Expanding the horizon of chemotherapeutic targets: From MDM2 to MDMX (MDM4)
Antonio
Macchiarulo
*a,
Nicola
Giacchè
a,
Andrea
Carotti
a,
Fabiola
Moretti
b and
Roberto
Pellicciari
a
aDipartimento di Chimica e Tecnologia del Farmaco, Università di Perugia, Via del Liceo 1, 06123 Perugia, Italy. E-mail: antonio@chimfarm.unipg.it; Fax: +39 075 585 5161; Tel: +39 075 585 5160
bIstituto di Neurobiologia e Medicina Molecolare CNR, Via del Fosso di Fiorano 64, 00143 Roma, Italy
First published on 28th February 2011
Abstract
Alterations of p53 signalling pathway is the most frequent event in human cancers. About 50% of these, albeit showing wild-type p53, have flaws in the control mechanisms of p53 levels and activity. MDM2 and MDMX (MDM4) are the main negative regulators of p53. The relevance of MDM2 on the regulation of p53 levels and activity has fostered the development of strategies aimed at restoring p53 functions by blocking the physical interaction between MDM2 and p53. As a consequence, a number of different small molecules and peptidomimetics have been disclosed in the last decade as inhibitors of MDM2/p53 interaction. Recent studies, however, have thrust MDMX into the limelight as an additional chemotherapeutic target, suggesting the presence of a more complex relationship between MDM2, MDMX and p53. In this review article, we report key aspects of MDMX-mediated regulation of p53, recent advances in the structural characterization of the protein, and the progress made so far in the medicinal chemistry of MDMX ligands.
1. Introduction
Almost half of human tumours harbour defects in the regulation of oncosuppressive functions of p53.1–3 These are mostly ascribed to altered cellular levels of its negative modulators, namely MDM2 and MDMX (also named MDM4), that constitute the core module of the p53 regulatory network.4MDM2, in particular, is a member of the superfamily of the E3 RING domain-containing ligases that mediate the proteasomal degradation of numerous proteins, including themselves.5 Upon binding to p53, MDM2 promotes the ubiquitylation of the protein, thereby tagging it for proteasomal degradation. In addition, MDM2 is also able to inhibit transcriptional activity of p53. On the basis of such evidence, MDM2 has been pursued as a relevant drug target in the last decade in cancer therapy,6 aiming to restore p53 functions through the blockage of MDM2 activity.7,8 As a result of this intense research activity, a number of peptides and small molecules have been disclosed as being able to block the formation of the MDM2–p53 complex, or to inhibit MDM2 E3 ligase activity.9–12 To our knowledge, however, few MDM2 ligands have made headway in clinical settings, with the MDM2 antagonist nutlin-3 (1) and the MDM2 E3 ligase inhibitor JNJ-26854165 being the only compounds studied in phase I clinical trials (ClinicalTrials.gov identifiers: NCT01143740, NCT00676910).13Unlike MDM2, MDMX has only recently attracted a great deal of interest in the scientific community, with an ever growing number of studies providing evidence of peculiar roles for this protein.14,15
MDMX is the closest analogue protein of MDM2 and a major negative regulator of p53. Overexpression of the MDMX gene has been reported in 40% of tumour cell lines.16 Furthermore, MDMX overexpression has been observed within different types of tumour,17 particularly in human retinoblastomas.18 It is noteworthy that MDMX and MDM2 are unable to compensate the cell for the loss of the other, and they regulate non-overlapping functions of p53.19–26
In this review, we provide the readers with a brief summary of the breakthroughs that have thrust MDMX into the limelight as an additional therapeutic target in cancer disease. Structural and conformational aspects of MDMX will be discussed to shed light on the similarities and dissimilarities with its analogue MDM2. Finally, the advances in the medicinal chemistry of MDMX will be highlighted, focusing on key aspects of lead generation and optimization.
2. MDMX as therapeutic target in cancer disease
The role of MDMX as a p53 inhibitor was shown at its discovery by Jochemsen's group.27 Later on, numerous studies investigated the molecular functions of MDMX towards p53, especially to understand the distinctive properties of MDMX in comparison to MDM2. This has led to various models, the most widespread of which recognizes a double function of MDMX: a direct inhibition of p53 transcriptional activity and a supporting/strengthening activity towards MDM2-mediated degradation of p53. Overall, these data have supported the hypothesis that MDMX is an oncogenic protein and therefore a cancer therapeutic candidate. This hypothesis is indeed corroborated by numerous studies providing evidence of MDMX overexpression in human tumours (Table 1),28–39 in some cases significantly correlated with worse clinicopathological features of tumours.34,40–42| Tumour type | Incidence | Detection method | Ref. |
|---|---|---|---|
| a qRT-PCR: Quantitative real time-PCR. b CGH: Comparative genomic hybridization. c IHC: Immunoistochemistry. d FISH: Fluorescence in situ hybridization. e ISH: In situ hybridization. f WB: Western Blot. | |||
| Astrocytic glioma | 27% | qRT-PCR a | 32 |
| Bladder Ca | 17% | CGH b, qRT-PCR, IHCc | 41 |
| BREAST Ca | 41/218 (19%) | FISH d, ISHe | 37 |
| BREAST Ca | 182/990 (18%) | IHC | 36 |
| BREAST Ca | 7/359 (2%) | CGH, qRT-PCR | 39 |
| Chronic lymphocytic leukaemia | 43CLL vs. 15CTR (p < 0.0001) | qRT-PCR | 30 |
| Colon Ca | 5/27 (18%) | IHC | 37 |
| Glioma | 5/208 (2%) | PCR | 33 |
| Head neck squamous Ca | 28/56 (50%) | IHC | 40 |
| Hepatocarcinoma | 11/24 (46%) | WB f | 28 |
| Hepatoblastoma | 1/56 (2%) | FISH | 29 |
| Lung Ca | 16/88 (18%) | IHC | 37 |
| Neuroendrocrine tumor | 98/171 (57%) | IHC, qRT-PCR | 42 |
| Oligodendroglioma | 4/40 (10%) | qRT-PCR | 31 |
| Retinoblastoma | 32/49 (65%) | FISH, IHC | 18 |
| Retinoblastoma | 2/21 (9%) | qRT-PCR, WB | 35 |
| Radiation induced sarcoma | 3/36 (8%) | RT-PCR | 38 |
| Soft tissue sarcoma | 11/66 (17%) | qRT-PCR | 34 |
Moreover, the MDMX genomic locus resides in chromosome 1q32.1, a site frequently amplified in human tumors. Intriguingly, mouse models of MDMX overexpression have provided controversial data about the oncogenic potential of MDMX. Indeed, a study has reported spontaneous tumourigenesis in MDMX overexpressing mice,43 whereas another has reported the inability of MDMX to support spontaneous or myc-induced tumourigenesis,44 suggesting that the oncogenic potential of MDMX is modified by additional factors. Accordingly, higher levels of MDMX have been reported to be significantly associated to lower stage thyroid tumours,45 or to better prognosis in ovarian46 and prostate cancer,47 or even as a positive prognostic factor in breast cancer.36 Similarly, gene expression profiling showed downregulation of MDMX in metastatic brain tumours.48
Recent molecular studies have given support to these data by showing positive activities of MDMX under stress conditions towards p53 stability and apoptotic function,46,49–51 pointing to a complex regulation between MDMX and p53.
It will be interesting to ascertain the relevance of this regulation in the efficacy of MDMX ligands, especially in combination with classical p53-activating therapies (chemo- and radiotherapy).
3. Structural and conformational aspects of MDMX
Like MDM2, the sequence of MDMX comprises three domains (Fig. 1):27,52 (i) the N-terminal domain, (ii) the central domain containing a zinc finger motif, and (iii) the C-terminal RING domain. In the following sections, we discuss what is currently known about each of these domains, also in comparison to MDM2, focusing on the relationships between structure, conformation and functional properties. | ||
| Fig. 1 Functional domains of MDMX (a) and MDM2 (b) sequences. Three dimensional structures are shown in cartoons where such information is available: N-terminal domain (MDMX, pdb code: 3DAB; MDM2, pdb code: 1YCR); zinc finger (MDM2, pdb code: 2C6B); C-terminal RING domain (heterodimer MDMX/MDM2, pdb code: 2VJF). | ||
3.1 The N-terminal domain
The N-terminal domain of MDMX shows the highest sequence similarity with the related domain of MDM2 (53.6% identity). In this domain the p53 binding cleft is localized, with a number of hydrophobic residues forming three contiguous narrow pockets that host Phe19, Trp23 and Leu26 from the N-terminal transactivation domain of p53 (Fig. 2).53,54 | ||
| Fig. 2 Complex of MDMX with the N-terminal transactivation domain of p53 (pdb code: 3DAB). MDMX is shown with water accessible surface. Key interacting residues of p53 are shown and labelled. | ||
Long after the early crystallization study of MDM2,53 whose results provided the first insights into the structural basis of the interaction between the N-terminal domain of MDM2 and p53, additional efforts resulted in the crystallization of a number of MDMX structures.55–61
Remarkably, these studies unveiled how subtle replacements of residues and diverse conformational profiles of the side chains within the p53 binding site, while conserving the overall hydrophobic nature of the p53 binding cleft, may induce different fit adaptations of MDM2 and MDMX in response to ligand binding.
At this regard, Holak and coworkers first showed that the shape of the p53 binding site in the humanized zebrafish MDMX structure is different from that of MDM2 for the replacement of Leu54 (MDM2 numbering) with Met53 (MDMX numbering) as well as for the presence of a peculiar conformation of Tyr99 (Fig. 3).55,56 Because of the longer side chain of the methionine residue, indeed, the Leu54Met substitution makes the binding site of MDMX smaller than that of MDM2. Mutagenesis experiments provided further evidence of how such a mutation accounts for the lower potency of inhibition shown by nutlin-3 (1) towards MDMX with respect to MDM2.55
 | ||
| Fig. 3 Superposition of MDMX/p53 (pdb code: 2Z5T) and MDM2/p53 (pdb code: 1YCR) complexes. Key p53 interacting residues are shown and labelled in black. The conformations of Tyr100 and Leu54 of MDM2 are compared respectively with those of Met53 and Tyr99 of MDMX. The additional steric hindrance provided by Met53 to the p53 binding cleft is red circled. | ||
The shape of the p53 binding site in the humanized zebrafish MDMX is further restricted by the presence of a peculiar “closed conformation” adopted by Tyr99 that is at odds with the “open conformation” of the corresponding Tyr100 observed in the human MDM2 structure bound to p53.62 Additional crystallization studies of human synthetic MDMX and MDM2 structures bound to a dual nanomolar peptide inhibitor, termed PMI (5), unveiled how the peculiar conformation of Tyr99 accounts also for the different induced fit adaptation of MDMX to PMI binding.60 Indeed, it was discovered that the closed conformation of Tyr99, while promoting the formation of a hydrogen bond between its side chain and Ser11 of the inhibitor peptide, is able to induce the formation of an additional hydrophobic cleft that makes room to host the C-terminal Pro residue of PMI (5). In accordance with these observations, deletions of Ser11 and Pro12 of PMI, albeit ineffective in influencing PMI binding to MDM2, were able to reduce the binding of the inhibitor peptide to MDMX by almost ten-fold.
An early interpretation of these studies suggested that the closed conformation of Tyr99 was a conserved property of the MDMX structure, contributing to accounting for the differences, found to then, in ligand recognition between MDMX and MDM2.
Afterwards, the solution of two additional crystal structures of MDMX bound to p53 peptide analogues challenged this notion, providing further insights into the conformational aspects of the p53 binding site in MDMX.59 It was found, indeed, that the 6-chloro-tryptophan peptidomimetic of p53 (10), while inhibiting both MDM2 and MDMX in the nanomolar range of potency (see below), was able to induce a widening of the binding site in MDMX through the stabilization of an “open conformation” of Tyr99 (Fig. 4). Such an amazing result was further confirmed in the crystallization study of the human MDMX structure bound to a single domain antibody (4), wherein Tyr99 was still observed in an open conformation.61 Interestingly, molecular dynamics studies carried out by the same group revealed that the binding sites of all human MDMX structures were able to converge into a common conformation after the removal of ligands, thereby suggesting that the conformational differences seen in those crystals were originating from different ligand induced fit effects on the oncogenic protein. It is worth mentioning that more recent crystallization studies have shown that even MDM2 can adopt a “closed conformation” of Tyr100 in response to ligand binding.63
 | ||
| Fig. 4 Superposition of MDMX and MDM2 in complex with the 6-Cl-tryptophan peptidomimetic of p53 (pdb codes: 3FEA and 2GV2). Key interacting residues of peptidomimetic are shown and black labelled. The conformations of Tyr99 of MDM2 and Tyr100 of MDMX are shown. | ||
In line with these results, statistical analyses of long range trajectories resulting from 60 ns molecular dynamics simulations showed that the conformation of Tyr99 in MDMX, as well as Tyr100 in MDM2, is the hallmark of the unbound and ligand-bound states of the protein.64
As a consequence of these findings, MDMX is a structure endowed with a high degree of flexibility, as also evidenced by additional molecular dynamics studies.65,66
In this context, recent NMR studies on the free N-terminal domain of MDMX have provided evidence for an even more flexible structure with respect to MDM2.67 This has been ascribed to the “lid motif” of MDMX that, conversely to MDM2, does not fold over the p53 binding cleft in the unbound state. A poor conservation of residues along the lid motif sequences of MDMX and MDM2 would sustain this hypothesis, pinpointing different roles of this region in MDMX and MDM2 during ligand recognition.
3.2 The central domain
Beside the largely studied N-terminal domain, MDMX and MDM2 share the less conserved central domain that includes a zinc finger motif (41.9% sequence identity).The central domain of MDMX (residues 128–444) has been shown to mediate the repression activity of MDMX on E2F1 transactivation.68,69 This protein is a transcriptional factor belonging to the E2F family, and whose members have been involved in the transcriptional regulation of genes associated with cell cycle, proliferation, senescence and apoptosis.70 While the central domain of MDMX does not directly interact with E2F1, it seems to favour a cytoplasmic localization of that transcription factor when MDMX is overexpressed. Remarkably, the inhibition of E2F1 transactivation by MDMX occurs in a p53 and MDM2 independent manner, suggesting the presence of specific regulatory mechanisms for this oncogenic protein.
Although the solution structure of the MDM2 zinc finger motif was solved by multidimensional NMR,71 little information is available about its molecular function and much less is reported on this region for MDMX. While early works by Brown and coworkers suggested a role for the zinc finger motif in MDM2-induced G1 cell cycle arrest and cell growth inhibitory function,72 more recently it was reported that this motif is involved in mediating the interaction of MDM2 with ribosomal proteins such as L5 and L11, as well as promoting p53 degradation.73
In the central domain of MDMX, it has been also recognized a caspase cleavage signal (aa358–361, DVPD), identical in MDM2. This site is functional and the resulting cleavage has been recognized as a signal of MDMX inactivation.106
3.3 The C-terminal RING domain
Conversely to that in MDM2, the C-terminal RING domain of MDMX (51.3% sequence identity) does not have E3 ubiquitin ligase activity and, therefore, does not stimulate ubiquitination of p53.74–81 However, it is involved in the heterodimerization with MDM2, enhancing the MDM2-dependent ubiquitination and degradation of p53.78,81,82 Structural studies of the heterodimeric complex of the MDMX and MDM2 RING domains show that the interacting residues form an extended surface that is remotely localized from the E2 binding site.82 Accordingly, it has been suggested that additional conformational and structural aspects beyond the heterodimerization and E2 recruitment may be involved to promote an optimal E3 ligase activity of the heterodimer.17In this regard, recent mutagenesis experiments have identified two distinct regions of MDMX that, when replaced by the respective regions of MDM2, turn the protein into a functionally active ubiquitin ligase for p53 in cells.83 While the first region (residues 448–453) is localized in the dimer interface, the second comprises residues 465–480, with a single mutation (Asn448Cys) resulting in a significantly increased ability of MDMX to ubiquitinate p53 in vitro.
Likewise to that in MDM2, the RING domain of MDMX contains also a functional P-loop motif (residues 437–454) that binds nucleotide analogues in the low micromolar range, with the affinity towards ATP being greater than towards GTP.84
Although such interaction seems to be important for translocation of MDM2 to the nucleolus,85 its functional impact on MDMX, that lacks the nucleolus localization signal (NoLS), remains poorly understood. In this regard, it has been proposed that the binding of ATP to MDMX could allosterically regulate conformational changes of the whole protein, leading to alterations in the interaction with p53 and/or other binding partners.84
4. Medicinal chemistry of MDMX
At odds with MDM2, very few medicinal chemistry efforts have been devoted to the design and synthesis of specific MDMX antagonists. As a consequence, current data on MDMX ligands rely solely on comparative studies addressing the selectivity of peptides and small molecules that have been mostly developed as MDM2 inhibitors.In this part of the review, we provide a structured report of ligands for which activity data on MDMX are available (Tables 2 and 3). This is done by classifying ligands on the basis of their structural diversity in peptides (i) and small molecules (ii). While offering a useful way to discuss and comment on the classes of MDMX modulators, it furnishes an accessible way to retrieve information on the part of the readers.
| No. | Name | Sequence or structure | MDMX activity/μM | MDM2 activity/μM | Ref. |
|---|---|---|---|---|---|
| a Sequence of the CDR3 domain of VH9 interacting with MDMX. b X = 3,4-dichlorophenylalanine, O = pyrrolysine. c Stars indicate the points along the peptide where the hydrocarbon crosslink (staple) is created. | |||||
| p53-peptide | QETFSDLWKLLP | 1.128 (IC50) | 3 (IC50) | 54 | |
| 2 | 12/1 | MPRFMDYWEGLN | 1.65 (IC50) | 0.15 (IC50) | 54 |
| 3 | pDI | LTFEHYWAQLTS | 0.1 (IC50) | 0.01 (IC50) | 87,88 |
| 4 | VH9 | PWYPFMASKGSEDFDYa | 0.044 (KD) | 6 (KD) | 61 |
| 5 | PMI | TSFAEYWNLLSP | 0.004 (KD) | 0.003 (KD) | 60 |
| 6 | stingin 3 | CNCKAPETFLCYWRCL | 0.016 (KD) | 0.057 (KD) | 90 |
| 7 | DPMIβ | TAWYANFEKLLR | 2.4 (KD) | 0.034 (KD) | 91 |
| 8 | PMIAsn8Ala | TSFAEYWALLSP | 0.0024 (KD) | 0.00049 (KD) | 92 |
| 9 | β53-16 | OILEIXOIFE b | 0.155 (KD) | 0.027 (KD) | 94 |
| 10 | 8-merX |

|
0.036 (KD) | 0.007 (KD) | 59 |
| 11 | L-NAPA 25 |

|
2.7 (IC50) | 2.6 (IC50) | 95 |
| 12 | SAH-p53-8 | Ac-QSQQTF*NLWRLL*QN-NH2c | 0.002 (KD) | 0.055 (KD) | 98 |

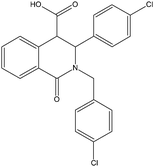




It should be mentioned that very recently a small molecule inhibitor of MDMX expression has been reported.86 Since it regulates MDMX activity through an indirect mechanism of action, the compound is not discussed in the following sections to further a comparative analysis of ligands targeting the N-terminal domain of MDMX and MDM2.
4.1 Peptides and peptidomimetics
The first comparative study reporting binding profiles of p53-derived peptides on MDMX and MDM2, dates back to 1999 with the work of Lane and coworkers.54 They investigated the interaction of MDMX and MDM2 with phage-derived peptides and full length p53, both in solid phase and solution phase.As a first result, the authors found the homogeneous and heterogeneous binding efficiencies of peptides when tested in solid and solution phase, respectively. Secondly, a preferential binding of larger phage-derived peptides, such as 15mer peptides, to MDM2 over MDMX was observed. Finally, the study disclosed a novel inhibitor, namely peptide 12/1 (2, Table 2), as able to block p53 binding to both MDMX and MDM2.
Almost one decade later, a research group led by Jiandong Chen at the H. Lee Moffitt Cancer Center reported the development of a second peptide inhibitor, namely pDI (3), as able to interact with MDMX and MDM2 (MDMX-IC50 = 100 nmol L−1; MDM2-IC50 = 10 nmol L−1).87 Using a delivery system based on a scaffold protein and recombinant adenovirus, the authors provided for the first time the proof of concept that the simultaneous disruption of p53 binding to MDM2 and MDMX by pDI (3) was able to efficiently suppress the growth of tumour xenografts in mice.88
Along with the first crystallization study of MDMX, Holak and coworkers reported a thorough profiling of peptides and small molecules as antagonists of MDMX.89 In particular, using ITC and NMR titration data, they proved a major affinity of p53 full length and p53 derived peptides towards MDMX with respect to MDM2. Further work by the same group resulted in the disclosure of additional crystal structures of MDM2 and MDMX in complex with pDI peptide (3).57 Despite the different shapes of binding cleft in MDM2 and MDMX, pDI (3) showed a similar alpha helix conformation in both complexes. Interestingly, this conformation was more extended than that of wild type p53, providing an explanation for the improved affinity of pDI (3) towards MDM2 and MDMX (MDM2-KD = 0.0036 μM, MDMX-KD = 0.0061 μM)
In 2009, the research team of Alan Fersht at the Centre for Protein Engineering (Medical Research Council, Cambridge, UK) reported the crystal structure of human MDMX bound to the single domain antibody VH9 (4).61 This peptide was endowed with an affinity constant in the nanomolar range of potency towards MDMX, and a micromolar affinity constant at MDM2 (MDM2-KD = 6 μM, MDMX-KD = 0.044 μM), as determined by ITC assay. Interestingly, the high affinity of VH9 (4) to MDMX was ascribed to smaller conformational rearrangements observed in the crystal structure of MDMX when bound to the single domain antibody with respect to p53.
In the same year, two papers published in the Proceedings of the National Academy of Sciences and Angewandte Chemie, International Edition, reported the discovery of novel potent peptide inhibitors of MDM2 and MDMX by researchers at the University of Maryland School of Medicine (Baltimore, USA). In the first work, the researchers disclosed PMI (5) as a potent inhibitor of MDM2 and MDMX, with KD values around 0.003 μM and 0.004 μM, respectively.60 Making a comparison, the authors tested the activity of pDI (4) in the same assay, observing KD values for this inhibitor of 0.019 μM towards MDM2 and 0.077 μM towards MDMX. Crystallization studies of PMI (5) in complex with MDM2 and MDMX provided explanations for the low nanomolar affinities of the peptide inhibitor. Again, the potency of PMI (5) was associated with the stabilization of a more extended alpha helix conformation with respect to p53. Further structural findings unveiled the presence of different conformational changes in MDM2 and MDMX structures. Indeed, as mentioned earlier, Tyr100 in MDM2 and Tyr99 in MDMX adopted diverse induced fit orientations. Consequently, the C-terminal residue Pro12 of PMI (5) was fully disordered in MDM2, while interacting with a newly formed hydrophobic pocket in MDMX. In the second work, the researchers elegantly reported the conversion of aparin, a bee venom neurotoxin, into a set of potent inhibitors of MDM2 and MDMX (stingin 1–5).90 This was accomplished by grafting four residues from PMI (5) into the scaffold of aparin peptide. As a result, they rationally designed five aparin-derived peptides that were endowed with binding affinities in the nanomolar range of potency. Remarkably, stingin 3 (6) was found to have the highest selectivity towards MDMX over MDM2 (MDM2-KD = 0.057 μM, MDMX-KD = 0.016 μM). Further work by the same group around the structure of PMI peptide (5) led to the design of the left-handed peptide DPMIβ (7), with the aim of improving the bioavailability of PMI (5).91 Albeit showing a binding affinity constant in the nanomolar range of activity towards MDM2, DPMIβ (6) proved to be a weaker ligand of MDMX (MDM2-KD = 0.034 μM, MDMX-KD = 2.4 μM). On the other hand, additional lead optimization efforts on PMI (5) resulted in a set of 35 p53- and PMI-derived mutants.92 The latter are of particular interest since the appraisal of their binding affinities at MDMX and MDM2 allowed a complete dissection of the energetic contributions of individual residues of PMI (5) and p53 to the interaction with the oncogenic proteins.
As a first result, researchers found that Phe3 and Trp7 of PMI (5) are the most critical interacting residues, with the latter being more important than the former insofar as contributing 0.85 kcal mol−1 and 0.37 kcal mol−1 more in binding free energies than Phe3 to MDM2 and MDMX, respectively.
Secondly, in agreement with previous findings,60 they observed how the substitution of Pro12 with alanine weakened PMI binding to MDMX by 0.3 kcal mol−1, while being ineffective for the binding to MDM2. Finally, they identified the Asn8Ala replacement as pivotal to convert PMI (5) into the most potent dual inhibitor of MDM2 and MDMX so far reported (8; MDM2-KD = 0.00049 μM, MDMX-KD = 0.0024 μM). MDM2 co-crystallization experiments of PMI bearing an Asn8Ala mutation (8) allowed to ascribe the potency of mutated peptide to the enhanced stability of its alpha helix conformation.
A research group in the Department of Chemistry at the Yale University reported the design of beta-peptide helices bearing non-natural side chains as MDM2 and MDMX ligands.93,94 Very briefly, beta-peptide helices feature a characteristic secondary structure, termed a 314-helix, that is endowed with a periodicity of 3.1-residues per turn.
As part of the results of their studies and assisted by molecular modelling calculations, they identified a beta-peptide, namely β53-16 (9), as a potent and dual inhibitor of MDM2 and MDMX (MDM2-KD = 0.027 μM and MDMX-KD = 0.155 μM). From a structural point of view, β53-16 (9) bears a meta-para-dichlorophenylalanine non-natural side chain at the central position which occupies the same hydrophobic pocket of MDM2 and MDMX as occupied by Trp23 of p53.
On the way to novel semi-synthetic p53 analogues, researchers at Novartis reported the design of a 8-mer peptide bearing a chlorine atom substituent at position 6 of Trp23 (10).59 Biological appraisals using isothermal titration calorimetry (ITC) and differential scanning fluorimetry (DSF) pinpointed that this peptide was a potent dual inhibitor of MDMX and MDM2, with KD values of 0.036 μM and 0.007 μM, respectively. As mentioned in the above paragraph, crystallization studies unveiled the presence of an open conformation of Tyr99 in MDMX upon binding of the 6-chloro-tryptophan peptidomimetic of p53. Furthermore, a cross-talk between the Trp23-pocket and Leu26-pocket (p53 numbering) of MDMX that was found to widen the latter site, allowing a deeper interaction with binding groups.
Additional dual inhibitors of MDM2 and MDMX comprise a series of N-acylpolyamine derivatives (NAPAs) that were designed as non-natural peptidomimetics of p53.95 Among the reported peptidomimetics, L-NAPA 25 (11) proved to be the best dual inhibitor, being endowed with IC50 values of 2.6 μM and 2.7 μM towards MDM2 and MDMX, respectively. Molecular modelling studies suggested the ability of L-NAPA 25 (11) to adapt its conformation with the respect to the binding pockets of MDM2 and MDMX, thereby providing an explanation for the equivalent inhibition of the two oncogenic proteins.
A more recent work described the use of a genetic selection system and encoded library of conformationally pre-organized peptides to perform functional profiling and unravel selective recognition features for MDMX and MDM2.96 As a result of structure–activity relationship studies on the most active peptides, the authors identified charged motifs as favoured for binding to MDMX, while neutral and aromatic-rich epitopes were favoured for binding to MDM2.
Finally, Walensky and coworkers reported the design of a p53 peptidomimetic by applying a chemical strategy known as hydrocarbon stapling.97,98 The strategy consisted of the formation of an all-hydrocarbon crosslink within a synthetic peptide in order to stabilize the alpha helix conformation, confer protease resistance, and promote cellular uptake. Accordingly, they disclosed SAH-p53-8 (12) that inhibited MDMX with a 25-fold selectivity over MDM2 in in vitro assays. Intravenous administration of SAH-p53-8 (12) in a murine xenograft model bearing cancer cells (JEG-3) characterized by nutlin-3 (1) resistance and MDMX overexpression, provided the proof of principle that MDMX inhibition efficiently suppresses tumour growthin vivo. Remarkably, this study also proposed a model establishing factors affecting cancer cell susceptibility to selective MDMX or MDM2 inhibitors, or to dual MDM2/MDMX inhibitors.98,99 Thus, it was advanced that selective MDMX inhibition would be optimal to treat cancer cells overexpressing MDMX along with endogenous elevated levels of p53. Conversely, treatment with selective MDMX inhibitors would be ineffective in cancer cells expressing low levels of p53. In such cases, depending on the low or high levels of MDMX selective MDM2 inhibitors or synergistic targeting of MDM2 and MDMX, respectively, would be optimal to rescue p53 tumour suppression function.
4.2 Small molecules
It is only recently that the growing interest in MDMX as an additional chemotherapeutic target has prompted the quest for small molecules with dual or selective inhibition profiles towards MDM2 and/or MDMX. As a result, while many of the early small molecule antagonists of MDM2 lack functional profiling at MDMX, some of them have only recently been tested in MDMX assay.Pioneering in this endeavour, Holak and coworkers firstly showed the slight selectivity of nutlin-3 (1, Table 3) towards MDM2 with respect to MDMX (MDM2-KD = 0.7 μM, MDMX-KD = 25 μM).89 The poor MDMX activity of nutlin-3 (1) was ascribed to the steric hindrance provided by Met53, since its replacement with the relative MDM2 residue, namely valine, was able to increase the affinity of the ligand towards the mutant MDMX protein. They also reported the inactivity at MDMX of another class of patented small molecules, namely NXN-6 derivatives (13; MDM2-KD = 2 μM, MDMX-KD = 500 μM).100
Further work by the same research group successfully resulted in the first crystal structure of the N-terminal domain of MDMX bound to a small molecule developed by Novartis researchers.63,101 Albeit the co-crystallized ligand (WK298, 14) showed a two-fold lower affinity at MDMX than MDM2 (MDMX-IC50 = 19.7 μM; MDM2-IC50 = 0.19 μM), the inspection of the complex revealed the presence of ligand induced conformations in the oncogenic protein. Consistent with the observations of Kallen et al., the chlorine substituent of the indole ring of WK298 (14) was found to force conformational rearrangements of Leu56, Met53, Leu98 and Leu102, thereby enlarging the central binding pocket (Trp23 binding pocket) of MDMX. However, in contrast to the open conformation observed by Kallen et al., the side chain of Tyr99 was found in an intermediate conformation between the open and closed orientations, suggesting the presence of different induced fits that are dependent on the specific interacting ligand.
Keeping with dual MDMX and MDM2 inhibition, a recent work reported that the main components of Bing De Ling, a highly valued traditional Chinese medicine remedy for cancer disease, inhibit the interaction of p53 with MDMX and MDM2, thereby accounting in part for the anti-tumour activity of this herbal formula.102
Finally, it is worth mentioning additional MDM2 small molecule inhibitors that showed a lack of activity towards MDMX inhibition. Accordingly, these compounds can be qualified as full selective MDM2 inhibitors and include some benzodiazepinedione derivatives103 and spiro-oxindoles, as exemplified by TDP665759 (15) and MI-219 (16), respectively.104 Remarkably, MI-219 (16) proved to be 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold selective over MDMX, showing a Ki of 5 nM at MDM2.
000-fold selective over MDMX, showing a Ki of 5 nM at MDM2.
On the other side, it is even more interesting to report the only small molecule so far disclosed as a selective MDMX inhibitor.105 In more detail, using a MDMX-p53 binding assay to screen an in house library of more than 290000 compounds, researchers at St. Jude Children's Research Hospital and Leiden University Medical Center discovered SJ-172550 (17) as able to selectively block the interaction of p53 with MDMX with an EC50 value of 0.84 μM. Cellular isobologram experiments with SJ-172550 (17) and nutlin-3 (1) showed additive effects of the two compounds in killing MDMX-amplified human retinoblastoma cells. Consistent with the regulatory function of MDMX in controlling p53 trancriptional activity but not p53 degradation, the exposure of MDMX-amplified retinoblastoma cells to SJ-172550 did not show significant accumulations of p53 levels, but apoptosis and p53-dependent cell death were observed. Mutagenesis experiments inspired by molecular docking of SJ-172550 (17) into the crystal structure of MDMX after removal of p53, provided an experimental validation to the proposed mechanism of inhibition of SJ-172550 (17) at the oncogenic protein. Thus, it was found that Met53Leu and His54Phe replacements were able to significantly decrease the activity of the compound, whereas Tyr66Ile and Gln58Asp had only marginal effects on SJ-172550 binding.
5. Conclusions
In recent years, several works have reinforced the idea that the simultaneous disruption of p53 interaction at MDM2 and MDMX is pivotal to develop more effective anti-tumour strategies.This notion, in particular, stems from evidence that MDM2 and MDMX have distinct and complementary modes of action in the regulation of the activity of p53. As a consequence, dual and selective MDMX inhibitors are being pursued with the aims of developing novel incisive p53-based anticancer therapies. Moreover, these inhibitors may contribute to unravelling and/or confirming specific functions of MDMX in its positive activity towards p53 activation. Interestingly, a recent work has proposed a framework for determining the optimal pharmacological treatment to rescue p53 tumour suppressor functions in cancer cells with MDMX and/or MDM2 inhibitors.
This model, in particular, has advanced that selective MDMX inhibitors would be beneficial in cancer cells expressing high levels of p53, whereas MDM2 inhibitors would prove effective in cancer cells expressing low levels of MDMX. Similarly, cancer cells with low levels of p53 as well as high levels of MDMX would be susceptible to dual MDM2 and MDMX inhibitors.98,99
In this context, a number of peptides and peptidomimetics have been recently disclosed as dual MDMX and MDM2 inhibitors, some of them being reported as active in the nanomolar range of potency. These data combine with recent structural characterizations of apo- and ligand-bound complexes of MDMX that, together with MDM2 structures, provide first insights into the structure–activity relationships that favour potency and/or selectivity towards the oncogenic proteins. Accordingly, it is found that the extension and stability of the alpha helix conformation of peptides is crucial to achieve potency, while the presence of either charged motifs or neutral and aromatic-rich epitopes may favour the binding to MDMX or MDM2, respectively.
As far as it concerns small molecules, however, the availability of dual and/or selective MDMX inhibitors is still poor compared to MDM2, with only one small molecule being reported as a selective modulator of MDMX. Filling this gap is instrumental to further validate MDMX as a therapeutic target and pave the way to the development of novel anticancer drug candidates. To this end, while hitherto disclosed peptides and peptidomimetics may supply medicinal chemists with interesting starting compounds for lead optimization strategies, the growing structural information on MDMX may provide additional clues to identify novel small molecules by structure-based design strategies as well as virtual screening protocols.
Acknowledgements
A.M., N.G., A.C. and R.P. acknowledge the financial support of the European Union. FP6 PRIORITY LSH-2005-2.2.0–8: Small-ligand libraries: improved tools for exploration and prospective anti-tumour therapy. DePPICT Project (Designing Therapeutic Protein-Protein Inhibitors for Brain Cancer Treatments) Contract number: LSHC-CT-2007- 037834 (http://www.deppict.eu/home.jsp). F.M. gratefully acknowledges the financial support of AIRC.References
- B. Vogelstein, D. Lane and A. J. Levine, Nature, 2000, 408, 307–310 CrossRef CAS.
- F. Toledo and G. M. Wahl, Nat. Rev. Cancer, 2006, 6, 909–923 CrossRef CAS.
- K. H. Vousden and D. P. Lane, Nat. Rev. Mol. Cell Biol., 2007, 8, 275–283 CrossRef CAS.
- G. M. Wahl, Cell Death Differ., 2006, 13, 973–983 CrossRef CAS.
- R. J. Deshaies and C. A. Joazeiro, Annu. Rev. Biochem., 2009, 78, 399–434 CrossRef CAS.
- J. D. Oliner, K. W. Kinzler, P. S. Meltzer, D. L. George and B. Vogelstein, Nature, 1992, 358, 80–83 CrossRef CAS.
- S. Patel and M. R. Player, Expert Opin. Invest. Drugs, 2008, 17, 1865–1882 CrossRef CAS.
- S. Shangary and S. Wang, Annu. Rev. Pharmacol., 2009, 49, 223–241 Search PubMed.
- F. Lu, S. W. Chi, D. H. Kim, K. H. Han, I. D. Kuntz and R. K. Guy, J. Comb. Chem., 2006, 8, 315–325 CrossRef CAS.
- Y. Lu, Z. Nikolovska-Coleska, X. Fang, W. Gao, S. Shangary, S. Qiu, D. Qin and S. Wang, J. Med. Chem., 2006, 49, 3759–3762 CrossRef CAS.
- A. L. Bowman, Z. Nikolovska-Coleska, H. Zhong, S. Wang and H. A. Carlson, J. Am. Chem. Soc., 2007, 129, 12809–12814 CrossRef CAS.
- M. P. Dickens, R. Fitzgerald and P. M. Fischer, Semin. Cancer Biol., 2010, 20, 10–18 CrossRef CAS.
- K. Kojima, J. K. Burks, J. Arts and M. Andreeff, Mol. Cancer Ther., 2010, 9, 2545–2557 CrossRef CAS.
- F. Mancini, G. Di Conza, O. Monti, A. Macchiarulo, R. Pellicciari, A. Pontecorvi and F. Moretti, Int. J. Biochem. Cell Biol., 2010, 42, 1080–1083 CrossRef CAS.
- J. C. Marine, M. A. Dyer and A. G. Jochemsen, J. Cell Sci., 2007, 120, 371–378 CrossRef CAS.
- Y. F. Ramos, R. Stad, J. Attema, L. T. Peltenburg, A. J. van der Eb and A. G. Jochemsen, Cancer Res., 2001, 61, 1839–1842 CAS.
- M. Wade and G. M. Wahl, Mol. Cancer Res., 2009, 7, 1–11 CrossRef CAS.
- N. A. Laurie, S. L. Donovan, C. S. Shih, J. Zhang, N. Mills, C. Fuller, A. Teunisse, S. Lam, Y. Ramos, A. Mohan, D. Johnson, M. Wilson, C. Rodriguez-Galindo, M. Quarto, S. Francoz, S. M. Mendrysa, R. K. Guy, J. C. Marine, A. G. Jochemsen and M. A. Dyer, Nature, 2006, 444, 61–66 CrossRef CAS.
- S. Francoz, P. Froment, S. Bogaerts, S. De Clercq, M. Maetens, G. Doumont, E. Bellefroid and J. C. Marine, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 3232–3237 CrossRef CAS.
- S. Xiong, C. S. Van Pelt, A. C. Elizondo-Fraire, G. Liu and G. Lozano, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 3226–3231 CrossRef CAS.
- J. C. Marine and A. G. Jochemsen, Biochem. Biophys. Res. Commun., 2005, 331, 750–760 CrossRef CAS.
- J. Parant, A. Chavez-Reyes, N. A. Little, W. Yan, V. Reinke, A. G. Jochemsen and G. Lozano, Nat. Genet., 2001, 29, 92–95 CrossRef CAS.
- D. Migliorini, E. Lazzerini Denchi, D. Danovi, A. Jochemsen, M. Capillo, A. Gobbi, K. Helin, P. G. Pelicci and J. C. Marine, Mol. Cell. Biol., 2002, 22, 5527–5538 CrossRef CAS.
- R. A. Finch, D. B. Donoviel, D. Potter, M. Shi, A. Fan, D. D. Freed, C. Y. Wang, B. P. Zambrowicz, R. Ramirez-Solis, A. T. Sands and N. Zhang, Cancer Res., 2002, 62, 3221–3225 CAS.
- S. N. Jones, A. E. Roe, L. A. Donehower and A. Bradley, Nature, 1995, 378, 206–208 CrossRef CAS.
- R. Montes de Oca Luna, D. S. Wagner and G. Lozano, Nature, 1995, 378, 203–206 CrossRef CAS.
- A. Shvarts, W. T. Steegenga, N. Riteco, T. van Laar, P. Dekker, M. Bazuine, R. C. van Ham, W. van der Houven van Oordt, G. Hateboer, A. J. van der Eb and A. G. Jochemsen, EMBO J., 1996, 15, 5349–5357 CAS.
- C. Schlaeger, T. Longerich, C. Schiller, P. Bewerunge, A. Mehrabi, G. Toedt, J. Kleeff, V. Ehemann, R. Eils, P. Lichter, P. Schirmacher and B. Radlwimmer, Hepatology, 2008, 47, 511–520 CAS.
- Y. Arai, S. Honda, M. Haruta, F. Kasai, Y. Fujiwara, J. Ohshima, F. Sasaki, A. Nakagawara, H. Horie, H. Yamaoka, E. Hiyama and Y. Kaneko, Genes Chromosomes Cancer, 2010, 49, 596–609 CAS.
- M. D. Bo, P. Secchiero, M. Degan, D. Marconi, R. Bomben, G. Pozzato, G. Gaidano, G. Del Poeta, F. Forconi, G. Zauli and V. Gattei, Br. J. Haematol., 2010, 150, 237–239.
- M. E. Alonso, M. J. Bello, D. Arjona, V. Martinez-Glez, J. M. de Campos, A. Isla, E. Kusak, J. Vaquero, M. Gutierrez, J. L. Sarasa and J. A. Rey, Am. J. Clin. Pathol., 2005, 123, 900–906 CrossRef CAS.
- D. Arjona, M. J. Bello, M. E. Alonso, A. Isla, J. M. De Campos, J. Vaquero, J. L. Sarasa, M. Gutierrez and J. A. Rey, Diagn. Mol. Pathol., 2005, 14, 224–229 CrossRef CAS.
- M. J. Riemenschneider, R. Buschges, M. Wolter, J. Reifenberger, J. Bostrom, J. A. Kraus, U. Schlegel and G. Reifenberger, Cancer Res, 1999, 59, 6091–6096 CAS.
- F. Bartel, J. Schulz, A. Bohnke, K. Blumke, M. Kappler, M. Bache, H. Schmidt, P. Wurl, H. Taubert and S. Hauptmann, Int. J. Cancer, 2005, 117, 469–475 CrossRef CAS.
- Y. Guo, S. Pajovic and B. L. Gallie, Biochem. Biophys. Res. Commun., 2008, 375, 1–5 CrossRef CAS.
- T. M. Abdel-Fatah, D. G. Powe, J. Agboola, M. Adamowicz-Brice, R. W. Blamey, M. A. Lopez-Garcia, A. R. Green, J. S. Reis-Filho and I. O. Ellis, J. Pathol., 2010, 220, 419–434 CAS.
- D. Danovi, E. Meulmeester, D. Pasini, D. Migliorini, M. Capra, R. Frenk, P. de Graaf, S. Francoz, P. Gasparini, A. Gobbi, K. Helin, P. G. Pelicci, A. G. Jochemsen and J. C. Marine, Mol. Cell. Biol., 2004, 24, 5835–5843 CrossRef CAS.
- N. Gonin-Laurent, N. S. Hadj-Hamou, N. Vogt, C. Houdayer, M. Gauthiers-Villars, C. Dehainault, X. Sastre-Garau, S. Chevillard and B. Malfoy, Oncogene, 2007, 26, 6106–6112 CrossRef CAS.
- G. Jonsson, J. Staaf, J. Vallon-Christersson, M. Ringner, K. Holm, C. Hegardt, H. Gunnarsson, R. Fagerholm, C. Strand, B. A. Agnarsson, O. Kilpivaara, L. Luts, P. Heikkila, K. Aittomaki, C. Blomqvist, N. Loman, P. Malmstrom, H. Olsson, O. T. Johannsson, A. Arason, H. Nevanlinna, R. B. Barkardottir and A. Borg, Breast Cancer Res., 2010, 12, R42 CrossRef.
- Y. A. Valentin-Vega, J. A. Barboza, G. P. Chau, A. K. El-Naggar and G. Lozano, Hum. Pathol., 2007, 38, 1553–1562 CrossRef CAS.
- A. Veerakumarasivam, H. E. Scott, S. F. Chin, A. Warren, M. J. Wallard, D. Grimmer, K. Ichimura, C. Caldas, V. P. Collins, D. E. Neal and J. D. Kelly, Clin. Cancer Res., 2008, 14, 2527–2534 CrossRef CAS.
- W. Hu, Z. Feng, I. Modica, D. S. Klimstra, L. Song, P. J. Allen, M. F. Brennan, A. J. Levine and L. H. Tang, Genes Cancer, 2010, 1, 360–368 Search PubMed.
- S. Xiong, V. Pant, Y. A. Suh, C. S. Van Pelt, Y. Wang, Y. A. Valentin-Vega, S. M. Post and G. Lozano, Cancer Res., 2010, 70, 7148–7154 CrossRef CAS.
- S. De Clercq, A. Gembarska, G. Denecker, M. Maetens, M. Naessens, K. Haigh, J. J. Haigh and J. C. Marine, Mol. Cell. Biol., 2010, 30, 5394–5405 CrossRef CAS.
- A. Prodosmo, S. Giglio, S. Moretti, F. Mancini, F. Barbi, N. Avenia, G. Di Conza, H. J. Schunemann, L. Pistola, V. Ludovini, A. Sacchi, A. Pontecorvi, E. Puxeddu and F. Moretti, J. Mol. Med., 2008, 86, 585–596 CrossRef CAS.
- F. Mancini, G. Di Conza, M. Pellegrino, C. Rinaldo, A. Prodosmo, S. Giglio, I. D'Agnano, F. Florenzano, L. Felicioni, F. Buttitta, A. Marchetti, A. Sacchi, A. Pontecorvi, S. Soddu and F. Moretti, EMBO J., 2009, 28, 1926–1939 CrossRef CAS.
- S. Nanni, C. Priolo, A. Grasselli, M. D'Eletto, R. Merola, F. Moretti, M. Gallucci, P. De Carli, S. Sentinelli, A. M. Cianciulli, M. Mottolese, P. Carlini, D. Arcelli, M. Helmer-Citterich, C. Gaetano, M. Loda, A. Pontecorvi, S. Bacchetti, A. Sacchi and A. Farsetti, Mol. Cancer Res., 2006, 4, 79–92 CrossRef CAS.
- V. M. Zohrabian, H. Nandu, N. Gulati, G. Khitrov, C. Zhao, A. Mohan, J. Demattia, A. Braun, K. Das, R. Murali and M. Jhanwar-Uniyal, Oncol. Rep., 2007, 18, 321–328 CAS.
- J. A. Barboza, T. Iwakuma, T. Terzian, A. K. El-Naggar and G. Lozano, Mol. Cancer Res., 2008, 6, 947–954 CrossRef CAS.
- F. Mancini and F. Moretti, Cell Cycle, 2009, 8, 3854–3859 CrossRef CAS.
- P. Tsvetkov, N. Reuven, C. Prives and Y. Shaul, J. Biol. Chem., 2009, 284, 26234–26242 CrossRef CAS.
- R. Honda, H. Tanaka and H. Yasuda, FEBS Lett., 1997, 420, 25–27 CrossRef CAS.
- P. H. Kussie, S. Gorina, V. Marechal, B. Elenbaas, J. Moreau, A. J. Levine and N. P. Pavletich, Science, 1996, 274, 948–953 CrossRef CAS.
- V. Bottger, A. Bottger, C. Garcia-Echeverria, Y. F. Ramos, A. J. van der Eb, A. G. Jochemsen and D. P. Lane, Oncogene, 1999, 18, 189–199 CrossRef CAS.
- G. M. Popowicz, A. Czarna, U. Rothweiler, A. Sszwagierczak, M. Krajewski, L. Weber and T. A. Holak, Cell Cycle, 2007, 6, 2386–2392 CrossRef CAS.
- G. M. Popowicz, A. Czarna and T. A. Holak, Cell Cycle, 2008, 7, 2441–2443 CAS.
- A. Czarna, G. M. Popowicz, A. Pecak, S. Wolf, G. Dubin and T. A. Holak, Cell Cycle, 2009, 8, 1176–1184 CrossRef CAS.
- E. Jacoby, A. Boettcher, L. M. Mayr, N. Brown, J. L. Jenkins, J. Kallen, C. Engeloch, U. Schopfer, P. Furet, K. Masuya and J. Lisztwan, Methods Mol. Biol., 2009, 575, 173–194 CrossRef CAS.
- J. Kallen, A. Goepfert, A. Blechschmidt, A. Izaac, M. Geiser, G. Tavares, P. Ramage, P. Furet, K. Masuya and J. Lisztwan, J. Biol. Chem., 2009, 284, 8812–8821 CAS.
- M. Pazgier, M. Liu, G. Zou, W. Yuan, C. Li, J. Li, J. Monbo, D. Zella, S. G. Tarasov and W. Lu, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4665–4670 CrossRef CAS.
- G. W. Yu, M. Vaysburd, M. D. Allen, G. Settanni and A. R. Fersht, J. Mol. Biol., 2009, 385, 1578–1589 CrossRef CAS.
- B. L. Grasberger, T. Lu, C. Schubert, D. J. Parks, T. E. Carver, H. K. Koblish, M. D. Cummings, L. V. LaFrance, K. L. Milkiewicz, R. R. Calvo, D. Maguire, J. Lattanze, C. F. Franks, S. Zhao, K. Ramachandren, G. R. Bylebyl, M. Zhang, C. L. Manthey, E. C. Petrella, M. W. Pantoliano, I. C. Deckman, J. C. Spurlino, A. C. Maroney, B. E. Tomczuk, C. J. Molloy and R. F. Bone, J. Med. Chem., 2005, 48, 909–912 CrossRef CAS.
- G. M. Popowicz, A. Czarna, S. Wolf, K. Wang, W. Wang, A. Domling and T. A. Holak, Cell Cycle, 2010, 9 DOI:10.4161/cc.9.6.10956.
- A. Carotti, A. Macchiarulo, N. Giacche and R. Pellicciari, Proteins: Struct., Funct., Bioinf., 2009, 77, 524–535 Search PubMed.
- A. Macchiarulo, N. Giacche, A. Carotti, M. Baroni, G. Cruciani and R. Pellicciari, J. Chem. Inf. Model., 2008, 48, 1999–2009 CrossRef CAS.
- T. L. Joseph, A. Madhumalar, C. J. Brown, D. P. Lane and C. Verma, Cell Cycle, 2010, 9 DOI:10.4161/cc.9.6.11067.
- M. C. Sanchez, J. G. Renshaw, G. Davies, P. N. Barlow and M. Vogtherr, FEBS Lett., 2010, 584, 3035–3041 CrossRef CAS.
- M. Wunderlich, M. Ghosh, K. Weghorst and S. J. Berberich, Cell Cycle, 2004, 3, 472–478 CAS.
- G. D. Strachan, K. L. Jordan-Sciutto, R. Rallapalli, R. S. Tuan and D. J. Hall, J. Cell. Biochem., 2003, 88, 557–568 CrossRef CAS.
- H. Z. Chen, S. Y. Tsai and G. Leone, Nat. Rev. Cancer, 2009, 9, 785–797 CrossRef CAS.
- G. W. Yu, M. D. Allen, A. Andreeva, A. R. Fersht and M. Bycroft, Protein Sci., 2006, 15, 384–389 CrossRef CAS.
- D. R. Brown, C. A. Thomas and S. P. Deb, EMBO J., 1998, 17, 2513–2525 CrossRef CAS.
- M. S. Lindstrom, A. Jin, C. Deisenroth, G. White Wolf and Y. Zhang, Mol. Cell. Biol., 2007, 27, 1056–1068 CrossRef CAS.
- M. W. Jackson and S. J. Berberich, Mol. Cell. Biol., 2000, 20, 1001–1007 CrossRef CAS.
- R. Stad, N. A. Little, D. P. Xirodimas, R. Frenk, A. J. van der Eb, D. P. Lane, M. K. Saville and A. G. Jochemsen, EMBO Rep., 2001, 2, 1029–1034 CrossRef CAS.
- H. Kawai, D. Wiederschain, H. Kitao, J. Stuart, K. K. Tsai and Z. M. Yuan, J. Biol. Chem., 2003, 278, 45946–45953 CrossRef CAS.
- M. Ghosh, K. Huang and S. J. Berberich, Biochemistry, 2003, 42, 2291–2299 CrossRef CAS.
- L. K. Linares, A. Hengstermann, A. Ciechanover, S. Muller and M. Scheffner, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 12009–12014 CrossRef CAS.
- M. V. Poyurovsky, C. Priest, A. Kentsis, K. L. Borden, Z. Q. Pan, N. Pavletich and C. Prives, EMBO J., 2007, 26, 90–101 CrossRef CAS.
- S. Uldrijan, W. J. Pannekoek and K. H. Vousden, EMBO J., 2007, 26, 102–112 CrossRef CAS.
- J. Gu, H. Kawai, L. Nie, H. Kitao, D. Wiederschain, A. G. Jochemsen, J. Parant, G. Lozano and Z. M. Yuan, J. Biol. Chem., 2002, 277, 19251–19254 CrossRef CAS.
- K. Linke, P. D. Mace, C. A. Smith, D. L. Vaux, J. Silke and C. L. Day, Cell Death Differ., 2008, 15, 841–848 CrossRef CAS.
- S. Iyappan, H. P. Wollscheid, A. Rojas-Fernandez, A. Marquardt, H. C. Tang, R. K. Singh and M. Scheffner, J. Biol. Chem., 2010, 285, 33065–33072 CrossRef CAS.
- C. Priest, C. Prives and M. V. Poyurovsky, Nucleic Acids Res, 2010 Search PubMed.
- M. V. Poyurovsky, X. Jacq, C. Ma, O. Karni-Schmidt, P. J. Parker, M. Chalfie, J. L. Manley and C. Prives, Mol. Cell, 2003, 12, 875–887 CrossRef CAS.
- H. Wang, X. Ma, S. Ren, J. K. Buolamwini and C. Yan, Mol. Cancer Ther., 2011, 10, 69–79 CrossRef CAS.
- B. Hu, D. M. Gilkes and J. Chen, Cancer Res., 2007, 67, 8810–8817 CrossRef CAS.
- A. Macchiarulo and R. Pellicciari, Expert Opin. Ther. Pat., 2009, 19, 721–726 Search PubMed.
- G. M. Popowicz, A. Czarna, U. Rothweiler, A. Szwagierczak, M. Krajewski, L. Weber and T. A. Holak, Cell Cycle, 2007, 6, 2386–2392 CrossRef CAS.
- C. Li, M. Pazgier, M. Liu, W. Y. Lu and W. Lu, Angew. Chem., Int. Ed., 2009, 48, 8712–8715 CrossRef CAS.
- M. Liu, M. Pazgier, C. Li, W. Yuan and W. Lu, Angew. Chem., Int. Ed. Engl., 2010, 49, 3649–3652 CAS.
- C. Li, M. Pazgier, W. Yuan, M. Liu, G. Wei, W. Y. Lu and W. Lu, J. Mol. Biol., 2010, 398, 200–213 CrossRef CAS.
- E. A. Harker, D. S. Daniels, D. A. Guarracino and A. Schepartz, Bioorg. Med. Chem., 2009, 17, 2038–2046 CrossRef CAS.
- J. Michel, E. A. Harker, J. Tirado-Rives, W. L. Jorgensen and A. Schepartz, J. Am. Chem. Soc., 2009, 131, 6356–6357 CrossRef CAS.
- R. Hayashi, D. Wang, T. Hara, J. A. Iera, S. R. Durell and D. H. Appella, Bioorg. Med. Chem., 2009, 17, 7884–7893 CrossRef CAS.
- S. Datta, M. E. Bucks, D. Koley, P. X. Lim and S. N. Savinov, Bioorg. Med. Chem., 2010, 18, 6099–6108 CrossRef CAS.
- F. Bernal, A. F. Tyler, S. J. Korsmeyer, L. D. Walensky and G. L. Verdine, J. Am. Chem. Soc., 2007, 129, 2456–2457 CrossRef CAS.
- F. Bernal, M. Wade, M. Godes, T. N. Davis, D. G. Whitehead, A. L. Kung, G. M. Wahl and L. D. Walensky, Cancer Cell, 2010, 18, 411–422 CrossRef CAS.
- J. C. Marine, Cancer Cell, 2010, 18, 399–400 CrossRef CAS.
- W. Lutz, Tetrahydro-isoquinolin-1-ones for the Treatment of Cancer. WO2006097323 Search PubMed.
- A. Boettcher, N. Buschmann, P. Furet, J. M. Groell, J. Kallen, J. Hergovich Lisztwan, K. Masuya, L. Mayr and A. Vaupel, 3-Imidazoyl-indoles for the treatment of proliferative diseases. WO2008119741 Search PubMed.
- Y. Zhang, H. Dong, Z. Li, S. Xiang, Y. Zhu, M. Zhang, J. Liu, W. Bai, S. V. Nicosia, J. Chen, R. J. Zhao and X. Zhang, Front. Biosci., 2010, E2, 221–230 CrossRef CAS.
- H. K. Koblish, S. Zhao, C. F. Franks, R. R. Donatelli, R. M. Tominovich, L. V. LaFrance, K. A. Leonard, J. M. Gushue, D. J. Parks, R. R. Calvo, K. L. Milkiewicz, J. J. Marugan, P. Raboisson, M. D. Cummings, B. L. Grasberger, D. L. Johnson, T. Lu, C. J. Molloy and A. C. Maroney, Mol. Cancer Ther., 2006, 5, 160–169 CrossRef CAS.
- S. Shangary, D. Qin, D. McEachern, M. Liu, R. S. Miller, S. Qiu, Z. Nikolovska-Coleska, K. Ding, G. Wang, J. Chen, D. Bernard, J. Zhang, Y. Lu, Q. Gu, R. B. Shah, K. J. Pienta, X. Ling, S. Kang, M. Guo, Y. Sun, D. Yang and S. Wang, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3933–3938 CrossRef CAS.
- D. Reed, Y. Shen, A. A. Shelat, L. A. Arnold, A. M. Ferreira, F. Zhu, N. Mills, D. C. Smithson, C. A. Regni, D. Bashford, S. A. Cicero, B. A. Schulman, A. G. Jochemsen, R. K. Guy and M. A. Dyer, J. Biol. Chem., 2010, 285, 10786–10796 CrossRef CAS.
- F. Gentiletti, F. Mancini, M. D'Angelo, A. Sacchi, A. Pontecorvi, A. G. Jochemsen and F. Moretti, Oncogene, 2002, 21, 867–877 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |