A digital microfluidic method for dried blood spot analysis†
Mais J.
Jebrail‡
a,
Hao
Yang‡
a,
Jared M.
Mudrik
a,
Nelson M.
Lafrenière
a,
Christine
McRoberts
b,
Osama Y.
Al-Dirbashi
bc,
Lawrence
Fisher
b,
Pranesh
Chakraborty
bc and
Aaron R.
Wheeler
*ad
aDepartment of Chemistry, University of Toronto, 80 St. George Street, Toronto, Ontario M5S 3H6, Canada. E-mail: aaron.wheeler@utoronto.ca; Fax: +416-946-3865; Tel: +416-946-3864
bNewborn Screening Ontario, Children's Hospital of Eastern Ontario, 401 Smyth Road, Ottawa, Ontario K1H 8L1, Canada
cDepartment of Pediatrics, Faculty of Medicine, University of Ottawa, 451 Smyth Road, Ottawa, Ontario K1H 8L1, Canada
dInstitute of Biomaterials and Biomedical Engineering, University of Toronto, 164 College Street, Toronto, Ontario M5S 3G9, Canada
First published on 25th August 2011
Abstract
Blood samples stored as dried blood spots (DBSs) are emerging as a useful sampling and storage vehicle for a wide range of applications. Unfortunately, the surging popularity of DBS samples has not yet been accompanied by an improvement in automated techniques for extraction and analysis. As a first step towards overcoming this challenge, we have developed a prototype microfluidic system for quantification of amino acids in dried blood spots, in which analytes are extracted, mixed with internal standards, derivatized, and reconstituted for analysis by (off-line and in-line) tandem mass spectrometry. The new method is fast, robust, precise, and most importantly, compatible with automation. We propose that the new method can potentially contribute to a new generation of analytical techniques for quantifying analytes in DBS samples for a wide range of applications.
Introduction
Dried blood spot (DBS) samples stored on filter paper are surging in popularity for applications ranging from screening for genetic disorders in newborn patients1 to point-of-care testing for infectious diseases2 to metabolic profiling in drug discovery and lead validation.3 The recent popularity of DBS samples is driven by the numerous advantages for this format relative to conventional samples, including small sample volumes (pin-prick in place of venous blood draw), reduced sample processing burden (no requirement of phlebotomist, centrifuge, freezer, etc.), uncomplicated transportation and cataloguing (samples are painlessly mailed and filed), and straightforward waste disposal (simple incineration rather than decontamination of sharps, etc.). There is great interest in developing new techniques for analyzing DBS samples,4 but despite this interest, there are few automated solutions available. In fact, most methods used today are manual, tedious, and slow.3We report here a new set of techniques useful for extracting and quantifying analytes for DBS samples. The methods are fast, use small amounts of reagents, and as far as we are aware, represent the first application of microfluidics to DBS samples. (Note that analytes manually extracted from DBS samples have been analyzed by microfluidic techniques,5 but the methods reported here are the first to accept a DBS directly as sample input.) The new methods are powered by digital microfluidics (DMF), a fluid-handling technique in which discrete droplets of samples and reagents are manipulated (i.e., dispensed from reservoirs, split, merged and mixed) on an open surface by applying a series of electrical potentials to an array of electrodes.6,7 Droplet actuation in such systems is driven by electromechanical forces8,9 generated on free charges in the droplet meniscus (in case of conductive liquids) or on dipoles inside of the droplet (in case of dielectric liquids).
The microfluidic DBS analysis methods described here were validated by application to quantifying analytes that are commonly measured as biomarkers for amino acid metabolism disorders in newborn patients: methionine (as a marker for homocystinuria10,11), phenylalanine (as a marker for phenylketonuria12,13), and tyrosine (as a marker for tyrosinemia11,14). These analytes are typically quantified using tandem mass spectrometry (MS/MS), and comprehensive testing represents a large public health undertaking around the world. For example, the Newborn Screening Ontario (NSO) facility at the Children's Hospital of Eastern Ontario10 uses MS/MS to evaluate DBS samples from approximately 140,000 patients each year, which necessitates a large laboratory that employs a large number of technologists and other staff-members.
In a typical screen for amino acid metabolism disorders in a newborn patient, a 3.2 mm diameter disc is punched from a DBS sample on filter paper, and the analytes are extracted, mixed with isotope-labelled internal standards, derivatized, and then reconstituted for analysis by tandem mass spectrometry (MS/MS). As shown in Fig. 1a, the derivatization step transforms each amino acid (AA) to its corresponding butyl ester (derivatized AA) that allows for a characteristic fragmentation pattern (neutral loss of 102) viacollision induced dissociation (CID). Fig. 1b–c contains representative primary (MS1) and secondary (MS2) mass spectra for phenylalanine, with peaks at m/z 222 and 120.
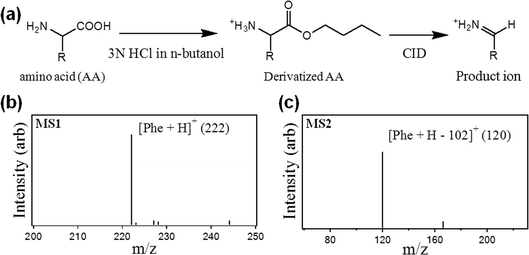 | ||
| Fig. 1 Processing dried blood spot samples for quantification of amino acids (AAs) by tandem mass spectrometry. (a) Reaction scheme involving derivatization of the extracted AA, followed by derivatization with n-butanol, followed by the formation of a product ion by collision induced dissociation (CID) in the mass spectrometer. (b)Mass spectrum generated from primary analysis (MS1) of derivatized phenylalanine (Phe). (c)Mass spectrum generated from the secondary analysis (MS2) of derivatized Phe showing the loss of 102 amu as a result of CID. | ||
The process flow shown in Fig. 1 is specific for newborn screening, but many different kinds of applications of DBS samples follow a similar scheme of extraction, derivatization, mixing with standards, purification, and analysis.1,4,15,16 We propose that the new methods reported here may represent a useful new tool for a wide range of applications of dried blood spots.
Results and discussion
Two related digital microfluidic methods were developed for analysis of dried blood spots to accommodate the potential for different sampling needs. As shown in Fig. 2, in method 1, a sample of liquid blood (5 μL) is spotted directly onto a device and allowed to dry. In method 2, a 3.2 mm diameter punch from filter paper bearing dried blood (3.1 μL) is deposited onto a device for analysis. In both methods, the detection and quantification is achieved with tandem mass spectrometry.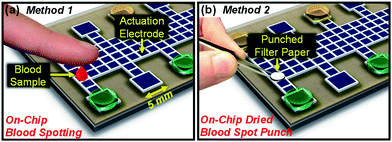 | ||
| Fig. 2 Two digital microfluidic methods designed to quantify amino acids in dried blood spot samples. In method 1 (a), a 5 μL droplet of blood is spotted directly onto the device surface and allowed to dry. In method 2 (b), a 3.2-mm diameter punch from filter paper bearing dried blood is positioned on the device surface. | ||
An experiment using method 1 is depicted in Fig. 3a. As shown, a blood sample is spotted onto the device, dried, extracted into methanol containing isotope-labelled standards, and the solvent is allowed to evaporate. The extract and standards are then derivatized, and the products are isolated by allowing the solvent to evaporate. The entire process requires 50 min to complete. Samples processed by method 1 were collected and analyzed off-line by nanoelectrospray ionization tandem mass spectrometry (nESI-MS/MS) to quantify AAs. Calibration curves with R2 greater than 0.996 (Fig. S1 in the online Supplementary Information†) were generated for Met, Phe, and Tyr by analyzing standards processed by DMF at known concentrations from the abundance ratio of each AA to its deuterated standard peak in the secondary (MS2) spectra.
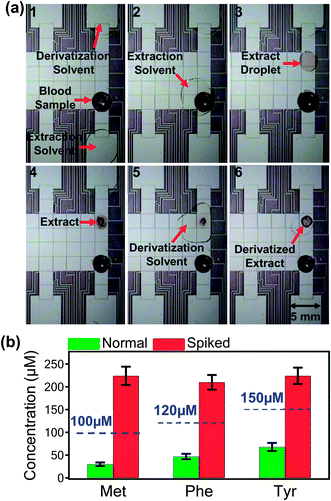 | ||
| Fig. 3 Analysis of amino acids in dried blood spots by digital microfluidic method 1. (a) Sequence of frames from a movie depicting several stages in sample processing by DMF including: (1) a dried blood sample prior to processing; (2) mixing and incubating an extractant droplet with the sample; (3) a droplet containing sample extract after translation away from the dried sample; (4) a dried extract; (5) mixing and incubating a derivatization reagent droplet with the dried extract; and (6) the dried, derivatized product. (b) Comparison of Met, Phe, and Tyr concentrations in normal (green) and spiked (red) blood samples as biomarkers for homocystinuria, phenylketonuria, and tyrosinemia, respectively. The dashed lines indicate the upper levels for normal concentrations in newborn blood samples. Each data point represents at least four replicate measurements, and error bars represent ±1 S.D. | ||
To evaluate the potential of DMF method 1 as a platform for discriminating between disease and healthy states, spiked blood samples (mimicking diseased states) and non-spiked blood samples (mimicking healthy state) were analyzed by mass spectrometry. Fig. 3b shows a comparison of measured concentration of AAs in normal and spiked blood samples. The dashed line indicates typical threshold values that correlate with homocysteinuria (100 μM Met)10,11phenylketonuria (120 μM Phe)12,13 and tyrosinemia (150 μM Tyr).11,14 As shown, the method is useful for distinguishing between these concentrations.
The new method was evaluated for a number of analytical performance parameters, including extraction efficiency, linear dynamic range, and detection limits. Two orthogonal tests were used to evaluate extraction efficiency: fluorescence and MS/MS. In the former, a fluorogenic dye was used to label standard samples dissolved in solvent before and after processing by DMF to determine the concentrations. In the latter, blood samples were spiked with AAs and the recovery efficiency was determined by comparing the measured AA concentration (endogenous plus spiked AA) vs. known concentration. As listed in Table 1, the two orthogonal methods (fluorescence and MS/MS) agree and reveal the new DMF technique to be highly efficient, with recoveries of ≥80% for each AA. More importantly, the precision of these measurements was high, with CVs ranging from 1 to 10%. And as shown in Figure S1 in the online supplementary information,† each measurement had a linear dynamic range of three orders of magnitude, and detection limits were 14.6, 6.0, and 15.2 ng for Met, Phe, and Tyr, respectively.
| Amino Acid | % Recovery by Fluorescence ± 1 S.D. (standards in solvent) | % Recovery by MS/MS ± 1 S.D. (standards in blood) |
|---|---|---|
| Methionine | 98 ± 10 | 100 ± 1 |
| Phenylalanine | 86 ± 9 | 85 ± 5 |
| Tyrosine | 82 ± 10 | 84 ± 7 |
Digital microfluidic method 2 (Fig. 2b) was developed to analyze punched discs from DBS samples on filter paper. A portion of an experiment is depicted in Fig. 4a. As shown, a droplet of extraction solvent was dispensed and driven to a 3.2 mm diameter filter paper punch, and the extract was then moved away for further processing (i.e., derivatization and solvent exchange, similar to Fig. 3a). After wetting, the filter paper punch remains adhered to the surface through friction and capillary forces, and fluids could be moved onto, through, and off-of the filter paper smoothly. The capacity to easily handle mesoscale solid samples like punched filter paper is a unique advantage of digital microfluidics relative to other miniaturization modalities. For example, 3.2-mm dia. filter paper punches are much larger than the dimensions of standard microchannels, which are moreover susceptible to clogging in applications involving biological samples.17 This is likely the reason why microfluidic devices reported previously for DBS sample analysis5 were used to analyze liquid extract (after off-chip sample processing), rather than the DBS samples, themselves.
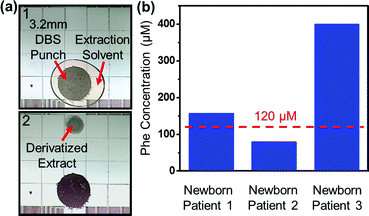 | ||
| Fig. 4 Analysis of amino acids in dried blood spots by digital microfluidic method 2. (a) Frames from a movie depicting sample processing of 3.2 mm diameter punch of a dried blood spot (DBS) on filter paper by digital microfluidics (DMF). (b) Graph of Phe concentrations measured by the digital microfluidic method in DBS punches from three NSO patients. As shown, patients 1 and 3 were correctly diagnosed as having phenylketonuria, and patient 2 was correctly identified as being unaffected. The dashed lines indicates the upper level for normal concentrations of Phe in newborn blood samples. | ||
To evaluate digital microfluidic method 2 relative to gold standard practices, a series of punches from blood samples containing various concentrations of Phe were processed by DMF method 2, and punches from the same samples were evaluated using the conventional newborn screening technique at NSO. As listed in Table 2, a paired t-test revealed no significant difference between the two data sets at a 95% confidence level. This result is notable, as the two sets of data were generated using different extraction techniques as well as different mass spectrometers (from different manufacturers) in different locations run by unique operators. To validate the new technique for application to clinical samples, DBS punches from three newborn patients of NSO were evaluated by the DMF method. As shown in Fig. 4b, the new technique correctly identified patients 1 and 3 as suffering from phenylketonuria, and patient 2 as being unaffected.
| Sample | Measured Phe concentration (μM) using DMF method 2 | Measured Phe concentration (μM) using standard NSO technique |
|---|---|---|
| 1 | 70 | 70 |
| 2 | 550 | 548 |
| 3 | 93 | 88 |
| 4 | 93 | 92 |
| 5 | 368 | 302 |
| 6 | 534 | 539 |
| 7 | 735 | 871 |
While the data described above were generated with microfluidic sample processing and off-line analysis, there may be some cases in which a fully integrated (processing + analysis) method is useful. To accommodate this potential need, a digital microfluidic platform was coupled directly to a nanoelectrospray emitter for in-line analysis by mass spectrometry. As shown in Figure S3 in the online Supplementary Information,† the central feature of this method (building on recent work with hybrid microfluidics18) is a vertical intersection between an array of DMF electrodes on top of a device, and a microchannel on the bottom of the device. A portion of an experiment is depicted in Fig. 5a. A blood sample was first spotted on the device as in DMF method 1 (note that the technique is also compatible with filter paper punches as in DMF method 2) and the AAs were extracted and derivatized as described above. The dried, derivatized sample was then resuspended and the droplet was actuated to the access hole such that it filled the channel below by capillary action. A nanoelectrospray was generated at a corner of the device19,20 (Fig. 5b) by applying a high voltage to the DMF top-plate electrode. Representative mass spectra generated from samples processed and analyzed on-chip are shown in Fig. 5c. The entire process requires ∼1 h from sampling to analysis, and requires only the hybrid DMF device and a mass spectrometer (i.e., no nanoflow pumps, capillary connections, or samplers). While digital microfluidics has been used to handle liquid samples for eventual analysis off-line by electrospray21–24 and MALDI25–31mass spectrometry in previous work, the device shown in Fig. 5 is the first reported to integrate digital microfluidics with in-line mass spectrometry.
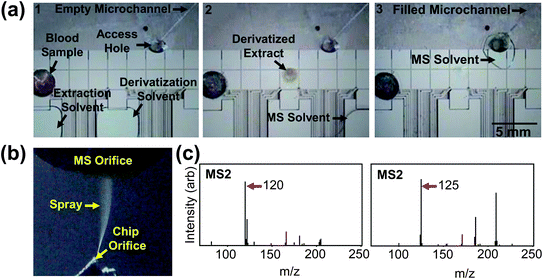 | ||
| Fig. 5 Processing and in-line analysis of amino acids in dried blood spots. (a) Frames from a movie (left-to-right) demonstrating derivatization and extraction of AA from dried blood, resolubilization in solvent, and analyte solution in spray microchannel. (b) Image of sample spraying from an integrated emitter. (c) MS2 spectra of Phe (left) and d5-Phe (right) generated from blood samples. | ||
The new microfluidic methods reported here have several advantages relative to conventional techniques. An obvious advantage is the potential for automation -- a single technologist using an instrument powered by digital microfluidics could likely do the work of several technologists using manual techniques. Furthermore, for the applications demonstrated here (for newborn screening), the digital microfluidic methods facilitate reduction in reagent use (20 μL vs. 170–450 μL14,32,33) and a reduction in analysis time (∼1 h vs. >3.5 h1,34). Another advantage of the new technique is the potential for direct integration with mass spectrometry (Fig. 5). Nanoflow electrospray ionization (nESI) is a finicky technique requiring operator expertise and vigilance to achieve reproducible results, which is, in part, the reason why all newborn DBS samples in Ontario are mailed to a single facility.10 Indeed, a recent editorial on this subject decried the inherent limitations of systems relying on “conventional electrospray plumbing including capillary tubes etc.”1 In the new method, there is no plumbing or tubes -- an integrated device is simply positioned in front of the mass spectrometer to generate nESI-MS/MS spectra.
Over the past two decades, MS/MS has revolutionized newborn screening, replacing the tests used historically for diagnosing amino acid disorders (i.e., Guthrie's bacterial inhibition35 and fluorometric36 and enzymatic37 tests). This is mainly because MS/MS facilitates the analysis of multiple analytes simultaneously in a single run in comparison to the traditional single-analyte assays, which improves efficiency and reduces costs.1,38 In this work, we present the first method to combine microfluidic sample handling and preparation with MS/MS for analysis of amino acids. We note that there are other metabolic and non-metabolic disorders (e.g., cystic fibrosis, congenital hypothyroidism, biotinidase deficiency, galactosemiaetc.) that are commonly screened in newborn patients that are not routinely detected by MS/MS. Instead, these diseases are evaluated by enzyme-, DNA-, or immunoassay-based tests38,39 and a variety of new technologies39,40 (including digital microfluidics41) are being developed to implement them, as well.
The current work demonstrates proof-of-concept for quantification of amino acids that are often measured for early diagnosis of diseases in newborn screening, but we propose that similar techniques might be useful for a wide range of applications in the future. For clinical analysis,1,2 blood samples could be spotted onto filter paper (Fig. 2b) or onto inexpensive DMF devices42,43 (or removable device coverings27) (Fig. 2a), after which they would be mailed or delivered to a laboratory for analysis. For pharmaceutical applications,3 built-in emitters (Fig. 5) for in-line analysis with mass spectrometry might be appropriate. We propose that the methods described here offer a flexible range of analysis modes that may be useful for the growing number of laboratories that are adopting dried blood spot samples for routine analysis.
Conclusions
Digital microfluidic methods were developed to extract analytes from dried blood spot samples, followed by mixing with standards, derivatization, and reconstitution for analysis by (in-line or off-line) tandem mass spectrometry. These new methods have the potential to contribute to a new generation of analytical techniques for quantifying analytes in DBS samples for a wide range of applications.Experimental
Study subjects
Liquid blood samples were collected after obtaining permission from a healthy adult male volunteer after a 10 h fasting period and were kept at −20 °C until analysis. Punches from dried blood spots from infants screened by NSO were obtained with permission from the infants' families and were stored at −80 °C until analysis.Reagents and materials
L-Methionine (Met), L-Phenylalanine (Phe), L-Tyrosine (Tyr), acetonitrile (ACN), acetone, methanol (MeOH), boric acid and fluorescamine were purchased from Sigma Chemical (Oakville, ON). Deuterated Methionine (Met-d3), Phenylalanine (Phe-d5), and Tyrosine (Tyr-d4) were obtained from Cambridge Isotope Laboratories (Andover, MA). Concentrated hydrochloric acid (HCl) was purchased from Fisher Scientific (Ottawa, ON) and n-butanol from ACP Chemicals (Montreal, QC). In all experiments, organic solvents were HPLC grade and deionized (DI) water had a resistivity of 18 MΩ·cm at 25 °C.Working solutions of all amino acids (AAs) (25, 50, 100 and 500 μM ea.) were prepared in DI water. For derivatization of extracted AAs, a 3 N HCl in n-butanol solution was used. For analysis of AAs in blood samples, the extracting solvent (MeOH) contained 50 μM of the appropriate deuterated AA (d3-Met, d5-Phe or d4-Tyr). For quantitative analysis of AA recovery from blood and for experiments mimicking diseased/healthy infant blood, samples were spiked with 200 μM of the appropriate AA (Met, Phe or Tyr). A series of 3.2 mm dia. punches of dried blood samples containing different concentrations of phenylalanine (see Table 2) were prepared by NSO staff.16
DMF device fabrication and operation
Details relating to device fabrication and operation can be found in the online Supplementary Information.†DMF-driven sample processing
For samples analyzed by digital microfluidic method 1 (Fig. 2a), 5–μL droplets of blood were spotted onto the bottom plate of a device and dried under ambient conditions for 2 h. The top plate was then affixed and two solvents were loaded into the appropriate reservoirs, including MeOH containing 50 μM of deuterated AA (extraction solvent), and 3 N HCl-butanol (derivatization reagent). A reservoir volume (10 μL) of extraction solvent was dispensed and driven by DMF to the dried sample and allowed to incubate (5 min). The extraction solvent was then actuated away from the sample and evaporated to dryness (∼15 min, room temperature) at a second site, after which a reservoir volume (10 μL) of derivatization solvent was dispensed to the dried extract and incubated for 15 min at 75 °C. Following the reaction, the top plate was removed and the solvent was allowed to evaporate (∼15 min, room temperature). For samples analyzed by method 2 (Fig. 2b), the process was identical to the above, but with two differences: (1) punches (3.2 mm dia.) from filter paper bearing dried blood were positioned on the bottom plate of a device in place of liquid blood, and (2) a larger volume (15 μL) of extraction solvent was used.DMF-driven sample analysis
Samples processed by digital microfluidics were analyzed by nanoelectrospray tandem mass spectrometry (nESI-MS/MS). For off-line analysis, extracted, derivatized samples (stored dry on device or in centrifuge tube until analysis) were reconstituted in 70 μL of acetonitrile/water (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v); samples originating from blood were, in addition, passed through PVDF membrane centrifuge-filters with 0.1 μm pore diameter (Millipore, ON). Such samples were injected into an LTQ Mass Spectrometer (Thermo Scientific) via a fused silica capillary transfer line (100 μm i.d.) mated to a New Objective Inc. (Woburn, MA) nanoelectrospray emitter (100 μm i.d. tapering to 50 μm i.d.) at a flow rate of 0.8 μL min−1, with an applied voltage of 1.7–1.9 kV and capillary temperature of 200 °C. Tandem MS/MS analysis was carried out by introducing 30% collision energy to the precursor ions and then the fragments over the m/z range of 100–300 were scanned. AA product ions detected in the second mass selection, which exhibit a loss of butylformate (HCOOC4H9, 102 m/z), were observed and used for quantification. Spectra were collected as an average of 50 acquisitions, and replicate spectra were obtained for DMF-derivatized samples of both control and blood. For samples processed and analyzed in-line, “hybrid” devices bearing an integrated nESI emitter were mounted on a 3-axis micromanipulator (Edmund Optics, NJ) positioned near the inlet of the LTQ MS. After sample processing, a spray was generated by applying 2.5–3.0 kV to a platinum wire inserted in the access hole, with parameters identical to the above. Sprays were generated and data collected from replicate experiments performed on five different hybrid devices.
:1 v/v); samples originating from blood were, in addition, passed through PVDF membrane centrifuge-filters with 0.1 μm pore diameter (Millipore, ON). Such samples were injected into an LTQ Mass Spectrometer (Thermo Scientific) via a fused silica capillary transfer line (100 μm i.d.) mated to a New Objective Inc. (Woburn, MA) nanoelectrospray emitter (100 μm i.d. tapering to 50 μm i.d.) at a flow rate of 0.8 μL min−1, with an applied voltage of 1.7–1.9 kV and capillary temperature of 200 °C. Tandem MS/MS analysis was carried out by introducing 30% collision energy to the precursor ions and then the fragments over the m/z range of 100–300 were scanned. AA product ions detected in the second mass selection, which exhibit a loss of butylformate (HCOOC4H9, 102 m/z), were observed and used for quantification. Spectra were collected as an average of 50 acquisitions, and replicate spectra were obtained for DMF-derivatized samples of both control and blood. For samples processed and analyzed in-line, “hybrid” devices bearing an integrated nESI emitter were mounted on a 3-axis micromanipulator (Edmund Optics, NJ) positioned near the inlet of the LTQ MS. After sample processing, a spray was generated by applying 2.5–3.0 kV to a platinum wire inserted in the access hole, with parameters identical to the above. Sprays were generated and data collected from replicate experiments performed on five different hybrid devices.
For samples processed by digital microfluidic method 1 (Fig. 2a), calibration plots (Fig. S1 in the Supplementary Information†) were generated by plotting the MS/MS intensity ratio of product ions from the extracted AAs from standards in methanol relative to the those of the internal standards (i.e., Met m/z 104:107, Phe m/z 120:125, and Tyr m/z 136:140) as a function of AA concentration in standard solutions (25–500 μM in DI water). Data points included in the calibration plots represent an average of at least 4 replicate measurements, and the data in each plot were fit with a linear regression. Blood samples were then evaluated (with on-chip derivatization and extraction, and measurement by MS/MS relative to internal standards, as above), and the values were compared to the calibration plots to determine the AA concentrations.
For samples processed by digital microfluidic method 2 (Fig. 2b), quantification was similar to that for method 1, except that standards were formed from adult male blood spiked with Phe to different concentrations (25–900 μM). 3.1 μL aliquots were pipetted onto pre-punched (3.2 mm dia.) discs of Whatman 903 filter paper (Whatman, NJ) and were allowed to dry. Calibration plots (Fig. S2 in the online Supplementary Information†) were formed with the assumption that endogenous blood samples contained 38 μM Phe (the measured concentration in the sample prior to spiking). Newborn blood samples (as DBS filter paper punches) from NSO were then analyzed as above and compared with the calibration plot to determine the Phe concentrations.
DMF-driven sample recovery determination
The % recovery of AAs in Method 1 (Fig. 2a) was evaluated quantitatively using (i) a fluorescence-based assay and (ii) MS/MS. For (i), control samples (Met, Phe or Tyr; 50 μM of each) were processed by DMF (as above), excluding the derivatization step. The dried extracts were diluted into 95 μL aliquots of borate buffer (20 mM, pH 8.5) in wells in a 96-well microplate. Upon addition of 5 μL of fluorescamine (5 mg mL−1 in acetone) the microplate was inserted into a fluorescence microplate reader (Pherastar, BMG Labtech, Durham, NC) equipped with a module for 390 nm excitation and 510 nm emission. The plate was shaken (5 s) and the fluorescence was measured. As a control, identical samples that had not been extracted were evaluated using the same fluorescent assay. To ensure that controls were processed in an identical manner relative to extracted samples, each control sample was spotted on a device, dried and then reconstituted in buffer for analyses. Four replicate measurements were made for each sample and control. For (ii), blood samples of known AA concentrations were spiked with 200 μM of AA standards and extracted (as above). Knowing the total concentration of AAs in blood spots (e.g. native methionine concentration plus spiked methionine), % recovery was obtained by comparing the concentration values (obtained from calibration curves) vs. the known values.Non-DMF sample processing and analysis
Amino acids were extracted and quantified from dried blood spot samples according to routine methods used at NSO.16Acknowledgements
We thank the Natural Sciences and Engineering Research Council (NSERC) and the province of Ontario for financial support. ARW thanks the Canada Research Chair (CRC) Program for a CRC.References
- D. H. Chace, J. Mass Spectrom., 2009, 44, 163–170 CrossRef CAS.
- A. Johannessen, Bioanalysis, 2010, 2, 1893–1908 CrossRef CAS.
- C. Arnaud, Chem. Eng. News, 2011, 89, 13–17 Search PubMed.
- P. Abu-Rabie and N. Spooner, Anal. Chem., 2009, 81, 10275–10284 CrossRef CAS.
- P. D. Rainville, Bioanalysis, 2011, 3, 1–3 CrossRef CAS.
- A. R. Wheeler, Science, 2008, 322, 539–540 CrossRef CAS.
- M. J. Jebrail and A. R. Wheeler, Curr. Opin. Chem. Biol., 2010, 14, 574–581 CrossRef CAS.
- T. B. Jones, Langmuir, 2002, 18, 4437–4443 CrossRef CAS.
- K. H. Kang, Langmuir, 2002, 18, 10318–10322 CrossRef CAS.
- Educational resource on blood spot collection developed by the Ontario Newborn Screening Program, October 2009, http://www.newbornscreening.on.ca/data/1/rec_docs/195_Unsatisfactory_Sample_Educational_Resource.pdf (accessed 7 January 2010).
- D. Matern, S. Tortorelli, D. Oglesbee, D. Gavrilov and P. Rinaldo, J. Inherited Metab. Dis., 2007, 30, 585–592 CrossRef CAS.
- H. L. Levy and S. Albers, Annu. Rev. Genomics Hum. Genet., 2000, 1, 139–177 CrossRef CAS.
- M. Zaffanello, C. Maffeis and G. Zamboni, J. Perinat. Med., 2005, 33, 246–251 CrossRef.
- C. Turgeon, M. J. Magera, P. Allard, S. Tortorelli, D. Gavrilov, D. Oglesbee, K. Raymond, P. Rinaldo and D. Matern, Clin. Chem., 2008, 54, 657–664 CAS.
- N. Spooner, R. Lad and M. Barfield, Anal. Chem., 2009, 81, 1557–1563 CrossRef CAS.
- O. Y. Al-Dirbashi, L. Fisher, C. McRoberts, K. Siriwardena, M. Geraghty and P. Chakraborty, Clin. Biochem., 2010, 43, 691–693 CrossRef CAS.
- Y. C. Toh, C. Zhang, J. Zhang, Y. M. Khong, S. Chang, V. D. Samper, D. Van Noort, D. W. Hutmacher and H. Yu, Lab Chip, 2007, 7, 302–309 RSC.
- M. W. L. Watson, M. J. Jebrail and A. R. Wheeler, Anal. Chem., 2010, 82, 6680–6686 CrossRef CAS.
- S. L. S. Freire, H. Yang and A. R. Wheeler, Electrophoresis, 2008, 29, 1836–1843 CrossRef CAS.
- J. S. Mellors, V. Gorbounov, R. S. Ramsey and J. M. Ramsey, Anal. Chem., 2008, 80, 6881–6887 CrossRef CAS.
- M. J. Jebrail and A. R. Wheeler, Anal. Chem., 2009, 81, 330–335 CrossRef CAS.
- M. J. Jebrail, A. H. C. Ng, V. Rai, R. Hili, A. K. Yudin and A. R. Wheeler, Angew. Chem., Int. Ed., 2010, 49, 8625–8629 CrossRef CAS.
- N. A. Mousa, M. J. Jebrail, H. Yang, M. Abdelgawad, P. Metalnikov, J. Chen, A. R. Wheeler and R. F. Casper, Sci. Transl. Med., 2009, 1, 1ra2 Search PubMed.
- M. J. Jebrail, V. N. Luk, S. C. C. Shih, R. Fobel, A. H. C. Ng, H. Yang, S. L. S. Freire and A. R. Wheeler, J. Vis. Exp., 2009, 33 DOI:10.3791/1603.
- H. Moon, A. R. Wheeler, R. L. Garrell, J. A. Loo and C. J. Kim, Lab Chip, 2006, 6, 1213–1219 RSC.
- A. R. Wheeler, H. Moon, C. J. Kim, J. A. Loo and R. L. Garrell, Anal. Chem., 2004, 76, 4833–4838 CrossRef CAS.
- H. Yang, V. N. Luk, M. Abeigawad, I. Barbulovic-Nad and A. R. Wheeer, Anal. Chem., 2009, 81, 1061–1067 CrossRef CAS.
- V. N. Luk and A. R. Wheeler, Anal. Chem., 2009, 81, 4524–4530 CrossRef CAS.
- W. C. Nelson, I. Peng, G. A. Lee, J. A. Loo, R. L. Garrell and C. J. Kim, Anal. Chem., 2010, 82, 9932–9937 CrossRef CAS.
- D. Chatterjee, A. Jimmy Ytterberg, S. U. Son, J. A. Loo and R. L. Garrell, Anal. Chem., 2010, 82, 2095–2101 CrossRef CAS.
- F. Lapierre, G. Piret, H. Drobecq, O. Melnyk, Y. Coffinier, V. Thomy and R. Boukherroub, Lab Chip, 2011, 11, 1620–1628 RSC.
- D. H. Chace, S. L. Hillman, D. S. Millington, S. G. Kahler, C. R. Roe and E. W. Naylor, Clin. Chem., 1995, 41, 62–68 CAS.
- T. H. Zytkovicz, E. F. Fitzgerald, D. Marsden, C. A. Larson, V. E. Shih, D. M. Johnson, A. W. Strauss, A. M. Comeau, R. B. Eaton and G. F. Grady, Clin. Chem., 2001, 47, 1945–1955 CAS.
- D. J. Dietzen, P. Rinaldo, R. J. Whitley, W. J. Rhead, W. H. Hannon, U. C. Garg, S. F. Lo and M. J. Bennett, Clin. Chem., 2009, 55, 1615–1626 CAS.
- R. Guthrie and A. Susi, Pediatrics, 1963, 32, 338 CAS.
- N. S. Gerasimova, I. V. Steklova and T. Tuuminen, Clin. Chem., 1989, 35, 2112–2115 CAS.
- S. Tachibana, M. Suzuki and Y. Asano, Anal. Biochem., 2006, 359, 72–78 CrossRef CAS.
- I. Sahai and D. Marsden, Crit. Rev. Clin. Lab. Sci., 2009, 46, 55–82 CrossRef CAS.
- N. S. Green and K. A. Pass, Nat. Rev. Genet., 2005, 6, 147–151 CrossRef CAS.
- S. F. Dobrowolski, R. A. Banas, E. W. Naylor, T. Powdrill and D. Thakkar, Acta Paediatr., 1999, 88, 61–64 CrossRef CAS.
- D. S. Millington, R. Sista, A. Eckhardt, J. Rouse, D. Bali, R. Goldberg, M. Cotten, R. Buckley and V. Pamula, Semin. Perinatol., 2010, 34, 163–169 CrossRef.
- M. Abdelgawad and A. R. Wheeler, Adv. Mater., 2007, 19, 133–137 CrossRef CAS.
- M. Abdelgawad and A. R. Wheeler, Microfluid. Nanofluid., 2008, 4, 349–355 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1lc20524b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2011 |