 Open Access Article
Open Access ArticleProteome-wide prediction of overlapping small molecule and protein binding sites using structure
Fred P.
Davis
*
Howard Hughes Medical Institute, Janelia Farm Research Campus, 19700 Helix Dr, Ashburn, VA 20147, USA. E-mail: davisf@janelia.hhmi.org; Tel: +1 571 209 4000 x3037
First published on 24th November 2010
Abstract
Small molecules that modulate protein–protein interactions are of great interest for chemical biology and therapeutics. Here I present a structure-based approach to predict ‘bi-functional’ sites able to bind both small molecule ligands and proteins, in proteins of unknown structure. First, I develop a homology-based annotation method that transfers binding sites of known three-dimensional structure onto protein sequences, predicting residues in ligand and protein binding sites with estimated true positive rates of 98% and 88%, respectively, at 1% false positive rates. Applying this method to the human proteome predicts 8463 proteins with bi-functional residues and correctly recovers the targets of known interaction modulators. Proteins with significantly (p < 0.01) more bi-functional residues than expected were found to be enriched in regulatory and depleted in metabolism functions. Finally, I demonstrate the utility of the method by describing examples of predicted overlap and evidence of their biological and therapeutic relevance. The results suggest that combining the structures of known binding sites with established fold detection algorithms can predict regions of protein–protein interfaces that are amenable to small molecule modulation. Open-source software and the results for several complete proteomes are available at http://pibase.janelia.org/homolobind.
Introduction
Small molecules that disrupt or stabilize protein–protein interactions can serve as chemical tools to dissect cellular signaling networks and drugs to treat disease.1,2 However, modulating interactions with small molecules is currently more challenging than traditional drug targets on single proteins, due to their unique physicochemical and structural properties. In contrast to small molecule binding sites, the average protein–protein interface is large, flat, and often lacks detectable cavities that typically bind small molecules.3 Despite these differences, recent structural and biophysical studies suggest that protein interactions may be more feasible targets than previously thought. For example, a small number of energetic ‘hot-spots’ often contribute disproportionately to the binding energetics of protein–protein interactions.4,5 This observation suggests that small molecule disruption of a few key residues could efficiently compete with protein interaction partners. In addition, protein interfaces can be flexible and contain cryptic cavities that are not present in the structure of a protein–protein complex, but can bind to small molecules.6 This observation suggests that even seemingly featureless interfaces may contain ‘druggable’ binding sites.2,7A combination of experimental2 and computational8 methods have been used to identify interaction modulators. For traditional targets, computational approaches for small molecule discovery typically begin with a crystal structure or homology model of the target protein. Next, a target site is identified using either pocket detection algorithms or the known location of an endogenous substrate. Finally, docking algorithms are used to virtually screen a small molecule library and identify candidate ligands. Virtual screening has been widely used to discover small molecule ligands, and recent work suggests it can be complementary to experimental high-throughput screens.9 This overall computational framework has also been applied, with some adaptations, to protein interaction targets.8 For example, the presence of cryptic cavities at protein interfaces has inspired the use of molecular dynamics simulations to sample the conformational space around protein–protein interfaces for transient druggable pockets that are then subjected to virtual screening.10
The identification of druggable sites on interaction targets is particularly challenging for two reasons. First, endogenous substrate binding sites, often used as starting points for traditional targets, are not typically available.3 Second, the flexible nature of protein interfaces can hide cryptic cavities in crystal structures of the target protein complex. Here I present an approach to predict druggable binding sites at protein interfaces, in proteins of unknown structure, by using structural information from homologs.
This approach builds on three related observations. First, proteins often physically sample conformational space in the same direction and magnitude as the conformational variability observed between homologs.11 This observation has been exploited in protein structure modeling and design procedures,12–14 and suggests that binding sites in homologous structures may complement molecular dynamics sampling for identifying cryptic druggable sites. Second, protein homologs often use similar surface regions to interact with their protein interaction partners.15,16 This observation has been useful in predicting binding sites for proteins of unknown function.16,17 Third, identifying ‘bi-functional’ positions that bind both ligands and proteins within families of protein structures recovers the targets of known interaction modulators, and can be used to predict the biological effects of small molecules.18 Here, I extend this approach to proteins of unknown structure, with the aim of predicting druggable interface regions that are suitable for follow-up with higher resolution, but more computationally demanding, methods.
I first describe a method to predict bi-functional sites in protein sequences of unknown structure and benchmark its performance on binding sites of known structure. Next, I use this method to predict bi-functional sites in several complete proteomes and examine their compositional and functional properties. I close by discussing the relevance of the results for small molecule modulation of protein interactions.
Results
Binding site prediction algorithm
The prediction algorithm uses a binding site library organized by domain family to annotate a target set of protein sequences annotated with domains (Fig. 1). Briefly, template small molecule (250–1000 Da) and protein binding sites of known three-dimensional structure were obtained from the LIGBASE19 and PIBASE20 databases, respectively (details in Materials and methods). These binding sites were projected onto SCOP domain family alignments obtained from the ASTRAL compendium.21,22 A subsequent redundancy removal procedure yielded 27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 152 small molecule, 2147 peptide, 23308 inter-molecular domain, and 8254 intra-molecular domain binding sites on 20037, 1875, 19846, and 7470 domains, respectively.
152 small molecule, 2147 peptide, 23308 inter-molecular domain, and 8254 intra-molecular domain binding sites on 20037, 1875, 19846, and 7470 domains, respectively.
 | ||
| Fig. 1 Overview of the method to predict overlapping ligand and protein binding sites. | ||
The boundaries and classification of domains in the target protein sequences were obtained from the SUPERFAMILY resource, which uses a hidden Markov model library of SCOP structural domains to annotate complete genomes.23 The ASTRAL alignments, described above, were then used to transfer template binding sites onto the SUPERFAMILY domains in the target protein sequences. The binding sites were transferred at sequence identity thresholds estimated to predict residues with a 1% false positive rate, using a benchmarking strategy described next.
Assessing the coverage and accuracy of the method
The performance of the method was characterized in terms of coverage and accuracy by cross-validating the domain family alignments annotated with binding sites. Coverage refers to the fraction of known binding residues that are aligned to at least one template binding site, regardless of sequence identity. Accuracy refers to the true and false positive rates of the method in predicting these covered residues, at varying thresholds of sequence identity. The coverage was estimated by determining the fraction of binding residues in each domain family that was aligned to a binding residue in at least one other family member. A range of coverage was observed, with an average of 88%, 71%, 74%, and 84% for ligand, peptide, inter-molecular domain, and intra-molecular domain binding residues (Fig. 2A). These estimates establish the maximum fraction of residues in known binding sites that would be predicted by a homology transfer procedure with a perfect scoring function. Next, I estimated the actual accuracy of the homology transfer procedure presented here, which uses the binding-site sequence identity as a scoring function.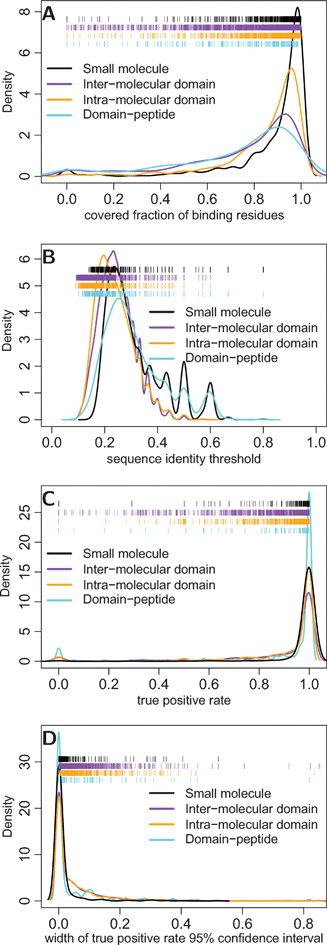 | ||
| Fig. 2 The coverage and accuracy of predicted binding residues. Coverage refers to the fraction of known binding residues in each family that align to a template binding site in a homologous protein; accuracy refers to the true and false positive rates in predicting these covered residues. (a) The distributions of binding site residue coverage per domain family is shown for each kind of binding site. (b) The distributions of sequence identity thresholds (per template binding site) estimated to achieve a maximum false positive rate of 1% and (c) the resulting true positive rates in predicting binding residues in each domain family. (d) The distribution of 95% confidence interval widths for true positive rates, estimated using Bayesian bootstrap with 500 replicates.24 | ||
The accuracy of the method was established by first determining sequence identity thresholds for each template binding site that would achieve a 1% false positive rate, as estimated on a simulated set of negative binding residues (see Materials and methods). A wide distribution of sequence identity thresholds was observed, with an average of 31% for ligand, 31% for peptide, 25% for inter-molecular domain, and 24% for intra-molecular domain binding sites (Fig. 2B). The corresponding true positive rates were then estimated in a family-wide fashion by determining the number of known (and covered) binding residues that passed the sequence identity thresholds determined above to achieve 1% false positive rates (Fig. 2C and 2D). The average true positive rates were estimated to be 98% for ligand, 89% for peptide, 88% for inter-molecular domain, and 91% for intra-molecular domain binding residues. These estimates are in concordance with published benchmarks of homology-transfer procedures.17
Bi-functional sites predicted in the human proteome
Having estimated the accuracy of the method, I used it to predict binding sites in the ENSEMBL human proteome containing 46591 proteins. Of the 64225 domains identified by SUPERFAMILY, significant similarities to ligand or protein binding sites were detected in 45541 domains (Table 1); 10561 of these domains contained residues with significant similarity to both ligand and protein binding sites.
| Type | # Proteins | # Domains (# Families) | # Residues |
|---|---|---|---|
| Input data | |||
| Complete proteome | 46591 |
— (—) | 23540008 |
| Annotated domains | 30712 |
64225 (1857) |
9119046 |
| Predicted binding sites | |||
| Peptide | 8091 | 11868 (200) |
516862 |
| Domain | 20990 |
42753 (1142) |
2166227 |
| Ligand | 10605 |
13074 (550) |
511993 |
| Bi-functional | 8463 | 10561 (442) |
294448 |
| All binding sites | 22916 |
45541 (1239) |
2499286 |
| Amino acid | Propensity at ligand-only residues | (95% confidence interval) | Propensity at protein-only residues | (95% confidence interval) | Propensity at bi-functional residues | (95% confidence interval) |
|---|---|---|---|---|---|---|
| A | 1.006 | (0.99, 1.022) | 0.891 | *(0.884, 0.896) | 0.875 | *(0.861, 0.889) |
| C | 0.974 | *(0.949, 1) | 1.147 | *(1.137, 1.157) | 0.753 | *(0.732, 0.77) |
| D | 0.878 | *(0.862, 0.896) | 1.044 | *(1.037, 1.051) | 1.111 | *(1.095, 1.128) |
| E | 0.675 | *(0.663, 0.69) | 1.085 | *(1.08, 1.092) | 0.971 | *(0.958, 0.986) |
| F | 1.311 | *(1.287, 1.334) | 0.912 | *(0.904, 0.918) | 1.093 | *(1.074, 1.11) |
| G | 1.392 | *(1.372, 1.411) | 1.049 | *(1.042, 1.054) | 1.235 | *(1.218, 1.25) |
| H | 1.058 | *(1.031, 1.084) | 1.121 | *(1.111, 1.131) | 1.003 | (0.982, 1.026) |
| I | 1.16 | *(1.139, 1.177) | 0.874 | *(0.867, 0.88) | 1.047 | *(1.032, 1.063) |
| K | 0.83 | *(0.815, 0.845) | 1.055 | *(1.049, 1.062) | 1.061 | *(1.047, 1.077) |
| L | 1.017 | *(1.004, 1.029) | 0.838 | *(0.834, 0.842) | 0.904 | *(0.894, 0.914) |
| M | 1.309 | *(1.277, 1.342) | 0.879 | *(0.871, 0.891) | 1.247 | *(1.218, 1.277) |
| N | 0.978 | *(0.956, 0.999) | 1.012 | *(1.002, 1.018) | 0.911 | *(0.891, 0.927) |
| P | 0.773 | *(0.757, 0.79) | 1.089 | *(1.082, 1.097) | 0.795 | *(0.781, 0.81) |
| Q | 0.697 | *(0.681, 0.715) | 1.064 | *(1.057, 1.072) | 0.814 | *(0.799, 0.83) |
| R | 0.899 | *(0.883, 0.917) | 1.094 | *(1.088, 1.102) | 1.128 | *(1.113, 1.145) |
| S | 0.98 | *(0.964, 0.996) | 1.027 | *(1.022, 1.033) | 0.903 | *(0.891, 0.916) |
| T | 1.054 | *(1.036, 1.073) | 1.082 | *(1.076, 1.09) | 0.977 | *(0.963, 0.992) |
| V | 1.018 | *(1.002, 1.033) | 0.894 | *(0.889, 0.899) | 0.98 | *(0.966, 0.994) |
| W | 1.081 | *(1.047, 1.116) | 1.096 | *(1.083, 1.111) | 0.958 | *(0.93, 0.989) |
| Y | 1.159 | *(1.134, 1.182) | 1.076 | *(1.066, 1.084) | 1.415 | *(1.391, 1.437) |
I next quantified the amino acid residue propensities of the predicted binding sites to facilitate comparison with bi-functional positions of known structure and previously described energetic hot-spots (eqn (2)). The predicted bi-functional residues exhibited a distinct amino acid residue propensity compared to predicted mono-functional residues (Fig. 3A; Table 2). The bi-functional residue propensities are mostly similar to those described previously for bi-functional positions of known structure.18 The most significant differences are that bi-functional positions of known structure exhibited enrichment for tryptophan and histidine, and near background levels of cysteine.
 | ||
| Fig. 3 The composition, frequency, and functional propensity of bi-functional residues predicted in the human proteome. (a) The residue type propensity (eqn (2)) at residues predicted to bind both ligands and proteins (black; n = 294448), bind ligands (cyan; n = 217545), or bind proteins (orange; n = 1987498) in comparison to all solvent-exposed residues (grey; n = 7236755). (b) The overlap observed between predicted ligand and protein binding residues. The maximum observed overlap score is 15.938 (not shown). (c) The function propensities of proteins with significantly (p < 0.01) higher (n = 3516) or lower (n = 624) number of bi-functional positions than expected by chance (eqn (3)). The statistical significance of the residue and function propensities was estimated by a bootstrap resampling procedure (Table 2 and 4). | ||
The bi-functional residue propensities are also similar in several respects to previously described energetic ‘hot-spots’.5,25 Hot-spot residues have been found to exhibit the following compositional trends: (1) enrichment for tryptophan, arginine and tyrosine, (2) under-representation of leucine, serine, threonine and valine, (3) over-abundance of isoleucine relative to leucine, and (4) preference for aspartate and asparagine over glutamate and glutamine.25 The predicted bi-functional residues exhibit all of these trends except for near-background levels of tryptophan and only slight enrichment for arginine (Fig. 3A).
To quantify the levels of overlap predicted between ligand and protein binding sites, an odds ratio was computed for each protein that considers the number of residues predicted to bind ligands (nl), proteins (np), or both ligands and proteins (nb), as well as the number of solvent-exposed residues (ns):
 | (1) |
| Most significant overlapping proteins | |||
|---|---|---|---|
| ID | Protein | Overlap | p-Value (left) |
| ENSP00000269577 | Topoisomerase (DNA) IIα | 6.093 | 2.2 × 10−16 |
| ENSP00000264998 | Transferrin | 5.130 | 2.2 × 10−16 |
| ENSP00000261266 | Protein tyrosine phosphatase, receptor type, B | 4.895 | 2.2 × 10−16 |
| ENSP00000359932 | TNNI3 interacting kinase | 4.809 | 2.2 × 10−16 |
| ENSP00000264331 | Topoisomerase (DNA) IIβ | 4.501 | 2.2 × 10−16 |
| ENSP00000231751 | Lactotransferrin | 4.495 | 2.2 × 10−16 |
| ENSP00000370076 | Baculoviral IAP repeat-containing protein 1 | 4.124 | 2.2 × 10−16 |
| ENSP00000371935 | ATP-binding cassette, sub-family C (CFTR/MRP), member 5 | 3.681 | 2.2 × 10−16 |
| ENSP00000261714 | Bleomycin hydrolase | 3.537 | 2.2 × 10−16 |
| ENSP00000319684 | Tensin 2 | 3.497 | 2.2 × 10−16 |
| Most significant non-overlapping proteins | |||
|---|---|---|---|
| ID | Protein | Overlap | p-Value (right) |
| ENSP00000223423 | Prostaglandin–endoperoxide synthase 1 | 0.094 | 9.78 × 10−16 |
| ENSP00000376187 | Discs, large homolog 1 | 0.000 | 2.771 × 10−15 |
| ENSP00000353047 | MAGUK p55 subfamily member 4 | 0.000 | 2.787 × 10−11 |
| ENSP00000295550 | Collagen, type VI, α3 | 0.000 | 8.347 × 10−11 |
| ENSP00000381234 | Cystathionine-β-synthase | 0.464 | 3.899 × 10−10 |
| ENSP00000241052 | Catalase | 0.416 | 1.125 × 10−9 |
| ENSP00000361049 | 3′-Phosphoadenosine 5′-phosphosulfate synthase 2 | 0.176 | 6.262 × 10−9 |
| ENSP00000376708 | von Willebrand factor A domain containing 2 | 0.000 | 3.026 × 10−8 |
| ENSP00000359210 | Dihydropyrimidine dehydrogenase | 0.679 | 3.728 × 10−8 |
| ENSP00000367937 | Lysyl-tRNA synthetase | 0.405 | 3.848 × 10−8 |
Functional significance of bi-functional residues
To explore the biological relevance of bi-functional residues, I next analyzed the functions of proteins with significantly greater or fewer such residues than expected by chance, using SUPERFAMILY function assignments of their component domains (eqn (3)).23 The proteins with greater overlap were most enriched in regulation and depleted in metabolism and information functions. Proteins with less overlap than expected were enriched in metabolism and depleted in intracellular and regulation processes (Fig. 3C; Table 4).| Function | Propensity of proteins with significantly low bi-functional positions | (95% confidence interval) | Propensity of proteins with significantly high bi-functional positions | (95% confidence interval) |
|---|---|---|---|---|
| Homo sapiens | (n = 624) | (n = 3516) | ||
| Information | 1.187 | (0.929, 1.442) | 0.795 | *(0.689, 0.897) |
| Metabolism | 1.528 | *(1.394, 1.658) | 0.445 | *(0.412, 0.479) |
| Extracellular processes | 1.075 | *(1.004, 1.154) | 1.184 | *(1.143, 1.227) |
| Intracellular processes | 0.381 | *(0.305, 0.465) | 1.002 | (0.932, 1.072) |
| Regulation | 0.807 | *(0.722, 0.89) | 1.271 | *(1.219, 1.322) |
| General | 0.559 | *(0.489, 0.64) | 1.009 | (0.959, 1.062) |
| Other | 3.91 | *(3.302, 4.509) | 0.507 | *(0.415, 0.609) |
| Drosophila melanogaster | (n = 338) | (n = 1902) | ||
| Information | 1.645 | *(1.201, 2.191) | 0.939 | (0.771, 1.141) |
| Metabolism | 1.93 | *(1.764, 2.08) | 0.575 | *(0.527, 0.628) |
| Extracellular processes | 0.59 | *(0.41, 0.787) | 0.693 | *(0.605, 0.788) |
| Intracellular processes | 0.142 | *(0.077, 0.215) | 1.238 | *(1.152, 1.341) |
| Regulation | 0.778 | *(0.648, 0.905) | 1.456 | *(1.365, 1.553) |
| General | 0.903 | (0.776, 1.047) | 1.087 | *(1.012, 1.168) |
| Other | 0.417 | *(0.154, 0.729) | 0.551 | *(0.401, 0.725) |
| Caenorhabditis elegans | (n = 303) | (n = 1678) | ||
| Information | 1.345 | (0.913, 1.852) | 0.661 | *(0.507, 0.833) |
| Metabolism | 1.916 | *(1.74, 2.109) | 0.482 | *(0.436, 0.532) |
| Extracellular processes | 1.521 | *(1.28, 1.771) | 1.082 | (0.972, 1.19) |
| Intracellular processes | 0.364 | *(0.224, 0.503) | 1.085 | (0.963, 1.212) |
| Regulation | 0.464 | *(0.365, 0.558) | 1.438 | *(1.358, 1.521) |
| General | 0.658 | *(0.513, 0.796) | 1.022 | (0.937, 1.113) |
| Other | 0.129 | *(0, 0.341) | 0.74 | *(0.545, 0.992) |
| Saccharomyces cerevisiae | (n = 124) | (n = 493) | ||
| Information | 1.118 | (0.667, 1.643) | 0.794 | (0.599, 1.033) |
| Metabolism | 1.672 | *(1.497, 1.855) | 0.563 | *(0.493, 0.644) |
| Extracellular processes | 0 | (0, 1) | 1.248 | (0, Inf) |
| Intracellular processes | 0.305 | *(0.125, 0.53) | 1.205 | *(1.012, 1.442) |
| Regulation | 0.649 | *(0.359, 0.991) | 1.733 | *(1.429, 2.072) |
| General | 0.503 | *(0.351, 0.675) | 1.324 | *(1.185, 1.484) |
| Other | 0.396 | (0, 1.524) | 0.699 | (0.22, 1.469) |
| Escherichia coli | (n = 117) | (n = 288) | ||
| Information | 1.181 | (0.642, 1.78) | 0.835 | (0.537, 1.207) |
| Metabolism | 1.088 | (0.945, 1.231) | 0.681 | *(0.595, 0.769) |
| Extracellular processes | 0 | (0, 1) | 0 | (0, 1) |
| Intracellular processes | 0.755 | (0.41, 1.153) | 1.134 | (0.831, 1.464) |
| Regulation | 1.869 | *(1.252, 2.583) | 0.627 | *(0.388, 0.928) |
| General | 0.609 | *(0.417, 0.824) | 1.789 | *(1.553, 2.028) |
| Other | 0.696 | (0, 1.625) | 0.798 | (0.277, 1.651) |
| NCBI viral sequence set | (n = 203) | (n = 2324) | ||
| Information | 0.546 | *(0.4, 0.697) | 1.119 | *(1.052, 1.182) |
| Metabolism | 1.444 | *(1.242, 1.664) | 0.491 | *(0.445, 0.535) |
| Extracellular processes | 0.146 | *(0, 0.515) | 0.274 | *(0.17, 0.403) |
| Intracellular processes | 1.043 | (0.758, 1.339) | 1.34 | *(1.225, 1.468) |
| Regulation | 0.826 | (0.479, 1.206) | 1.554 | *(1.382, 1.75) |
| General | 1.128 | (0.935, 1.347) | 1.178 | *(1.105, 1.253) |
| Other | 1.079 | (0.788, 1.382) | 0.858 | *(0.774, 0.953) |
These results largely agree with the functional analysis of bi-functional positions of known 3D structure, although the propensity values presented here are more statistically significant due to a larger sample size.18 The trends for the regulation and metabolism functions were similarly found in the previous analysis of protein families. The only significant difference is that the previous analysis found overlapping proteins to be enriched in intracellular processes, while that category is near-background in the present analysis. One possible reason for these differences is the level of analyses: the previous analysis was performed at the level of individual domains in contrast to the results presented here at the protein level, which consider all component domains.
These functional trends were further explored by predicting bi-functional residues in several other species. Similar trends were observed for the metabolism and regulation functions in nearly all the other proteomes tested: Mus musculus, Drosophila melanogaster, Caenorhabditis elegans, Saccharomyces cerevisiae, Escherichia coli, and the NCBI viral sequence set (Table 4). The sole exception was the reversal of the regulatory function in E. coli, with overlapping proteins exhibiting a depletion, while non-overlapping proteins were enriched.
Examples of overlapping binding site predictions
To determine the predictive utility of the method, I next examined examples of predicted overlap between ligand and protein binding sites. Below, I describe four examples of therapeutically relevant targets and small molecules. These examples involve protein–protein interactions from several distinct functional classes, including enzyme–substrate/inhibitor, regulatory, and structural interactions.
TopoIIa belongs to a broad family of proteins that share a homologous ATPase domain and includes several chemotherapeutic targets: bacterial DNA gyrase, topoisomerase IV, and topoisomerase VI are antibiotic targets; topoIIa and Hsp-90 are antineoplastic targets.27 Small molecules have shown cross-reactivity between these family members, and this feature has been exploited to discover inhibitors. For example, radicicol was initially discovered as an antifungal antibiotic and was later shown to inhibit both Hsp-90 and mammalian topoIIa.30 Novobiocin, a natural product with antibacterial and weak anti-mammalian topoisomerase activity, has been derivatized to yield selective Hsp-90 inhibitors.32 Both of these compounds act by binding to the ATP substrate pocket. In addition, radicicol prevents ATP-mediated topoisomerase VI homo-dimerization;27 a coumarin antibiotic structurally related to novobiocin interferes with Hsp90 dimerization.33 This example illustrates the utility of distant homologs for predicting binding sites: The folds are shared across prokaryotic and eukaryotic species, and the ligands exhibit cross-reactivity across this evolutionary range. It also indicates that even well-established drug classes – novobiocin was discovered over 50 years ago34 – that target traditional targets like enzyme active sites, may also disrupt protein interactions.
This example highlights the issue of ligand/family member specificity. XIAP, cIAP1, and cIAP2 all have three BIR domains, each of which interacts with different proteins. For example, XIAP interacts with caspase-9 through its BIR3 domain and with capsases-3 and -7 through its BIR2 domain.38 Most small molecules have been designed against BIR3, although at least one study has targeted BIR2. No small molecules have been described that bind to the BIR1 domain. Cross-reactivity has also been observed between XIAP BIR2 and BIR3.36 As expected, HOMOLOBIND predicted bi-functional residues for all three BIR domains of cIAP1, using mostly the same template ligand, peptide, and domain–domain binding sites. The prediction of binding specificity between homologous domains (both within the same protein and in different proteins) is largely beyond the scope of the method, which only aims to predict the binding site.
Several small molecules have been synthesized with varying selectivity for the anti-apoptotic Bcl-2 members including Bcl-2 itself, Bcl-XL, and Mcl-1. Recent studies suggest that both the Mcl-1/Bcl2A1 and the Bcl-2/Bcl-XL/Bcl-w sub-families of anti-apoptotic proteins must be inhibited for effective induction of cancer cell apoptosis.42 Given the structural similarity between Bcl-2 members, and the observed cross-reactivity of small molecules,43 the predicted Mcl-1 bi-functional residues are likely relevant targets.
This example again highlights the issue of specificity. The Bcl-2 ligand used to predict the ligand binding site on Mcl-1 is an acyl-sulfonamide compound designed to bind both Bcl-2 and Bcl-XL.44 Although this compound was not tested against Mcl-1, an earlier study testing a similar compound found weak Mcl-1 binding.45 Recently, small molecules have been designed with activity against multiple anti-apoptotic Bcl-2 members, including Bcl-2, Bcl-XL, Bcl-w, and Mcl-1.46 As mentioned previously, the method presented here does not aim to predict actual ligands, as this requires estimation of binding affinities using explicit structural models at a much higher resolution than the fold detection sequence alignments used here.
Discussion
I developed a homology transfer algorithm, HOMOLOBIND, to predict binding site residues (Fig. 1), characterized its coverage and accuracy (Fig. 2), used it to predict overlapping ligand and protein binding sites in the human proteome (Table 1 and 3), described the compositional and functional properties of these bi-functional residues (Fig. 3; Table 2 and 4), and illustrated the utility of the results for identifying protein interface regions amenable to small molecule modulation (Fig. 4 and 5). I now describe possible extensions to the method and follow-up analyses to further characterize the relevance of predicted bi-functional residues for modulating protein interactions. | ||
| Fig. 4 The method correctly recovers the targets of known interaction modulators.2 Predicted binding sites are depicted as colored tick-marks within larger boxes representing SUPERFAMILY domain annotations. Template ligand and protein binding sites are shown as ribbon diagrams, produced by UCSF Chimera. | ||
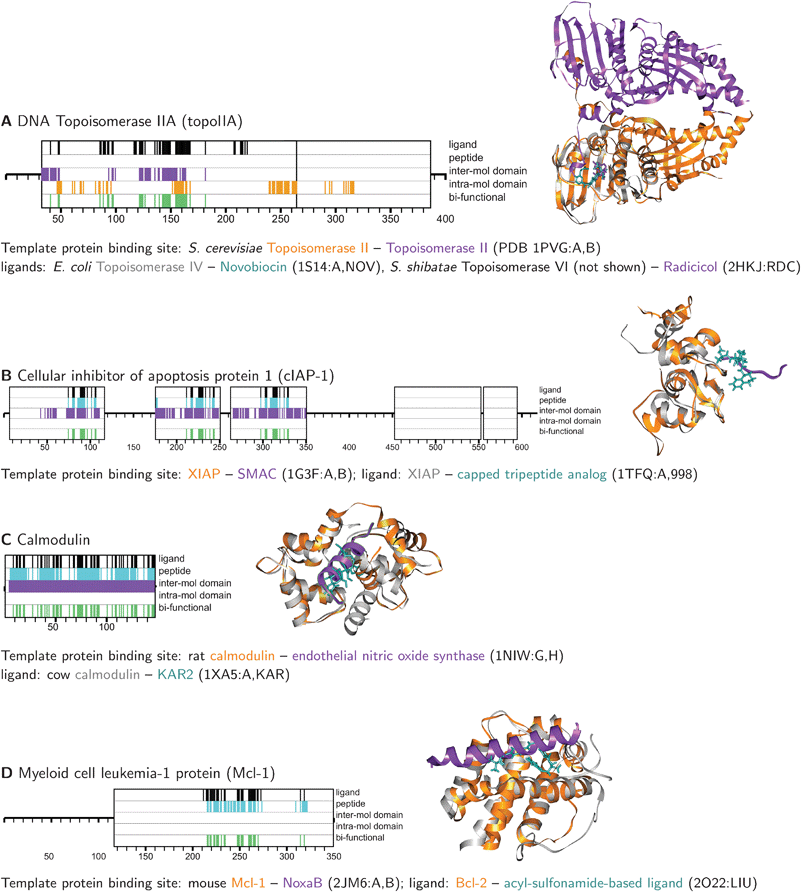 | ||
| Fig. 5 Examples of overlap predicted between ligand and protein binding sites on human proteins. Predicted binding sites are depicted as colored tick-marks within larger boxes representing SUPERFAMILY domain annotations. Template ligand and protein binding sites are shown as ribbon diagrams, produced by UCSF Chimera. Full-length topoIIA is nearly 1600 residues; only the first 400 residues, where binding sites were predicted, are shown. | ||
Homology transfer of binding sites has been extensively demonstrated in a variety of systems and applied to the annotation of protein sequences, structures, and their interactions.17,47–49 Beyond its specific application to the prediction of bi-functional residues, HOMOLOBIND is a systematically benchmarked method that integrates several well-established protein structure resources to facilitate comprehensive prediction of binding sites in complete proteomes. The underlying structural domain assignment algorithm (SUPERFAMILY) has been rigorously benchmarked, the domain definitions and classification (SCOP) are considered gold standard, and the binding site libraries (PIBASE, LIGBASE) are comprehensive and regularly updated. The automated nature of the method makes it suitable for large-scale studies of binding sites. As illustrated above, it can be run on a genomic scale using either the pre-computed genomic domain assignments available from SUPERFAMILY, or domain assignments made for a newly sequenced genome using SUPERFAMILY software.
Relevance for modulating protein interactions
The primary goal in developing HOMOLOBIND has been for use as a proteome-wide first-pass to identify potentially druggable protein interface residues. Small molecules that have successfully targeted protein interactions fall into three broad categories: (1) competitive inhibitors that prevent the binding of a protein by competing with the binding site, (2) allosteric inhibitors that bind to a region distinct from the protein interface but still inhibit protein interaction, possibly through a conformational change, and (3) interfacial inhibitors that bind to an interface and stabilize the protein complex in a functionally inactive state.3,50 The method presented here is of course only applicable to competitive and interfacial inhibitors, because it relies on overlapping protein and ligand binding sites. Predicting druggable allosteric sites requires the identification of allosteric pathways that can communicate conformational changes from a distal site to the interface.51The bi-functional residues predicted with HOMOLOBIND are suitable candidates for evaluation with higher resolution computational methods that are also more computationally expensive.8 For example, a first step would be to build an explicit structural model of the target protein by comparative modeling26 and high resolution refinement techniques.52 Next, flexible docking algorithms could be used to predict small molecules that bind to the predicted bi-functional region.53 Finally, computationally validated targets and predicted ligands could then be evaluated using the extensive repertoire of experimental biophysical and biochemical techniques used to identify and evaluate interaction inhibitors.3
Experimental high-throughput screening data, such as bioassay data available through ChEMBL (http://www.ebi.ac.uk/chembldb/) and PubChem,54 might also be useful in evaluating the predictions. In the most simple usage, bioassay data could be searched to determine if ligands whose template binding sites were used to annotate a target protein sequence in fact bind to the target. Although clearly confirmatory, this scenario is likely to occur in only a few cases, as HOMOLOBIND only predicts binding sites rather than actual ligands, due to the complexity and difficulty in predicting ligand specificity. However, even in the absence of experimental data for a particular ligand, the bioactivity profiles for the target and template protein sequences might be useful as a pharmacological similarity measure, to complement the sequence similarity thresholds that HOMOLOBIND uses to make the binding site predictions. The (untested) hypothesis is that if a pair of proteins bind similar or identical ligands, then binding sites transferred by homology from one to the other protein are likely to be correct. For example, several Mcl-1 inhibitors in PubChem also demonstrate activity against both Bcl-2 and Bcl-XL (e.g., CIDs 406171 and 1002248). Although these cross-reactive compounds don't include the actual ligands used for the HOMOLOBIND prediction, this cross-reactivity gives more confidence in the Mcl-1 binding site predicted by homology from Bcl-2 (Fig. 5D). The concept of ligand-based protein similarity has been successfully used to develop statistical models of polypharmacology and to predict off-target effects of drugs.55,56
Future directions
The main limit of the method's coverage, as with any homology-based method, is its reliance on homologous template binding sites of known structure. Recent work suggests that remote structural neighbors beyond the family or superfamily levels can provide useful information for predicting binding sites.16 More generally, tools that identify local structural similarity rather than full-length domain similarity could further harness available binding site structures.57 However, even at the current domain family level of similarity detection, the coverage and accuracy of the method will naturally increase as the binding site library is updated to reflect newly determined three-dimensional structures of binding sites.Besides predictions for individual proteins, the proteome-wide compositional and functional properties of bi-functional residues, and the similar functional trends observed across a wide phylogenetic range, suggest that these residues are a biologically relevant phenomena (Fig. 3A and C; Table 2 and 4). Analyzing their biophysical properties could further clarify their relevance for modulating protein interactions. In particular, the relationship of bi-functional residues to energetic hot-spot residues remains an open question. The predicted bi-functional residues described here, as well as the structurally characterized bi-functional positions reported previously,18 exhibit residue propensities similar in many ways to energetic hot-spots (Fig. 3A).5,25 Comparing HOMOLOBIND predictions to experimentally observed58 and computationally predicted hot-spots59–61 will establish how frequently bi-functional residues are also energetic hot-spots.
Another property that has been observed at successfully targeted protein interfaces is that the conformation ‘captured’ by a small molecule is often distinct from that involved in the protein–protein interaction.6,10 The flexibility of bi-functional residues can be investigated using temperature factors from crystallographically determined structures, order parameters of structures determined by nuclear magnetic resonance spectroscopy, or through molecular dynamics simulations. If a clear difference in flexibility is observed at bi-functional residues, this feature might provide an additional means to predict and evaluate the relevance of bi-functional residues. At the extreme of flexibility, the past decade has seen a growing number of studies that demonstrate the importance of intrinsic disorder in protein interaction networks.62 Tools that have been developed to predict protein binding residues in disordered proteins could shed light on the occurrence of bi-functional residues in disordered regions.63
The distinctive features of bi-functional residues, such as their residue propensities, suggest an alternative template-independent approach for their prediction. Previous studies have explored the utility of structural, physicochemical, and evolutionary features for predicting protein binding sites directly from sequence or structure.64 A similar approach might be useful for predicting bi-functional sites in proteins whose structures are unknown, do not have detectable similarity to a protein of known structure, or for which template binding sites are not available. Although the predictive accuracy of these feature-based methods remains to be explored in the context of bi-functional positions, such a method would be complementary to homology- and physics-based predictions.
In summary, I have presented a method that aims to maximize the utility of experimentally determined protein structures for identifying potentially druggable regions of protein–protein interfaces. The method is implemented in open-source software that integrates with well-established protein structure resources. The results provide a protein structure resource for targeting interactions and is complementary to a growing number of computational methods that catalog, characterize, and predict small molecule interaction modulators.8,10,65–70
Materials and methods
Binding site library
The binding-site library contains small molecule ligand, peptide, and protein domain binding sites of known three-dimensional structure. These sites were extracted from LIGBASE19 and PIBASE,20 comprehensive databases of binding sites observed in the Protein Data Bank (PDB), as previously described.18458 ligand, 4553 peptide, 81014 inter-molecular domain, and 35042 intra-molecular domain binding sites on 22463, 3845, 51847, 29317 domains to the final template library of 27152 ligand, 2147 peptide, 23308 inter-molecular domain, and 8254 intra-molecular domain binding sites on 20037, 1875, 19846, and 7470 domains, respectively. Binding site similarity was computed as: (alignment positions shared by the two binding sites)/(positions in either binding site). The clustering was done with respect to alignment position, rather than amino acid identity, to achieve a conservative representation of structural diversity. The practical utility of the redundancy removal procedure was to reduce the computational expense of benchmarking and performing the homology transfer procedure described below.
Homology-transfer of binding sites
The binding sites are transferred to target protein sequences in two steps. First, family-level domain assignments are obtained for the target sequences from SUPERFAMILY (v 1.73).23 For each domain assignment, SUPERFAMILY provides an alignment of the target sequence to a SCOP domain of known structure. This alignment is combined with the ASTRAL alignment of the corresponding domain family. Next, the binding sites that were previously mapped onto these ASTRAL alignments are transferred onto the target sequences. The sequence identity of the target sequence to the binding site template is computed across the putative binding site. Sequence identity thresholds are then imposed on the transferred binding sites. These thresholds are calibrated through a statistical analysis of the template binding site library, described below, to predict binding site residues with an estimated false positive rate ≤1%.The procedure is implemented in a Perl program, HOMOLOBIND, that is licensed under GPL v3 and runs on a single CPU or a Sun Grid Engine computing cluster. The program takes as input a SUPERFAMILY assignment file describing the domains found in a set of target protein sequences. The output is a list of target residues with similarities to a template binding site. HOMOLOBIND can also generate diagrams depicting the locations of predicted binding sites relative to the SUPERFAMILY domain architecture of a target protein. Annotation of all human proteins with a SUPERFAMILY domain assignment (n = 30712) takes 2.5 h on a single 2 GHz Xeon processor. HOMOLOBIND is compatible with the current SUPERFAMILY version (v 1.73), and its binding site library will be updated as SUPERFAMILY transitions to newer versions of SCOP.
Assessment of prediction coverage and accuracy
All analysis was done using the non-redundant set of binding sites that are used as templates. The coverage was assessed in a family-specific manner by counting the fraction of ligand-binding residues that were aligned to another family member with a corresponding binding site residue, irrespective of the sequence identity. This analysis establishes the scope of the method.The accuracy of the method was characterized by the false and true positive rates achieved as a function of the sequence identity threshold used to transfer binding sites from templates onto target sequences. Transferring binding sites without imposing a sequence identity threshold would yield a low false negative rate, correctly identifying all (correctly aligned) binding residues, but at the cost of a high false positive rate. At the other extreme of a 100% sequence identity threshold, all predicted residues would be correct (low false positive rate), but many real binding site residues would be omitted (high false negative rate). Ideally, these error rates would be estimated using a benchmark set of protein sequences where all individual residues are systematically known either to be involved (positive set) or not involved (negative set) in binding ligands and proteins. Such an ideal benchmark set is of course not available because of the vast number of possible ligand and protein binding partners and the currently sparse experimental sampling of this space.
The best source for positive binding residues are the PDB structures used in the template library. A negative set of residues that are not involved in binding is more difficult to construct because the absence of binding in the PDB for a particular protein residue does not rule out all possible binding events. An artificial negative set was constructed using a sequence-shuffling method originally developed for fold recognition,72 and later applied to protein–protein interaction prediction,73 that performs comparably to a more physical model of structural sampling. A set of negative binding residues was defined for each template binding site by creating shuffled sequences (n = 10000) from the family-wide alignment, while preserving gap structure, and selecting those shuffled residues in alignment columns corresponding to the actual template binding site. These negative binding residues were used during benchmarking to estimate the false positive rates achieved for each template binding site, at varying sequence identity thresholds.
Briefly, sequence identity thresholds were first established for each binding site that achieved a 1% residue-level false positive rate (FPR) and the resulting true positive rates (TPR) were estimated for each family by cross-validation. FPR was calculated by counting the number of negative binding residues that passed progressively higher sequence identity thresholds. The lowest sequence identity value that achieved a FPR ≤ 1% was chosen as the threshold. If such a threshold was not identified for a binding site, it was not used as a template. This happened for 18 ligand, 0 peptide, 20 inter-molecular domain, and 8 intra-molecular domain binding sites.
These binding site-specific sequence identity thresholds were then used to estimate the corresponding TPR in a family-wide fashion by cross-validation of the template binding sites. Briefly, each sequence in the family with a binding site was annotated using the other template binding sites in the family at the previously established sequence identity thresholds. Known binding residues that were aligned to template binding residues were considered the positive set: those that passed the sequence identity threshold were considered true positives, and the remainder considered false negatives. Confidence intervals for the TPR were estimated using Bayesian bootstrap resampling with 500 replicates.24
Computing residue type propensities
Residue type propensities were calculated for ligand binding-only, protein binding-only, and bi-functional residues relative to all solvent-exposed residues: | (2) |
Computing function propensities
Function propensities were calculated for proteins with significantly fewer or greater number of bi-functional residues than expected, relative to all proteins predicted to have both ligand and protein binding sites. The propensity of function i was computed by considering the fraction of domains in each protein set assigned function i by SUPERFAMILY: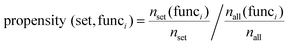 | (3) |
Acknowledgements
I thank Julian Gough (Univ. Bristol) for help with SUPERFAMILY, Ursula Pieper and Andrea Rossi (UCSF) for maintenance of LIGBASE, Dorothea Emig (MPI Saarbrucken) for discussion, Sean Eddy (HHMI Janelia) for comments on the manuscript, and Goran Ceric for managing Janelia's high-performance computing resources.References
- T. Berg, Curr. Opin Drug Discov. Devel., 2008, 11, 666–674 Search PubMed.
- J. A. Wells and C. L. McClendon, Nature, 2007, 450, 1001–1009 CrossRef CAS.
- M. R. Arkin and J. A. Wells, Nat. Rev. Drug Discovery, 2004, 3, 301–317 CrossRef CAS.
- T. Clackson and J. A. Wells, Science, 1995, 267, 383–386 CrossRef CAS.
- A. A. Bogan and K. S. Thorn, J. Mol. Biol., 1998, 280, 1–9 CrossRef CAS.
- C. D. Thanos, W. L. DeLano and J. A. Wells, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15422–15427 CrossRef CAS.
- J. C. Fuller, N. J. Burgoyne and R. M. Jackson, Drug Discovery Today, 2009, 14, 155–161 CrossRef CAS.
- B. O. Villoutreix, K. Bastard, O. Sperandio, R. Fahraeus, J. L. Poyet, F. Calvo, B. Deprez and M. A. Miteva, Curr. Pharm. Biotechnol., 2008, 9, 103–122 CAS.
- R. S. Ferreira, A. Simeonov, A. Jadhav, O. Eidam, B. T. Mott, M. J. Keiser, J. H. McKerrow, D. J. Maloney, J. J. Irwin and B. K. Shoichet, J. Med. Chem., 2010, 53, 4891–4905 CrossRef CAS.
- S. Eyrisch and V. Helms, J. Med. Chem., 2007, 50, 3457–3464 CrossRef CAS.
- R. Elber and M. Karplus, Science, 1987, 235, 318–321 CAS.
- B. Qian, A. R. Ortiz and D. Baker, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15346–15351 CrossRef CAS.
- A. Leo-Macias, P. Lopez-Romero, D. Lupyan, D. Zerbino and A. R. Ortiz, Biophys. J., 2005, 88, 1291–1299 CrossRef CAS.
- G. D. Friedland, N. A. Lakomek, C. Griesinger, J. Meiler and T. Kortemme, PLoS Comput. Biol., 2009, 5, e1000393 CrossRef.
- D. Korkin, F. P. Davis and A. Sali, Protein Sci., 2005, 14, 2350–2360 CrossRef CAS.
- Q. C. Zhang, D. Petrey, R. Norel and B. H. Honig, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 10896–10901 CrossRef CAS.
- M. A. Marti-Renom, A. Rossi, F. Al-Shahrour, F. P. Davis, U. Pieper, J. Dopazo and A. Sali, BMC Bioinf., 2007, 8(Suppl 4), S4 CrossRef.
- F. P. Davis and A. Sali, PLoS Comput. Biol., 2010, 6, e1000668 CrossRef.
- A. C. Stuart, V. A. Ilyin and A. Sali, Bioinformatics, 2002, 18, 200–201 CrossRef CAS.
- F. P. Davis and A. Sali, Bioinformatics, 2005, 21, 1901–1907 CrossRef CAS.
- A. G. Murzin, S. E. Brenner, T. Hubbard and C. Chothia, J. Mol. Biol., 1995, 247, 536–540 CrossRef CAS.
- J. M. Chandonia, G. Hon, N. S. Walker, L. L. Conte, P. Koehl, M. Levitt and S. E. Brenner, Nucleic Acids Res., 2004, 32, D189–D192 CrossRef CAS.
- D. Wilson, R. Pethica, Y. Zhou, C. Talbot, C. Vogel, M. Madera, C. Chothia and J. Gough, Nucleic Acids Res., 2009, 37, D380–D386 CrossRef CAS.
- D. Rubin, Ann. Stat., 1981, 9, 130–134 CrossRef.
- I. S. Moreira, P. A. Fernandes and M. J. Ramos, Proteins: Struct., Funct., Bioinf., 2007, 68, 803–812 Search PubMed.
- A. Sali and T. L. Blundell, J. Mol. Biol., 1993, 234, 779–815 CrossRef CAS.
- K. D. Corbett and J. M. Berger, Nucleic Acids Res., 2006, 34, 4269–4277 CrossRef CAS.
- S. Bellon, J. D. Parsons, Y. Wei, K. Hayakawa, L. L. Swenson, P. S. Charifson, J. A. Lippke, R. Aldape and C. H. Gross, Antimicrob. Agents Chemother., 2004, 48, 1856–1864 CrossRef CAS.
- S. Classen, S. Olland and J. M. Berger, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10629–10634 CrossRef CAS.
- D. Gadelle, M. Graille and P. Forterre, Biochem. Pharmacol., 2006, 72, 1207–1216 CrossRef CAS.
- P. Hussy, G. Maass, B. Tummler, F. Grosse and U. Schomburg, Antimicrob. Agents Chemother., 1986, 29, 1073–1078 CAS.
- J. A. Burlison, L. Neckers, A. B. Smith, A. Maxwell and B. S. Blagg, J. Am. Chem. Soc., 2006, 128, 15529–15536 CrossRef CAS.
- R. K. Allan, D. Mok, B. K. Ward and T. Ratajczak, J. Biol. Chem., 2006, 281, 7161–7171 CrossRef CAS.
- H. Hoeksema, J. L. Johnson and J. W. Hinman, J. Am. Chem. Soc., 1955, 77, 6710–6711 CrossRef CAS.
- S. Fulda, ACS Chem. Biol., 2009, 4, 499–501 CrossRef CAS.
- E. C. LaCasse, D. J. Mahoney, H. H. Cheung, S. Plenchette, S. Baird and R. G. Korneluk, Oncogene, 2008, 27, 6252–6275 CrossRef CAS.
- C. Ndubaku, E. Varfolomeev, L. Wang, K. Zobel, K. Lau, L. O. Elliott, B. Maurer, A. V. Fedorova, J. N. Dynek, M. Koehler, S. G. Hymowitz, V. Tsui, K. Deshayes, W. J. Fairbrother, J. A. Flygare and D. Vucic, ACS Chem. Biol., 2009, 4, 557–566 CrossRef CAS.
- A. D. Schimmer, S. Dalili, R. A. Batey and S. J. Riedl, Cell Death Differ., 2006, 13, 179–188 CrossRef CAS.
- I. Horvath, V. Harmat, A. Perczel, V. Palfi, L. Nyitray, A. Nagy, E. Hlavanda, G. Naray-Szabo and J. Ovadi, J. Biol. Chem., 2005, 280, 8266–8274 CAS.
- T. Kirikae, F. Kirikae, Y. Oghiso and M. Nakano, Infect Immun., 1996, 64, 3379–3384 CAS.
- A. Molnar, K. Liliom, F. Orosz, B. G. Vertessy and J. Ovadi, Eur. J. Pharmacol., Mol. Pharmacol. Sect., 1995, 291, 73–82 Search PubMed.
- M. Vogler, D. Dinsdale, M. J. Dyer and G. M. Cohen, Cell Death Differ., 2009, 16, 360–367 CrossRef CAS.
- J. Wei, J. L. Stebbins, S. Kitada, R. Dash, W. Placzek, M. F. Rega, B. Wu, J. Cellitti, D. Zhai, L. Yang, R. Dahl, P. B. Fisher, J. C. Reed and M. Pellecchia, J. Med. Chem., 2010, 53, 4166–4176 CrossRef CAS.
- M. Bruncko, T. K. Oost, B. A. Belli, H. Ding, M. K. Joseph, A. Kunzer, D. Martineau, W. J. McClellan, M. Mitten, S. C. Ng, P. M. Nimmer, T. Oltersdorf, C. M. Park, A. M. Petros, A. R. Shoemaker, X. Song, X. Wang, M. D. Wendt, H. Zhang, S. W. Fesik, S. H. Rosenberg and S. W. Elmore, J. Med. Chem., 2007, 50, 641–662 CrossRef CAS.
- M. D. Wendt, W. Shen, A. Kunzer, W. J. McClellan, M. Bruncko, T. K. Oost, H. Ding, M. K. Joseph, H. Zhang, P. M. Nimmer, S. C. Ng, A. R. Shoemaker, A. M. Petros, A. Oleksijew, K. Marsh, J. Bauch, T. Oltersdorf, B. A. Belli, D. Martineau, S. W. Fesik, S. H. Rosenberg and S. W. Elmore, J. Med. Chem., 2006, 49, 1165–1181 CrossRef CAS.
- M. Nguyen, R. C. Marcellus, A. Roulston, M. Watson, L. Serfass, S. R. M. Madiraju, D. Goulet, J. Viallet, L. Belec, X. Billot, S. Acoca, E. Purisima, A. Wiegmans, L. Cluse, R. W. Johnstone, P. Beauparlant and G. C. Shore, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 19512–19517 CrossRef CAS.
- K. A. Snyder, H. J. Feldman, M. Dumontier, J. J. Salama and C. W. Hogue, BMC Bioinf., 2006, 7, 152 CrossRef.
- U. Pieper, N. Eswar, B. M. Webb, D. Eramian, L. Kelly, D. T. Barkan, H. Carter, P. Mankoo, R. Karchin, M. A. Marti-Renom, F. P. Davis and A. Sali, Nucleic Acids Res., 2009, 37, D347–D354 CrossRef CAS.
- M. N. Wass and M. J. Sternberg, Proteins, 2009, 77, 147–151 Search PubMed.
- Y. Pommier and J. Cherfils, Trends Pharmacol. Sci., 2005, 26, 138–145 CrossRef CAS.
- N. Halabi, O. Rivoire, S. Leibler and R. Ranganathan, Cell, 2009, 138, 774–786 CrossRef CAS.
- E. Krieger, K. Joo, J. Lee, J. Lee, S. Raman, J. Thompson, M. Tyka, D. Baker and K. Karplus, Proteins, 2009, 77(Suppl. 9), 114–122 Search PubMed.
- H. Fan, J. J. Irwin, B. M. Webb, G. Klebe, B. K. Shoichet and A. Sali, J. Chem. Inf. Model., 2009, 49, 2512–2527 CrossRef CAS.
- Y. Wang, E. Bolton, S. Dracheva, K. Karapetyan, B. A. Shoemaker, T. O. Suzek, J. Wang, J. Xiao, J. Zhang and S. H. Bryant, Nucleic Acids Res., 2010, 38, D255–D266 CrossRef CAS.
- M. J. Keiser, B. L. Roth, B. N. Armbruster, P. Ernsberger, J. J. Irwin and B. K. Shoichet, Nat. Biotechnol., 2007, 25, 197–206 CrossRef CAS.
- M. J. Keiser, V. Setola, J. J. Irwin, C. Laggner, A. I. Abbas, S. J. Hufeisen, N. H. Jensen, M. B. Kuijer, R. C. Matos, T. B. Tran, R. Whaley, R. A. Glennon, J. Hert, K. L. Thomas, D. D. Edwards, B. K. Shoichet and B. L. Roth, Nature, 2009, 462, 175–181 CrossRef CAS.
- G. J. Kleywegt, J. Mol. Biol., 1999, 285, 1887–1897 CrossRef CAS.
- K. S. Thorn and A. A. Bogan, Bioinformatics, 2001, 17, 284–285 CrossRef CAS.
- T. Kortemme and D. Baker, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14116–14121 CrossRef CAS.
- O. Keskin, B. Ma and R. Nussinov, J. Mol. Biol., 2005, 345, 1281–1294 CrossRef CAS.
- N. Tuncbag, O. Keskin and A. Gursoy, Nucleic Acids Res., 2010, 38(Suppl.), W402–W406 CrossRef CAS.
- A. K. Dunker, M. S. Cortese, P. Romero, L. M. Iakoucheva and V. N. Uversky, FEBS J., 2005, 272, 5129–5148 CrossRef CAS.
- B. Meszaros, I. Simon and Z. Dosztanyi, PLoS Comput. Biol., 2009, 5, e1000376 CrossRef.
- H. X. Zhou and S. Qin, Bioinformatics, 2007, 23, 2203–2209 CrossRef CAS.
- A. P. Higueruelo, A. Schreyer, G. R. Bickerton, W. R. Pitt, C. R. Groom and T. L. Blundell, Chem. Biol. Drug Des., 2009, 74, 457–467 CrossRef CAS.
- C. Reynes, H. Host, A. C. Camproux, G. Laconde, F. Leroux, A. Mazars, B. Deprez, R. Fahraeus, B. O. Villoutreix and O. Sperandio, PLoS Comput. Biol., 2010, 6, e1000695 CrossRef.
- O. Sperandio, C. H. Reynes, A. C. Camproux and B. O. Villoutreix, Drug Discovery Today, 2010, 15, 220–229 CrossRef CAS.
- L. Parthasarathi, F. Casey, A. Stein, P. Aloy and D. C. Shields, J. Chem. Inf. Model., 2008, 48, 1943–1948 CrossRef CAS.
- F. P. Casey, E. Pihan and D. C. Shields, J. Chem. Inf. Model., 2009, 49, 2708–2717 CrossRef CAS.
- D. M. Kruger and H. Gohlke, Nucleic Acids Res., 2010, 38(Suppl.), W480–W486 CrossRef.
- D. Dimitropoulos, J. Ionides and K. Henrick, Curr. Protoc. Bioinf., 2006, ch. 14, Unit14.3 Search PubMed.
- F. Melo, R. Sanchez and A. Sali, Protein Sci., 2002, 11, 430–448 CAS.
- F. P. Davis, H. Braberg, M. Y. Shen, U. Pieper, A. Sali and M. S. Madhusudhan, Nucleic Acids Res., 2006, 34, 2943–2952 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |