-linear manner to lipophilicity, the analysis of this property by Ritchie and MacDonald gives interesting insights which can be summarised as “the fewer aromatic rings in an oral drug candidate the better” with less than three being suggested as an appropriate target number.9 Continuing this theme of the dangers of too much sp2 or aromatic character in molecules, Humblet and colleagues have shown that the survival rate of compounds through the drug discovery process is enhanced by an increase in the fraction of sp3 hybridised carbon atoms and the number of chiral centres present.10 In addition to giving access to a greater diversity of compounds to explore it seems likely that one of the benefits of chirality in a drug is that it leads to increased complexity (and hence potential potency through appropriate complementarity) to a specific target without increasing the molecular weight of ligands.12 Clear evidence that a chiral motif (i.e. increased complexity) is contributing greater molecular recognition can be found by Eudismic analysis within a series.13
Lipophilicity is well known to be the antithesis of solubility and lack of solubility has been a consistent problem for medicinal chemists.19,20 As noted earlier improved formulation methodologies can somewhat mitigate this situation. However relying on formulation to get insoluble compounds on board is likely only to aggravate the body to work harder to eliminate them. The usual response of the body to lipophilic xenobiotics is to try to make them more polar viametabolism so they can be excreted. Medicinal chemists are then faced with the need to make their lipophilic and insoluble compounds more metabolically stable to prolong their duration of actions. This can put enormous demands on a compounds profile, especially if once a day dosing is being sought after as the target product profile. Blocked metabolism means the body will need to find more extreme ways of removing the compound, often inducing more high potential and thus intrinsically more reactive and toxic species.
It is clear from the above discussion that increasing MW and lipophilicity in our hunt for potency, results in molecules that could be described as obese in that they have become too large and too lipophilic for their own good. This addiction to what I term molecular obesity represents a high risk to the future “health” of the molecule as a drug candidate.21 As with medical obesity, which is measured by Body Mass Index (BMI), medicinal chemists have developed their own indices such as Ligand Efficiency LE (= binding affinity/number of heavy atoms)19 and Lipophilic Ligand Efficiency LLE5 to help identify and control the effects of molecular obesity, which have clearly caused the premature death of far too many drug candidates in recent years. Additional indices continue to emerge in the literature, all with the aim of restricting the tendency to Molecular Obesity. One such index is the LELP index, which combines aspects of LE and LLE into a single index by dividing logP by LE. It thus represents the price of ligand efficiency paid for with logP.16
Two excellent recent reviews include further in depth discussion of physicochemical related attrition issues and are recommended for further reading.26,27
Driving potency through molecular obesity
So if excess molecular weight and lipophilicity are not conducive to the ultimate success of compounds in the drug discovery process why is it that we so readily find ourselves at the limits or beyond of what is the statistically safer zone. One of the key drivers that leads to this situation is our perceived need for potency, bordering on an addiction, and the fact that potency can often be most easily gained by increasing the number of interactions our ligands make and/or by increasing lipophilicity. We like potency in our molecules for a number of reasons and it is worth examining why this is.One of the basic tenets of medicinal chemistry is that increasing ligand potency leads to increased specificity and hence to an improved therapeutic index.11 One of the corollaries of our study into molecular complexity and the probability of finding hits is that finding appropriate interactions by increasing matched complexity is essential for optimising weak leads to give increased potency.12 However if complexity is introduced unnecessarily it does nothing to build the specificity rather it can introduce the opportunity for recognition at other targets.
High potency is also often perceived to allow reduction in the size of dosage and thus helps reduce the cost of goods. While potency can compensate for low bioavailability in that the small portion of, for example, a poorly absorbed drug that does get into the circulation will at least have a chance of being efficacious if it has high molar potency. However this brings a high risk in that the part of the high dosage that is not being effectively utilised is available to cause off target issues.
So while there are many reasons why potency is a good thing, the problem is how we go about achieving it. Again it is the power of looking at larger and more cross-series data than are usually published in a specific medicinal chemistry case history that is revealing. Ladbury and colleagues have shown that the Free Energy of interaction for synthetic ligands correlates well with a ligand's hydrated apolar surface area (ie lipophilicity) that is buried in the interaction leading to the crystallographically observed complex.14 This phenomenon is even more relevant when the total buried surface area is deconvoluted into the apolar and polar parts, where it is the apolar contributions that dominate. This shows that, at least for this data set which is representative of a wide range of complexes typical of drug discovery programmes, we tend to use the easy gains of potency by adding lipophilicity.
Thermodynamic considerations in lead optimisation are also prominent in the work of Ferenczy and Keseru. They have introduced the term Size Independent Enthalpic Efficiency (SIHE) to help monitor the contribution of the enthalpy to potency as lead optimisation progresses. Thus monitoring thermodynamic profiles during optimisation should enable a more appropriate balance between enthalpic and entropic potency contributions to be achieved rather than just relying on logP as a surrogate.15 These and other authors have also highlighted that inappropriate medicinal chemistry can just as easily lead to poor compounds whether the starting point comes from HTS or a fragments approach.16 The problem with the proliferation of so many “rules” is the trend to slavishly apply them without really understanding their required context for use and subsequent limitations.
In other surveys of specific and cross-series data it is also apparent that increasing molecular weight and complexity tend to correlate with increased potency.17 This is consistent with adding either specific or non-specific interactions but is also likely to be a consequence of the predilection of medicinal chemists, who invariably trained as synthetic organic chemists, to build molecules rather than take them apart. Interestingly the equivalent of retro-synthetic analysis, which is so critical to planning good syntheses, has only recently become more embedded into the medicinal chemistry sphere in terms of fragmenting hits to find the most ligand efficient and smallest critical part.18
A recent study by Waring and colleagues of 9598 AstraZeneca compounds has shown that as molecules are made larger then they need more lipophilicity to be permeable through cell membranes. Thus the apparent twin drivers of potency (mw = more interactions and logP = increased permeability) are seen to be not truly independent variables in relation to bioavailabilty but increasingly linked as compounds get larger.19 In Lipinski's rule of five, the cut off of MW of 500 is consistent with the experimentally observed upper limit of permeability of compounds through membranes without invoking active transport. What Waring's work now clearly shows is that the space below 500 is not binary, in the sense of being permeable with MW less than 500, but that it has an increasing logP demand as 500 is approached.
Another reason why it is all too easy to increase logP in the early stage of drug discovery projects is that if the initial assay is a very specific target assay (eg enzyme or artificially constructed complex) then hits found there are often rapidly progressed down a screening cascade that has a cellular and likely more phenotypic assay facing the nascent leads. In order to gain cellular potency it is all too easy to add lipophilicity to get a compound into the right compartment. This tool compound may have short term benefits of demonstrating cellular efficacy but it is equally too easy to then just forge ahead with this “fattened ligand” in the desire to make further speedy progress towards the project's next milestones. We should pause at this stage to re-evaluate the properties of the now current lead compounds. Of course, in previous eras of drug discovery when in vivo testing was often much earlier in the discovery process this issue did not exist, because compounds that either showed no activity through lack of bioavailability or toxicity through inappropriate mode of action or side effects were detected and dropped very early.
Behavioural addictions in drug discovery
While a typical medicinal chemistry publication will set out to show that a very logical process was followed towards achieving the project's objectives, it is clear that by any objective or subjective measure, the path that a drug discovery project actually follows through a multi-dimensional property space is non-linear. Fig. 1 illustrated this by showing a pathway that could be taken from a start point X1 which has been found by some potency based screening process. From there on, there are numerous pathways (one of which is illustrated) that could be taken to finally reach Xn which is the candidate drug. Whether there is overlap of the zones in any given project as implied in Fig. 1 is only found out by trial and error, although experience with a related target may well inform on such target tractability.We have seen that potency can have an unhealthy connection to molecular obesity, so if we wish to avoid the impact of molecular obesity then we need to ensure that the initial conditions in terms of MW and logP are fully explored to find those that are best suited to living with potency as a strange attractor. This is very much the raison d'etre of Fragment Based Drug Discovery – this and other strengths of this approach are highlighted in Table 2.
1. The ![[c with combining low line]](https://www.rsc.org/images/entities/char_0063_0332.gif) ![[o with combining low line]](https://www.rsc.org/images/entities/char_006f_0332.gif) ![[m with combining low line]](https://www.rsc.org/images/entities/char_006d_0332.gif) ![[b with combining low line]](https://www.rsc.org/images/entities/char_0062_0332.gif) ![[i with combining low line]](https://www.rsc.org/images/entities/char_0069_0332.gif) ![[n with combining low line]](https://www.rsc.org/images/entities/char_006e_0332.gif) ![[a with combining low line]](https://www.rsc.org/images/entities/char_0061_0332.gif) ![[t with combining low line]](https://www.rsc.org/images/entities/char_0074_0332.gif) ![[r with combining low line]](https://www.rsc.org/images/entities/char_0072_0332.gif) ![[l with combining low line]](https://www.rsc.org/images/entities/char_006c_0332.gif) ![[e with combining low line]](https://www.rsc.org/images/entities/char_0065_0332.gif) ![[x with combining low line]](https://www.rsc.org/images/entities/char_0078_0332.gif) ![[p with combining low line]](https://www.rsc.org/images/entities/char_0070_0332.gif) ![[s with combining low line]](https://www.rsc.org/images/entities/char_0073_0332.gif) ![[f with combining low line]](https://www.rsc.org/images/entities/char_0066_0332.gif) ![[h with combining low line]](https://www.rsc.org/images/entities/char_0068_0332.gif) ![[y with combining low line]](https://www.rsc.org/images/entities/char_0079_0332.gif) means that fragments can sample more of the available chemistry space at that level of complexity than is possible with more complex molecules.22 means that fragments can sample more of the available chemistry space at that level of complexity than is possible with more complex molecules.22 |
2. At ![[w with combining low line]](https://www.rsc.org/images/entities/char_0077_0332.gif) ![[g with combining low line]](https://www.rsc.org/images/entities/char_0067_0332.gif) ![[u with combining low line]](https://www.rsc.org/images/entities/char_0075_0332.gif) ![[d with combining low line]](https://www.rsc.org/images/entities/char_0064_0332.gif) even though they may be harder to detect. More complex molecules are statistically more likely to have more “clashes” and thus not fit/bind.12 even though they may be harder to detect. More complex molecules are statistically more likely to have more “clashes” and thus not fit/bind.12 |
3. ![[M with combining low line]](https://www.rsc.org/images/entities/char_004d_0332.gif) ![[k with combining low line]](https://www.rsc.org/images/entities/char_006b_0332.gif) and so starting small with fragments is the natural feedstock for - . and so starting small with fragments is the natural feedstock for - . |
4. ![[B with combining low line]](https://www.rsc.org/images/entities/char_0042_0332.gif) compounds more lead-like starting points are found which enhance the chances of successful lead optimisation campaigns. compounds more lead-like starting points are found which enhance the chances of successful lead optimisation campaigns. |
| 5. in the initial lead, only the necessary interactions are built in to the compound as it is optimised. This should help ensure good developability properties of the resulting candidates. This is also relevant if polypharmacology is needed.23 |
Another important way to diminish the negative effects of potency is to be more realistic about what level of potency we should aspire to. Again surveys of literature data can help reset expectations. Overington et al.'s analysis of a data set of known drugs shows that the median affinity for current small-molecule drugs is ca. 20 nM (pIC50 = 7.7) – see Fig. 5.24 In an excellent analysis of lead/drug pairs, Perola highlights many of the challenges of successful lead optimisation discussed in this current perspective. The successful drug examples discussed show a mean pIC50 of ca. 8 for the final drugs if the micromolar compound Topetecan is excluded.25 When Topotecan (pKi 5.5) is included in Perola's full set the mean value is 6.7. Unpublished in house data suggests that for oral drugs the affinity median also shifts to less potency being required (pIC50 = 6.7). These average levels of potency for successful drugs are considerably lower than the often aspired to pKi or pIC50 values of greater than or equal to 9. Different target classes (eg ion channelsvs. GPCRs) will effect the aspirational potency at the outset of a project but as efficacy is, at the end of the day, what is needed, greater emphasis on the factors that can enable overall drug efficacy need to be more to the fore in the earliest stages rather than just potency at the target. Thus, while the desire for potency is understandable, the tendency to choose the most potent compounds in lead selection and then let it remain the primary driver in early stage lead optimisation remains a strong attractor and must be resisted.25 The fact that potency is often easy to measure (once the assay is established) can often mean that this is the data most likely first returned to the team. The team (or individual) then tends to react to it by making synthesis decisions without waiting for a fuller profile of data on which to make a more informed decision as to what to make next. So reducing the desire for potency in favour of better ADME properties is one way of subjugating the potency strange attractor and its potentially fatal relationship to molecular obesity. Technology to be able to routinely measure the cellular concentration of ligands in cells of interest would also go a long way to helping to do more effective SAR work in cellular assays.
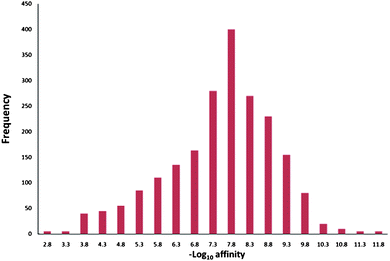 | ||
| Fig. 5 Potencies of marketed small molecule drugs at their target macromolecule (redrawn with data estimated from ref. 24). | ||
More potent molecules should be more selective because the complexity of interactions at the target of interest reduces the probability of their being effective in interactions at off targets.12 From this perspective it therefore seems that increasing MW should be a good thing in that it leads to selectivity. However we have seen that passage through cell membranes required for cellular activity requires ever increasing amounts of lipophilicity as MW increases. It seems possible that it is this concomitant increase in logP that destroys (by increasing the local concentration of the drug in membranes) what selectivity that may have been anticipated to be gained from increased complexity. It should also be remembered that lipophilicity is low grade in its discriminatory powers of molecular recognition, compared to the directionality of, for example, hydrogen bonds.
Of the data sets and final potencies discussed by Perola and others, we have no way of knowing whether a better drug could have been found with less potency. This is because the expensive process of drug discovery only allows us to test very few exemplars in the clinic and if those selected had potency as an early pre-requisite, then the number of less potent but possibly better drugs that we have missed will frustratingly remain an unknowable unknown, although the evidence increasingly points to there being many.
The evolution of fit molecules
Our mission as medicinal chemists is to maximise the use of what is known, and yet to understand the risks of what is not fully known and then develop strategies to integrate them, whilst always recognising that we are ultimately under the apparently chaotic influence of a strange attractor landscape. While medicinal chemists will always aspire to act as intelligent designers of new molecules, it is clearly the processes of molecular evolution that allows the fittest compounds to survive and progress to being worthy of being ultimately labelled as a drug. What allows a compound to survive is fitness and this is clearly only partly understood in any given circumstance, so ensuring we do use what we increasingly understand is necessary but not sufficient for ultimate success.In the same way that this perspective began with Philip Ball's observations on the similarity of aspects of drug discovery to economics, it seems appropriate to close with an observation on parallels with another science. It is interesting to look at the processes of drug discovery from the point of view of evolutionary biology terminology. Table 3 contains a suggested concordance of a number of terms from evolutionary biology with those that we use in drug discovery. The parallels and consequences of the similarities in the table are left for the reader to muse on. However one obvious one is that it is the balance between speciation and extinction that determines the outcomes of biological evolution, and similarly it is the balance of success and failure that will continue to determine the fate of the drug discovery industry. Our ability to improve the odds through understanding how to use more effectively what we do know is a key factor in this.
| Evolutionary Biology Terminology | Medicinal Chemistry and Drug Discovery Terminology |
|---|---|
| Chromosomes | Molecular structures that can be modified |
| Phenotype | Properties of the structure in a biological setting |
| Neutral Changes and Drift | Bio-isosterism or ability to replace parts of structure with other entities while maintaining biological activity that may later reveal new opportunities |
| Fitness | Quality of a lead or lead series to be optimised as a drug and the maintenance of this state. |
| Parallel evolution | Multiple lead series for same target |
| Mutations and Crossover | Structural changes made by a medicinal chemist |
| Genetic Pool | Size and Diversity of Screening Collection |
| Rate of random genetic drift | Rate at which analogues can be made |
| Rate of phenotypic change | Rate of progress towards the properties desirable for a new drug |
| Speciation | Drugs that make it all the way to help patients |
| Extinction | Compounds that fail to make it and add to the cost of drug discovery |
Summary
Molecular obesity and its inappropriate use to drive potency and get ligands through membranes have been killing too many drug discovery projects. Starting with the smallest possible lead (ie fragments) and striving to maintain their fitness through the use of various indices is now accepted as a key approach in a more holistic approach to contemporary drug discovery. The absolute need for potency should not be as dominant an attractor as we often allow it to become at the expense of other characteristics of a good drug.There will always be some compounds that make it all the way to drugs and which lie outside of the known preferred space for likely success. However unless truly forced to by circumstances that are fully understood, it is not appropriate to set out with the mentality that my project will be the “exception that proves the rule” as a risk mitigation strategy.
Note added in proof
A comprehensive review has recently been published on the issues involved in the balance of in vitro potency, ADMET and physicochemical parameter. In particular the authors highlight the poor correlation between in vitro potency and human dose required for efficacy.28Acknowledgements
I would like to thank many present and former colleagues at GSK and colleagues in the wider community involved in drug discovery for their contributions to the ideas expressed in this perspective – while they may not all agree with my views expressed here many have engaged in active debate at meetings. In particular I would like to thank Andrew Leach, Darren Green, Ian Churcher, Andrew Brewster, Paul Gleeson, Andrew Hopkins and Paul Leeson. I am also very grateful for the input of George Keseru at Gideon Richter in Hungary for suggestions on an earlier version of this manuscript and the referees for their helpful suggestions.References
- S. M. Paul, D. S. Mytelka, C. T. Dunwiddie, C. C. Persinger, B. H. Munos, S. R. Lindborg and A. L. Schacht, Nat. Rev. Drug Discovery, 2010, 9, 203 CAS.
- (a) P. Ball. Nature News, Online 5th April 2010, http://www.nature.com/news/2010/100405/full/news.2010.157.html Search PubMed; (b) A. W. Lo, M. T. Mueller, Social Sciences Review Network, March 12, 2010, http://ssrn.com/abstract%3D1563882 Search PubMed.
- S. H. Kellert, In the Wake of Chaos: Unpredictable Order in Dynamical Systems, University of Chicago Press, 1993 Search PubMed.
- I. Kola and J. Landis, Nat. Rev. Drug Discovery, 2004, 3, 711 CrossRef CAS.
- P. D. Leeson and B. Springthorpe, Nat. Rev. Drug Discovery, 2007, 6, 881 CrossRef CAS.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3 CrossRef.
- J. D. Hughes, J. Blagg, D. A. Price, S. Bailey, G. A. DeCrescenzo, R. V. Devraj, E. Ellsworth, Y. M. Fobian, M. E. Gibbs, R. W. Gilles, N. Greene, E. Huang, T. Krieger-Burke, J. Loesel, T. Wager, L. Whiteley and Y. Zhang, Bioorg. Med. Chem. Lett., 2008, 18, 4872 CrossRef CAS.
- M. P. Gleeson, J. Med. Chem., 2008, 51, 817 CrossRef CAS.
- T. J. Ritchie and S. J. Macdonald, Drug Discovery Today, 2009, 14, 1011 CrossRef CAS.
- F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752 CrossRef CAS.
- A. Hopkins, J. Mason and J. Overington, Curr. Opin. Struct. Biol., 2006, 16, 127 CrossRef CAS.
- M. M. Hann, A. R. Leach. and G. Harper, Journal of Chemical Information and Computer Sciences, 2001, 41(3), 856 Search PubMed.
- M. E. Bunnage, J. P. Mathias, A. Wood, D. Miller and S. D. A. Street, Bioorg. Med. Chem. Lett., 2008, 18, 6033 CrossRef CAS.
- T. S. Olsson, M. A. Williams, W. R. Pitt and J. E. Ladbury, J. Mol. Biol., 2008, 384, 1002 CrossRef CAS.
- (a) G. G. Ferenczy and G. M. Keserű, Drug Discovery Today, 2010, 15, 919 CrossRef CAS; (b) G. G. Ferenczy and G. M. Keserű, J. Chem. Inf. Model., 2010, 50, 1536 CrossRef CAS.
- G. M. Keserü and G. M. Makara, Nat. Rev. Drug Discovery, 2009, 8, 203 CrossRef.
- P. Selzer, H.-J. Roth, P. Ertl and A. Schuffenhauer, Curr. Opin. Chem. Biol., 2005, 9, 310 CrossRef CAS.
- J. Bajorath, Nat. Rev. Drug Discovery, 2002, 1, 882 CrossRef CAS.
- M. J. Waring and C. Johnstone, Bioorg. Med. Chem. Lett., 2009, 19, 2844 CrossRef CAS.
- A. P. Hill and R. J. Young, Drug Discovery Today, 2010, 15, 648 CrossRef CAS.
- The term Lead Obesity has previously been introduced by Gyorgy Keseru – see for instance Meeting Report in Drugs of the Future, 2010, 35, 143–153. The term Molecular Obesity introduced here could be considered more generic as a term applicable to more than just leads.
- (a) A. R. Leach, M. M. Hann, J. N Burrows and E. J. Griffen, Mol. BioSyst., 2006, 2, 429 RSC; (b) L. C. Blum and J.-L. Reymond, J. Am. Chem. Soc., 2009, 131, 8732 CrossRef CAS.
- R. Morphy and Z. Rankovic, Drug Discovery Today, 2007, 12, 156 CrossRef CAS.
- J. P. Overington, B. Al-Lazikani and A. L. Hopkins, Nat. Rev. Drug Discovery, 2006, 5, 993–996 CrossRef CAS.
- E. Perola, J. Med. Chem., 2010, 53, 2986 CrossRef CAS.
- M. J. Waring, Expert Opin. Drug Discovery, 2010, 5(3), 235–248 Search PubMed.
- P. D. Leeson and J. R. Empfield, Annu. Rep. Med. Chem., 2010, 45, 393–407 Search PubMed.
- M. P. Gleeson, A. Hersey, D. Montanari and J. Overington, Nat. Rev. Drug Discovery, 2011, 10, 197–208 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |