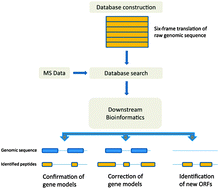
With the onset of modern DNA sequencing technologies, genomics is experiencing a revolution in terms of quantity and quality of sequencing data. Rapidly growing numbers of sequenced genomes and metagenomes present a tremendous challenge for bioinformatics tools that predict protein-coding regions. Experimental evidence of expressed genomic regions, both at the RNA and protein level, is becoming invaluable for genome annotation and training of gene prediction algorithms. Evidence of gene expression at the protein level using mass spectrometry-based proteomics is increasingly used in refinement of raw genome sequencing data. In a typical “proteogenomics” experiment, the whole proteome of an organism is extracted, digested into peptides and measured by a mass spectrometer. The peptide fragmentation spectra are identified by searching against a six-frame translation of the raw genomic assembly, thus enabling the identification of hitherto unpredicted protein-coding genomic regions. Application of mass spectrometry to genome annotation presents a range of challenges to the standard workflows in proteomics, especially in terms of proteome coverage and database search strategies. Here we provide an overview of the field and argue that the latest mass spectrometry technologies that enable high mass accuracy at high acquisition rates will prove to be especially well suited for proteogenomics applications.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?
