Comparison of two- and three-dimensional activity landscape representations for different compound data sets
Preeti
Iyer
,
Mathias
Wawer
and
Jürgen
Bajorath
*
Department of Life Science Informatics, B-IT, LIMES Program Unit Chemical Biology and Medicinal Chemistry, Rheinische Friedrich-Wilhelms-Universität, Dahlmannstr. 2, D-53113, Bonn, Germany. Tel: +49-228-2699-306; Fax: +49-228-2699-341; E-mail: bajorath@bit.uni-bonn.de
First published on 30th November 2010
Abstract
Modeling of activity landscapes provides a basis for the analysis of structure–activity relationships (SARs) in large compound data sets. Activity landscape models enable visual access to SAR features. Regardless of their specific details, these models generally have in common that they integrate molecular similarity and potency relationships between active compounds. Different two-dimensional (2D) landscape representations have been introduced and recently also the first detailed three-dimensional (3D) model. Herein we compare advanced 2D and 3D activity landscape models for compound data sets having different SAR character. Although the compared 2D and 3D representations are conceptually distinct, it is found that global SAR features of compound data sets can be equally well deduced from them. However, local SAR information is often captured in different ways by these representations. Since these 2D and 3D landscape modeling tools have been made freely available, the analysis also provides guidelines for how to best utilize these alternative landscape representations for practical SAR analysis.
Introduction
The study of compound structure–activity relationships plays a central role in medicinal chemistry, and a variety of computational approaches are employed to analyze and predict SARs in a qualitative or quantitative manner.1–9 Among these approaches is the modeling of activity landscapes for compound data sets.10,11 Generally, an activity landscape can be understood as any representation that integrates similarity and potency relationships between compounds sharing the same biological activity.11 Different types of 2D activity landscape representations have been introduced.6,11 In addition, 3D landscape models have also been discussed6,11 that can be rationalized as a 2D projection of a chemical reference space (where proximity of compounds is an indicator of similarity) with compound potency added as the third dimension. Such 3D landscape views then essentially describe biological activity response surfaces as a consequence of changes in chemical structure (i.e. “walks” in chemical space12).The most prominent feature of activity landscapes, however they might be represented, are activity cliffs10,11,13 that are formed by structurally similar compounds or analogs with large differences in potency. Activity cliff regions are associated with high SAR information content11,13,14 because in these regions small changes in compound structure lead to significant potency effects.
Although several 2D activity landscape representations of varying complexity are currently available, only hypothetical 3D activity landscapes have been discussed until recently when the first detailed 3D landscape model has been introduced for the analysis of compound data sets.15 Alternative activity landscape representations have thus far rarely been compared for given compound data sets to better understand how they capture SAR information in detail and how they might differ.
Therefore, we have generated conceptually distinct 2D and 3D activity landscape views of different compound data sets, and using different molecular representations, in order to compare how these models convey SAR information. The results presented herein show that distinct landscape models often capture SAR information contained in compound data sets in a comparable manner, but also reveal unique features of 2D and 3D landscape representations and their SAR information content. Furthermore, our analysis also provides some practical guidelines for the complementary use of different activity landscape models in medicinal chemistry.
Methods
3D activity landscapes
The generation of 3D activity landscapes followed a previously reported protocol.15 Euclidean distance relationships between compounds (constituting a coordinate-free chemical reference space) were calculated using the MACCS16 or, alternatively, Molprint2D17 fingerprint and projected onto a 2D plane using multidimensional scaling (MDS)18 as a dimension reduction technique. Compound coordinates were normalized to range from 0 to the maximum observed pairwise chemical dissimilarity in a given compound data set, such that the range of the planar coordinates (and hence the size of the landscape plots) reflected the overall chemical dissimilarity within the data set. Compound potency information was then added as a third dimension and potency values were interpolated using the Krige function19 to create coherent surfaces. To simplify the comparison of different landscapes, all representations were displayed from the same view point (azimuth: 60°, co-latitude: 45°). The potency axis was consistently scaled for all data sets, ranging from the lowest (3.46) to the highest (11.72) log-potency value observed for all compound activity classes. Furthermore, x- and y-axes ranged from the lowest (0.00) to the highest values of pairwise chemical distances of all activity classes (MACCS: 9.27, Molprint2D: 9.79). The surface of the activity landscapes was colored according to interpolated potency values (surface elevation) using a color gradient from green (lowest potency) to red (highest potency). The value range mapped onto the color gradient was determined by the highest minimal and the lowest maximal interpolated potency values of all activity classes, ranging from a log-potency of 5.79 (green) to 9.19 (red). In addition, landscape transparency15 was adjusted to reflect the density of experimental potency measurements. Grid points with close proximity to original data points (compounds) were colored opaque and those furthest away from data point were fully transparent. Hence, regions of interpolated surface area not populated with data points appear in white.Network-like similarity graphs
A Network-like similarity graph (NSG)20 is a 2D activity landscape model that represents similarity and potency relationships in a compound data set as an annotated graph. In NSGs, nodes represent individual molecules that are connected by edges if their structural similarity exceeds a predefined threshold. Here, dissimilarity relationships corresponding to similarity threshold criteria were also expressed by Euclidean distances to enable a direct comparison with 3D landscape models. Euclidean distance thresholds of 4.90 and 5.29 were used for MACCS and Molprint2D, respectively (corresponding to pairwise MACCS Tanimoto similarity21 of 0.65), which also yielded a comparable edge density in all NSGs. Nodes were colored by potency corresponding to the color scheme of 3D landscapes based on a continuous gradient from green (log-potency ≤ 5.57) via yellow to red (log-potency ≥ 10.05). The lower and upper boundary of the gradient corresponded to the highest minimal and the lowest maximal potency values of all activity classes. Nodes were scaled in size based on a numerical SAR analysis function, the local SAR Index (SARI),4,20 which determines the contribution of an individual compound to global SAR discontinuity. Generally, SAR discontinuity is introduced when small changes in compound structure are accompanied by large changes in potency. Thus, in NSGs, large nodes indicate that the potency of a compound substantially differs from that of its structural neighbors. For NSG display, a graphical layout algorithm is applied that places multiple densely connected compounds in close vicinity and separates weakly connected regions from each other.22Compound activity classes
For activity landscape comparison, five data sets consisting of 112–209 compounds active against different targets were taken from the MDDR,23 as summarized in Table 1. These data sets were selected based on global SARI4 calculations to represent different degrees of SAR heterogeneity.| Abbreviation | Activity class | No. of compounds | Potency range/nM |
|---|---|---|---|
| a For the five activity classes used in the study, the number of compounds and their potency range are reported. | |||
| ACH | Acetylcholinesterase | 112 | 0.02–85000 |
| COX | Cyclooxygenase 2 | 149 | 0.09–50000 |
| 5HT | 5-HT reuptake | 129 | 0.01–2700 |
| PH4 | Phosphodiesterase IV | 209 | 0.0025–348000 |
| THR | Thrombin | 172 | 0.0019–30000 |
Results and discussion
Two-dimensional activity landscape representations
In Fig. 1, alternative 2D landscape views are shown that illustrate their conceptual diversity. All these representations have in common that they integrate structure and potency relationships that are present in compound data sets, albeit in different ways. Structure–activity similarity (SAS) maps10 have probably been the first explicit 2D activity landscape representation. SAS maps compare structure and activity similarity of active compounds in a pairwise manner. Activity similarity is expressed by potency differences. These maps delineate regions of high SAR discontinuity (i.e. high structural and low activity similarity) and high SAR continuity (i.e. low structural and high activity similarity) as well as nondescript regions that contain only very little SAR information (e.g. where both structure and activity similarity are low). Also shown is a color coded 2D projection of chemical reference space (providing the basis for 3D landscape modeling; see Methods) that captures compound distance relationships and mirrors potency distributions. Thus, the information provided is similar to SAS maps, but the representations are distinctly different. In addition, a so-called structure–activity landscape index (SALI) graph5 is shown that follows a different design idea and represents a potency-directed compound network. SALI graphs largely focus on identifying activity cliffs of different magnitude and delineating cliff pathways for different SALI score threshold values. Furthermore, in network-like similarity graphs,20 compounds are also represented as nodes and undirected edges indicate pairwise similarity relationships. In NSGs, color coding provides potency information and scaling of nodes the contribution of each individual compound to local SAR discontinuity (i.e. the larger the node, the higher the degree of discontinuity a compound introduces). Thus, prominent activity cliffs are indicated by connected pairs of large red and green nodes. Different from SALI graphs and other representations, NSGs are designed to explore both global and local SAR features in compound data sets and identify compound subsets that form different SARs patterns not limited to activity cliffs. As such, NSGs probably represent the most detailed 2D activity landscape views that are available at present.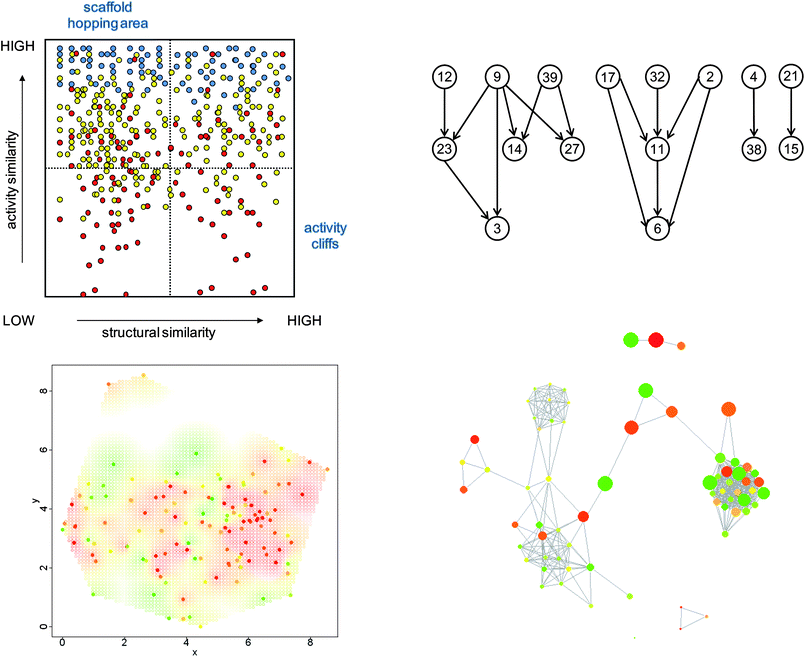 | ||
| Fig. 1 Alternative 2D activity landscape representations. Four conceptually different graphical representations for SAR analysis are shown. SAS maps (upper left) represent all pairs of compounds of a data set as points in a scatter plot organized by the level of structural and activity similarity for each compound pair. The color of points reflects whether the potency level of the more potent compound of the pair is high (blue), intermediate (yellow), or low (red). In all other representations, points or nodes represent individual molecules. In SALI graphs (upper right), the nodes are connected by edges if they form an activity cliff exceeding a predefined SALI score. The plot in the lower left section was generated by mapping compound dissimilarity values to 2D coordinates using multi-dimensional scaling. The gradient from green to red reflects potency from low to high values. The same color code is applied for the nodes in an NSG (lower right). Here, edges between nodes are drawn if the similarity of the corresponding compounds exceeds a predefined threshold. In addition, nodes are scaled in size according to local SAR discontinuity contributions of the corresponding compounds. | ||
Three-dimensional activity landscapes
In Fig. 2, an idealized 3D activity landscape and a 3D landscape calculated for a specific compound data set are shown. Idealized activity landscapes have frequently been utilized to illustrate important SAR characteristics including activity cliffs as well as rugged regions and smooth regions that represent SAR discontinuity and continuity, respectively. Hence, in such 3D landscape views, differences in landscape topology can be readily associated with different SAR behaviour. Despite the intuitive nature of 3D activity landscapes, 3D landscape models for actual compound data sets have only recently been introduced.15 The exemplary data set-based activity landscape shown in Fig. 2 mirrors characteristic topological features depicted in idealized landscapes.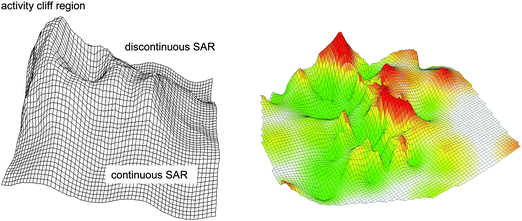 | ||
| Fig. 2 Hypothetical and compound data-based 3D activity landscapes. The landscape on the left represents an idealized heterogeneous activity landscape that contains different local SAR regions. High peaks correspond to activity cliffs, rugged regions represent discontinuous local SARs (small changes in structure are accompanied by significant potency effects) and smooth regions continuous local SARs (gradual changes in structure are accompanied by small changes in potency). On the right, a 3D landscape model calculated on the basis of an actual compound data set (acetylcholinesterase inhibitors in Table 1) is shown. The surface of the activity landscapes is colored according to interpolated potency values (surface elevation) using a color spectrum from green to red. | ||
Comparison of NSGs and 3D landscape models
Despite their different design, the 2D and 3D landscape models presented herein have common features. Both NSGs and 3D landscapes rely on the assessment of global molecular similarity (other similarity measures would of course also be possible). Furthermore, both 2D and 3D landscape models capture global SAR features and local SAR environments. We have compared these representations in detail for five different compound activity classes summarized in Table 1. In each case, activity landscapes were generated with two different molecular representations; MACCS keys, a structural fragment fingerprint, and Molprint2D, a combinatorial fingerprint that captures layered topological atom environments. For each compound data set, the two pairs of corresponding NSGs and 3D models are shown in Fig. 3.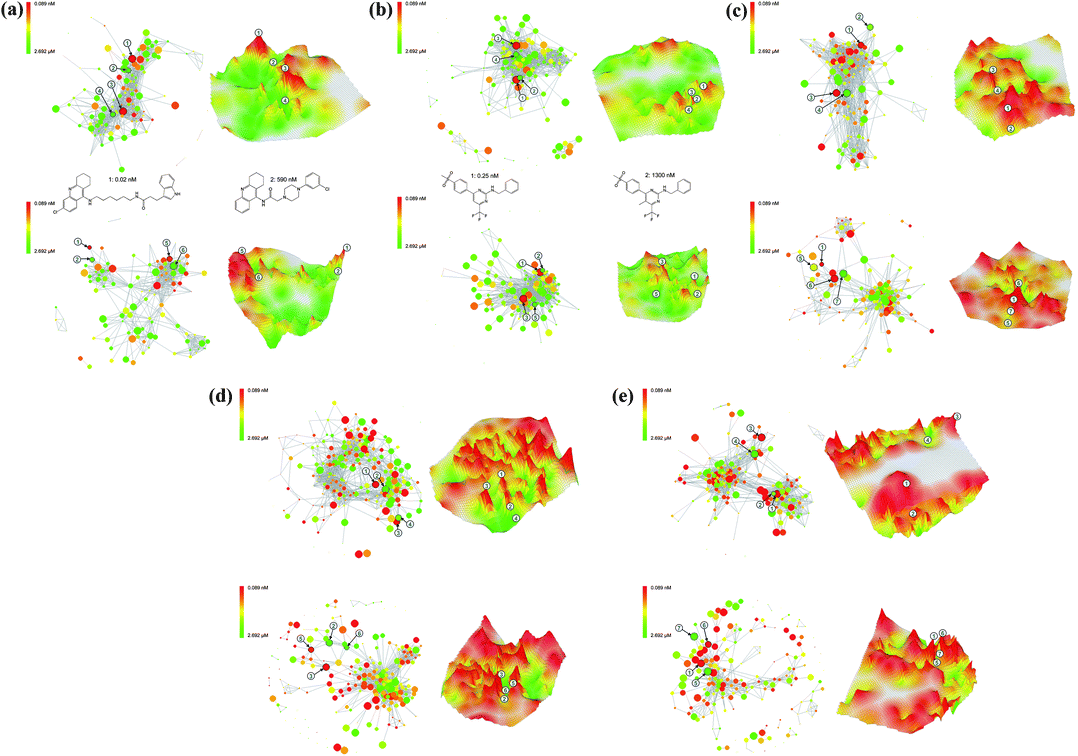 | ||
| Fig. 3 Comparison of NSGs and 3D activity landscape models. NSG and 3D landscape representations calculated on the basis of either MACCS (top) or Molprint2D fingerprint distances (bottom) are presented for five different compound data sets. Corresponding positions of selected compounds in 2D and 3D representations are indicated by numbers. Exemplary compound structures are also displayed. Landscape representations are shown for sets of (a) acetylcholinesterase inhibitors (ACH), (b) cyclooxygenase 2 inhibitors (COX), (c) serotonin reuptake inhibitors (5HT), (d) phosphodiesterase 4 inhibitors (PH4), and (e) thrombin inhibitors (THR). | ||
The general observation can be made that both 2D and 3D activity landscape features were in part significantly influenced by the chosen fingerprint representation, in accord with earlier findings.15 For all compound classes, activity cliffs produced by the alternative molecular representations often differed, due to the fact that the two fingerprints often accounted for compound similarity relationships in different ways. In this context, it should be noted that the topology of NSGs is only determined by pairwise similarity relationships and the graphical layout algorithm that separates densely connected compound clusters for visualization. Hence, in NSGs, fingerprint-specific differences in the distribution of similarity values were rather obvious.
Another general observation has been that NSGs and 3D landscape models provided essentially equivalent global views of global SAR characteristics of different data sets, despite representation-dependent differences. For example, in Fig. 3a, the NSGs and 3D landscapes of class ACH (the MACCS-based 3D model of this data set is also shown in Fig. 2) both display coexisting continuous and discontinuous regions interspersed with activity cliffs. The COX landscapes in Fig. 3b are characterized by the presence of many structurally similar compounds with relatively low potency and a higher degree of global continuity. By contrast, the 5HT landscapes in Fig. 3c are much more discontinuous in nature (here, fingerprint-dependent topology differences are striking) and contain much larger activity cliffs. Moreover, all PH4 landscapes in Fig. 3d are characterized by the presence of many activity cliffs, which represent their most significant feature. Similarly, the THR landscapes in Fig. 3e are also strongly discontinuous in nature. In this case, MACCS (but not Molprint2D) introduced a notable compound clustering effect that can be well appreciated in both the NSG and the corresponding 3D model. The NSG displays a clear separation of densely connected clusters and the 3D landscape contains a large area of purely interpolated white surface separating two compound subsets.
Taken together, the comparisons shown in Fig. 3 revealed that differences in SAR information content between analyzed compound data sets and their SAR characteristics were well accounted for by both 2D and 3D activity landscape representations. Moreover, these landscape views could also be used as a diagnostic for chosen molecular representations. For example, in the case of THR, comparison of the MACCS- and Molprint2D-based landscapes clearly indicated that the choice of one or the other fingerprint would lead to substantial differences in the analysis of structure–activity relationships.
Mapping of activity cliffs
We next focused our analysis on the comparison of activity cliffs in our 2D and 3D landscape models. NSGs are based on calculated pairwise compound distances and measured potency values, whereas 3D activity landscapes are based on projected compound distances and interpolated potency values. Hence, the data structure underlying 3D landscapes is in principle more approximate in nature than the NSG data structure. However, the topology of 3D landscapes provides a particularly intuitive access to prominent activity cliffs and we therefore selected large cliffs from these 3D landscape views and then mapped these cliffs to NSGs. The results are also shown in Fig. 3. With no exception, prominent activity cliffs selected from 3D activity landscape models were also found to be large activity cliffs in NSGs. Thus, corresponding activity cliffs in 3D models and NSGs were of comparably large magnitude. For activity classes with moderate or low global SAR discontinuity (ACH and COX) the largest activity cliffs were easily identified in both 2D and 3D representations. For the remaining classes with increasingly high discontinuity, many comparably large activity cliffs were observed in both 2D and 3D landscape representations.Differences in SAR information content
The comparisons in Fig. 3 also reveal a number of differences in the way NSGs and 3D activity landscapes capture SAR information. For example, similarity relationships between individual compounds and their local SAR contributions are only provided by NSGs. Furthermore, NSG topology is ultimately determined by edge connectivity, whereas 3D landscapes are based on explicit pairwise compound distances. This difference generally results in stronger compound clustering effects observed in NSGs. Furthermore, the topology of NSGs is not affected by “structural outliers” that do not form similarity relationships to other compounds, whereas such outliers might influence the topology of 3D models. By contrast, areas of sparse SAR data are much easier to identify in 3D landscape models as purely interpolated surface area, which provides a basis for further directed compound data collection. Moreover, 3D landscape models are better suited to quickly focus on the most prominent activity cliffs in a data set than NSGs, in particular, in the presence of strong global SAR discontinuity. This is the case because both surface elevation and the color gradient of 3D landscape models mark prominent activity cliffs. However, the shape of large-magnitude activity cliff regions is the most difficult surface area to interpolate and hence 3D landscapes contain little interpretable information concerning the immediate environment of activity cliffs. This information is provided in detail in NSGs, which suggests the complementary use of 3D activity landscape models and NSGs for the detailed analysis of activity cliff regions in compound data sets. Because the intrinsically different layout of NSGs and 3D landscape models, it is usually difficult to delineate corresponding regions (compound subsets) in these representations. However, once prominent activity cliffs have been identified in 3D landscape models, they can directly be mapped to NSGs where their local SAR environments can then be analyzed in detail and from which other attractive candidate compounds can be selected.Conclusions
Activity landscape models are attractive tools for graphical SAR analysis of large compound data sets. Once generated, such graphical representations can be easily navigated and interpreted by medicinal chemists. Activity landscapes can be modeled in two or three dimensions. However, while idealized 3D landscape models have already been used for considerable time to rationalize SAR characteristics, only recently detailed 3D activity landscape models have been derived for sets of known active compounds. These models are obtained by dimension reduction of computational chemical reference spaces followed by interpolation of potency surfaces, and their most attractive feature for visual analysis is their intuitive topology. Herein, we have compared in detail 3D landscape models for different data sets with corresponding network-like similarity graphs, which provide a distinctly different access to SAR data. For model comparison, we have established a consistent data representation scheme. Global SAR characteristics and differences between data sets could be well appreciated on the basis of both 2D and 3D landscape models. Also, especially the 3D landscape models clearly showed how much SAR analysis might depend on the chosen molecular representations (here different types of fingerprints). Accordingly, these models can be used as a diagnostic tool to better understand how to represent data sets for meaningful SAR analysis. For example, for a systematic exploration of SARs in large data sets, representations are preferred that produce contiguous landscape surfaces and do not result in strong compound clustering effects. Furthermore, both 2D and 3D landscape models could be applied to monitor evolving compound data sets and gain insights into the progression of SAR trends. Moreover, especially 3D landscape models might also be utilized to identify under-sampled SAR regions in data sets.Importantly, we found that large-magnitude activity cliffs identified in 3D landscape models consistently corresponded to large activity cliffs in NSGs. For a detailed analysis of activity cliffs, 3D and 2D landscapes are best used in a complementary manner because prominent activity cliffs are easily identified in 3D models and details of their SAR environments can then be extracted from NSGs.
The NSG tools are publicly available as part of the SARANEA software24 and programs to generate 3D activity landscape models are also freely available (both can be obtained viahttp://www.lifescienceinformatics.uni-bonn.de/; see the Downloads section). The comparative activity landscape analysis presented herein should be helpful to further study specific features of 2D and 3D landscapes and also provides some guidelines how to utilize these landscape representations for practical SAR analysis.
References
- D. T. Manallack, D. D. Ellis and D. J. Livingstone, J. Med. Chem., 1994, 37, 3758–3767 CrossRef CAS.
- D. E. Patterson, R. D. Cramer, A. M. Ferguson, R. D. Clark and L. E. Weinberger, J. Med. Chem., 1996, 39, 3049–3059 CrossRef CAS.
- E. X. Esposito, A. J. Hopfinger and J. D. Madura, Methods Mol. Biol., 2004, 275, 131–214 CAS.
- L. Peltason and J. Bajorath, J. Med. Chem., 2007, 50, 5571–5578 CrossRef CAS.
- R. Guha and J. H. Van Drie, J. Chem. Inf. Model., 2008, 48, 646–658 CrossRef CAS.
- L. Peltason and J. Bajorath, Future Med. Chem., 2009, 1, 451–466 Search PubMed.
- A. R. Leach, V. J. Gillet, R. A. Lewis and R. Taylor, J. Med. Chem., 2010, 53, 539–558 CrossRef CAS.
- H. Geppert, M. Vogt and J. Bajorath, J. Chem. Inf. Model., 2010, 50, 205–216 CrossRef CAS.
- M. Wawer and J. Bajorath, J. Chem. Inf. Model., 2010, 50, 1395–1409 CrossRef CAS.
- V. Shanmugasundaram and G. M. Maggiora, Proceedings of 222nd American Chemical Society National Meeting, Division of Chemical Information, 2001 Search PubMed ; abstract no. 77.
- A. M. Wassermann, M. Wawer and J. Bajorath, J. Med. Chem., in press DOI:10.1021/jm100933w.
- R. van Deursen and J.-L. Reymond, ChemMedChem, 2007, 2, 636–640 CrossRef CAS.
- G. M. Maggiora, J. Chem. Inf. Model., 2006, 46, 1535–1535 CrossRef CAS.
- G. M. Maggiora and V. Shanmugasundaram, Methods Mol. Biol., 2011, 672, 39–100 CAS.
- L. Peltason, P. Iyer and J. Bajorath, J. Chem. Inf. Model., 2010, 50, 1021–1033 CrossRef CAS.
- MACCS Structural Keys; Symyx Software: San Ramon, CA, 2005 Search PubMed.
- A. Bender, H. Y. Mussa, R. C. Glen and S. Reiling, J. Chem. Inf. Comput. Sci., 2004, 44, 170–178 CrossRef CAS.
- I. Borg and P. J. F. Groenen, Modern Multidimensional Scaling. Theory and Applications, 2nd ed.; Springer: New York, NY, 2005 Search PubMed.
- N. Cressie, Statistics for Spatial Data, revised ed.; Wiley: New York, NY, 1993 Search PubMed.
- M. Wawer, L. Peltason, N. Weskamp, A. Teckentrup and J. Bajorath, J. Med. Chem., 2008, 51, 6075–6084 CrossRef CAS.
- P. Willett, J. M. Barnard and G. M. Downs, J. Chem. Inf. Comput. Sci., 1998, 38, 983–996 CrossRef CAS.
- T. M. J. Fruchterman and E. M. Reingold, Software: Pract. Exper., 1991, 21, 1129–1164 Search PubMed.
- MDL Drug Data Report (MDDR); Symyx Software: San Ramon, CA, 2005 Search PubMed.
- E. Lounkine, M. Wawer, A. M. Wassermann and Bajorath, J. Chem. Inf. Model., 2010, 50, 68–78 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |