DNA-templated assembly of droplet-derived PEG microtissues†
Cheri Y.
Li
b,
David K.
Wood
a,
Caroline M.
Hsu
b and
Sangeeta N.
Bhatia
*a
aDivision of Health Sciences and Technology, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA, USA. E-mail: sbhatia@mit.edu; Fax: +1 617 324-0740; Tel: +1 617 324-0221
bDepartment of Chemical Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA, USA
First published on 21st July 2011
Abstract
Patterning multiple cell types is a critical step for engineering functional tissues, but few methods provide three-dimensional positioning at the cellular length scale. Here, we present a “bottom-up” approach for fabricating multicellular tissue constructs that utilizes DNA-templated assembly of 3D cell-laden hydrogel microtissues. A flow focusing-generated emulsion of photopolymerizable prepolymer is used to produce 100 μm monodisperse microtissues at a rate of 100 Hz (105 h−1). Multiple cell types, including suspension and adherently cultured cells, can be encapsulated into the microtissues with high viability (∼97%). We then use a DNA coding scheme to self-assemble microtissues “bottom-up” from a template that is defined using “top-down” techniques. The microtissues are derivatized with single-stranded DNA using a biotin–streptavidin linkage to the polymer network, and are assembled by sequence-specific hybridization onto spotted DNA microarrays. Using orthogonal DNA codes, we achieve multiplexed patterning of multiple microtissue types with high binding efficiency and >90% patterning specificity. Finally, we demonstrate the ability to organize multicomponent constructs composed of epithelial and mesenchymal microtissues while preserving each cell type in a 3D microenvironment. The combination of high throughput microtissue generation with scalable surface-templated assembly offers the potential to dissect mechanisms of cell–cell interaction in three dimensions in healthy and diseased states, as well as provides a framework for templated assembly of larger structures for implantation.
Introduction
The three-dimensional microscale architecture of living tissues provides vital environmental cues, including extracellular matrix, soluble factors and cell–cell interactions.1,2Paracrine and autocrine cell signaling are critical factors guiding tissue development3,4 and maintenance,5,6 and dysregulation of these cues contributes to the pathogenesis of diseased states such as cancer.7–9 Understanding and emulating these cell–cell interactions has been shown to be critical in engineering functional tissues in both 2D10–13 and 3D14–16 systems. In 3D culture, top-down approaches for organizing multiple cell types such as dielectrophoresis,17,18 photopatterning,19,20 and microfabrication21 provide high-precision control over cell placement, but are challenging to scale-up for the assembly of mesoscale tissues.In contrast, bottom-up methods, wherein small tissue building blocks are assembled into larger structures, have potential for creating multicellular constructs in a facile, scalable fashion.22–26 Living tissues are comprised of repeating units on the order of hundreds of microns; therefore, synthetic microtissues comprised of cell-laden hydrogels in this size range27 represent appropriate fundamental building blocks of such bottom-up methods. Synthetic microtissues of this size have been previously assembled in packed-bed reactors22,28 or by hydrophobic/hydrophilic interactions24,29 but without the ability to specify the placement of many different microtissues relative to one another. One potential method for controlled assembly of heterostructures would be to incorporate the specificity of biomolecular interactions with surface templating to direct assembly. This approach could allow for scalable patterning of multiple cell types into arbitrary architectures with high precision.
In this work, we harness the well-characterized molecular recognition capabilities of DNA to achieve rapid templated assembly of multiple microtissue types (Fig. 1). This method is enabled by the high-throughput production of spherical cell-laden microtissues from a microfluidically derived, monodispersed emulsion of a photocurable hydrogel. Cell-laden microtissues are derivatized with single-stranded oligonucleotides and integrated with custom DNA microarray templates. Orthogonal DNA sequences are used to specify the assembly of multiple cell types over large (∼mm) length scales with high capture efficiency. This fusion of “bottom-up” (templated assembly) and “top-down” (microfluidics and robotic spotting) approaches allows for unprecedented control over mesoscale tissue microarchitecture and exemplifies the potential of integrating disparate fabrication strategies.
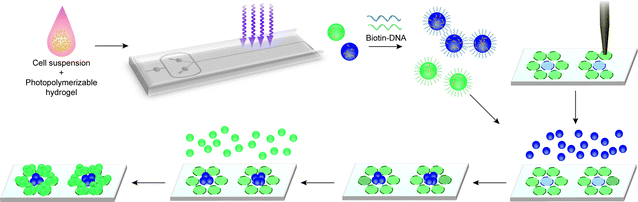 | ||
| Fig. 1 Schematic of microtissue encapsulation, functionalization, and DNA-templated self-assembly. Cells are injected with a photopolymerizable hydrogel prepolymer into a high-throughput microfluidic encapsulation device. Droplets of the cell–prepolymer mixture are exposed to UV on-chip to form streptavidin-containing microtissues which are then coated with 5′-biotin terminated oligonucleotides. Encoded microtissues containing different cell types are seeded on a DNA microarray template which directs the binding of microtissues to specific spots on the templating surface, attaining sequential DNA-templated patterning of cell-laden microtissues. | ||
Materials and methods
Device fabrication
Microfluidic device masters were fabricated on 4 inch silicon wafers using standard photolithographic methods, with a SU-8 2050 photoresist (Microchem, MA) spin coated at 1200 rpm to create 125 μm tall features. Masters were coated with trichloro perfluorooctyl silane (Sigma-Aldrich) for 1 h in a vacuum desiccator prior to casting polydimethylsiloxane (PDMS, Dow Corning) devices. Cured devices with inlet holes made by a 20G dispensing needle (McMaster-Carr) were bonded to glass slides following air plasma treatment. In order to ensure a hydrophobic surface for droplet generation, Aquapel (PPG Industries) was briefly injected into the device and flushed out with nitrogen.Ligand conjugation
Acrylate–PEG–RGDS peptide was prepared as previously described.14 To conjugate streptavidin with acrylate groups, streptavidin was dissolved in 50 mM sodium bicarbonate (pH 8.5) at 0.8 mg ml−1. Amine-reactive acrylate–PEG–SVA (3.4 kDa, Laysan) was added at a 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio and allowed to react with the protein at room temperature for 2 hours. Conjugated acrylate–PEG–streptavidin was purified from unconjugated PEG by washing in PBS with a 30000 MWCO spin filter (Millipore). The acrylate–PEG–streptavidin conjugate was then reconstituted to 38 μM streptavidin in PBS, sterile filtered, and stored at −20 °C.
:1 molar ratio and allowed to react with the protein at room temperature for 2 hours. Conjugated acrylate–PEG–streptavidin was purified from unconjugated PEG by washing in PBS with a 30000 MWCO spin filter (Millipore). The acrylate–PEG–streptavidin conjugate was then reconstituted to 38 μM streptavidin in PBS, sterile filtered, and stored at −20 °C.
Microtissue polymerization
Irgacure-2959 initiator (Ciba) was dissolved at 100 mg ml−1 in n-vinyl pyrrolidinone accelerator (Sigma-Aldrich) to make photoinitiator working solution. The basic 2× concentrated prepolymer solution consists of 20% w/v poly(ethylene glycol) diacrylate (PEG-DA, 20 kDa, Laysan) and 2% v/v of photoinitiator working solution. Additional prepolymer ingredients included 38 μM of acrylate–PEG–streptavidin conjugate, 10 mM acrylate–PEG–RGDS, and/or 1% v/v of fluorescent microspheres (2% solids, Invitrogen) as markers.The final 2× prepolymer solution was injected into the microencapsulation device in parallel with, for cell-free microtissues, a 1:1 diluting stream of PBS. Syringe pumps were used to control the flow rates of the aqueous phases and the oil phase, which consists of the perfluoro polyether, Fomblin (Y-LVAC, Solvay Solexis), and 0–2 w/v% Krytox 157 FSH surfactant (DuPont). Prepolymer droplets were gelled on-chip by exposure to 500 mW cm−2 of 320–390 nm UV light (Omnicure S1000, Exfo) for an approximately one second residence time under typical flow conditions. Cell-free microtissues were collected in handling buffer (PBS with 0.1% v/v Tween-20), allowed to separate from the oil phase, and washed on a 70 μm cell strainer to remove un-polymerized solutes.
Bead hybridization
To stain for the surface-availability of ssDNA bound on microtissues, 1 μm NeutrAvidin biotin-binding beads (yellow-green, Invitrogen) were coated with the complementary 5′-biotin-DNA (IDT). The original suspension of beads (1% solids) was diluted 1:10 with BlockAid blocking solution (Invitrogen), sonicated for 5 minutes, and then incubated with a final concentration of 4 μM 5′-biotin-DNA for 1 hour at room temperature. Beads were then washed three times in PBS by centrifugation at 2000 × g. DNA-functionalized microtissues were incubated overnight on a room-temperature shaker with coated beads resuspended to 0.1% solids in BlockAid.
Microarray spotting
Microarray templates were printed in-house using a contact-deposition DNA spotter (Cartesian Technologies) with a 946MP10 pin (Arrayit). Complementary pairs of single-stranded oligonucleotides used to functionalize microtissues and template their assembly are listed below and consist of a poly-A linker followed by a heterogeneous 20 nucleotide sequence. The 20-nucleotide binding regions of A and A′ are complementary, B and B′, etc. Sequences were modified with 5′-amino groups for microarray spotting, and 5′-biotin groups for microtissue functionalization.| Label | Sequence |
|---|---|
| A | 5′-AAAAAAAAAAGCCGTCGGTTCAGGTCATA-3′ |
| A′ | 5′-AAAAAAAAAAATATGACCTGAACCGACGGC-3′ |
| B | 5′-AAAAAAAAAAAGACACGACACACTGGCTTA-3′ |
| B′ | 5′-AAAAAAAAAATAAGCCAGTGTGTCGTGTCT-3′ |
| C | 5′-AAAAAAAAAAGCCTCATTGAATCATGCCTA-3′ |
| C′ | 5′-AAAAAAAAAATAGGCATGATTCAATGAGGC-3′ |
| D | 5′-AAAAAAAAAATAGCGATAGTAGACGAGTGC-3′ |
| D′ | 5′-AAAAAAAAAAGCACTCGTCTACTATCGCTA-3′ |
5′-Amino oligonucleotides (IDT) for templating were dissolved in 150 mM phosphate buffer (pH 8.5) at concentrations up to 250 μM, and spotted on epoxide coated slides (Corning) at 70% RH. Patterned slides were then incubated for 12 hours in a 75% RH saturated NaCl chamber, blocked for 30 minutes in 50 mM ethanolamine in 0.1 M Tris with 0.1% w/v SDS (pH 9), and rinsed thoroughly with deionized water.
DNA-directed assembly
Microtissues containing PEG–streptavidin were incubated with 1 nmol of 5′-biotin oligonucleotides per 10 μl of packed microtissues for one hour at room temperature or overnight at 4 °C. Un-bound oligonucleotides were removed by washing microtissues on a 70 μm cell strainer or using 100000 MWCO spin filters. Multi-well chambers (ProPlate, Grace Bio-Labs) were assembled over templating slides, and DNA-functionalized microtissues were seeded in a concentrated suspension over the microarray patterns. Microtissues quickly settled into a monolayer, which was visually confirmed under a microscope. Unbound microtissues were washed off the template by gently rinsing the slide with several ml of handling buffer. Capture efficiency was quantified by the average capture density over replicate spots on a slide, divided by the average seeding density of settled microtissues in a 4× microscope field of view. The percent of maximum packing fraction was calculated as the ratio of capture density to the theoretical density of close-packed circles.
Cell culture
J2-3T3 fibroblasts were cultured in Dulbecco's Modified Eagle Medium (DMEM, Invitrogen) with 10% bovine serum (Invitrogen), 10 U ml−1penicillin (Invitrogen), and 10 mg ml−1streptomycin (Invitrogen). TK6 lymphoblasts (suspension culture) and A549 lung adenocarcinoma cells were cultured in RPMI 1640 with L-glutamine (Invitrogen) and 10% fetal bovine serum (Invitrogen), 10 U ml−1penicillin, and 10 mg ml−1streptomycin. All cells were cultured in a 5% CO2 humidified incubator at 37 °C.Cell encapsulation
Prior to encapsulation, adherent cells (J2-3T3 and A549) were detached with 0.25% trypsin–EDTA (Invitrogen). Cell pellets were resuspended at cell densities between 10 × 106cells per ml and 30 × 106cells per ml in an isopycnic injection medium consisting of 20% v/v OptiPrep (Sigma-Aldrich) in serum-free DMEM. Isopycnic cell suspensions were injected into microencapsulation devices in place of the diluting stream of PBS, along with 2× prepolymer solution. Gelled microtissues were collected and handled in culture media. To assess cell viability after 3 hours, microtissues stained with calcein AM (1:200, 1 mg ml−1 in DMSO, Invitrogen) and ethidium homodimer (1:400, 1 mg ml−1 in DMSO, Invitrogen) for 15 minutes at 37 °C. Alternatively, microtissues for DNA-templated assembly were marked with CellTracker Green CMFDA (1:200, 5 mg ml−1 in DMSO, Invitrogen) or CellTracker Blue CMAC (1:200, 5 mg ml−1 in DMSO, Invitrogen) for 1 hour at 37 °C.
Imaging and visualization
Images were acquired with a Nikon Ellipse TE200 inverted fluorescence microscope, a CoolSnap-HQ Digital CCD Camera, and MetaMorph Image Analysis Software. NIH software ImageJ was used to uniformly adjust brightness/contrast, and pseudocolor, merge, and quantify images. Confocal images were acquired with an Olympus FV1000 multiphoton microscope and Olympus Fluoview software. NIS-Elements software was used to pseudocolor and reconstruct maximum intensity, slice, and volume views.Results and discussion
High-throughput microtissue fabrication
One factor restricting the application of bottom-up assembly to tissue engineering has been the low throughput of typical microtissue fabrication approaches to date, many of which are batch processes.22,27,30 We first sought to design a microfluidic chip to rapidly produce uniform microtissues. Droplets generated by flow focusing of aqueous/oil phases are monodisperse and amenable to photopolymerization.31 Thus, we fabricated a device to shear photopolymerizable poly(ethylene glycol) diacrylate (PEG-DA) prepolymer containing cells into droplets in oil for downstream gelation by UV-light (Fig. 2a). Concentrated pre-polymer was injected into the microencapsulation device as a separate stream from the cell suspension (PBS for cell-free microtissues), where the two aqueous streams were designed to meet before reaching a flow-focusing junction (Fig. 2b and Movie S1†). With a 60 μm nozzle, shear forces were sufficient to disperse the aqueous combination into droplets that passed through a corrugated serpentine channel32 to thoroughly mix the cell–prepolymer solution (Fig. 2c). The droplets were then polymerized by UV irradiation for 1 second during transport to the outlet. Resulting microtissues were uniformly spherical and monodisperse (Fig. 2d). We observed that by adjusting aqueous vs. oil phase flow rates (Fig. 2e) and oil-phase surfactant concentrations (Fig. 2f), we could finely control droplet diameter, and hence microtissue size between 30 and 120 μm.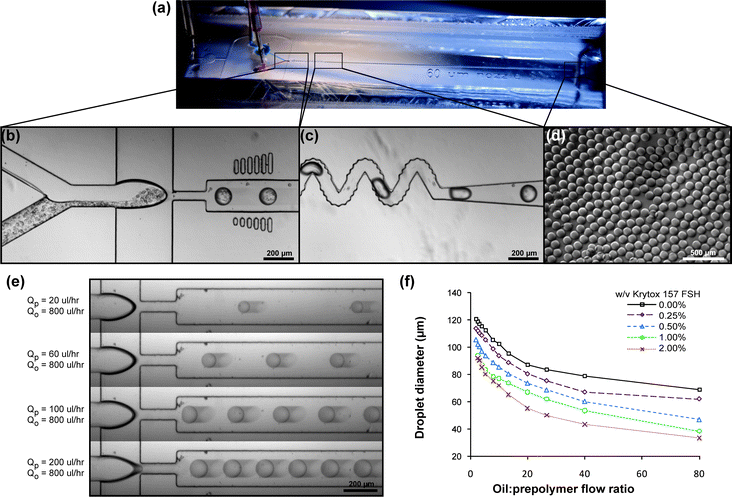 | ||
| Fig. 2 Microencapsulation device. (a) Overview of device showing two aqueous input streams (red and blue) dispersed by shear flow from an oil stream into droplets that mix (purple) and travel down the UV-exposure channel. (b) Prepolymer (2× concentrated) and a cell suspension meet and flow into a 60 μm droplet generating nozzle. Vertical columns on either side of the channel provide visual references (50–100 μm below, 100–150 μm above) for real-time adjustment of the droplet size. See ESI† for a movie. (c) Droplets pass through a bumpy serpentine mixer section to thoroughly disperse cells in prepolymer and are then polymerized by UV irradiation from a curing lamp. (d) Microtissues collected from the device (6000 min−1) are spherical and monodisperse. (e) Microtissue size is controlled by the relative flow rates of the combined aqueous phase (QP) and the continuous oil phase (QO), and increases with prepolymer:oil flow ratio. (f) Adding small amounts of Krytox 157 FSH fluorosurfactant into the oil decreased droplet diameter at all flow ratios, allowing higher prepolymer flow rates for a given microtissue size. | ||
At a typical prepolymer flow rate of 200 μl h−1, our device was capable of achieving a production throughput of 6000 microtissues per min (∼105 h−1), two orders of magnitude faster than other continuous systems such as stop-flow lithography33 (∼103 h−1) or batch fabrication processes.27 Microtissue fabrication by microfluidic droplet photopolymerization provides precise control over microtissue shape and size, whereas photolithographic27 and molding22,24 techniques do not produce spherical gels and can suffer from resolution limits. Planar microtissue surfaces tend to adhere non-specifically to hydrophilic surfaces due to the high water content (>90%)34 of the hydrogel material, whereas the low contact area of spherical microtissues reduces capillary adhesion during both handling and assembly. Droplet-based gels have previously been made using agarose35 or alginate;36 here, we chose a PEG hydrogel material for its biocompatibility and biochemical versatility. PEG–diacrylate hydrogels have high water content, are non-immunogenic and resistant to protein adsorption, and can be easily customized with degradable linkages, adhesive ligands, and other biologically or chemically active factors.37
Microtissue functionalization with surface-encoding DNA
Having established a method to uniformly produce microtissues, we next sought to modify our microtissues with streptavidin for binding biotinylated DNA. To accomplish this, streptavidin was incubated with amine-reactive acrylate–PEG–SVA (3.4 kDa). Following purification, the acrylate-decorated streptavidin was then mixed into the prepolymer and covalently bound into the acrylate–PEG–acrylate hydrogel network during gelation by acrylate polymerization (Fig. 3a). Cell-free PEG–SA microtissues containing conjugated acrylate–PEG–streptavidin were stained to verify biotin-binding capacity using biotin-4-fluorescein. We also confirmed the surface-availability of streptavidin with an anti-streptavidin antibody, which was size restricted to only the surface of the microtissue (∼7 nm mesh size34). Both biotin fluorescence and antibody staining intensities increased with the volumetric concentration of conjugated streptavidin (Fig. 3b).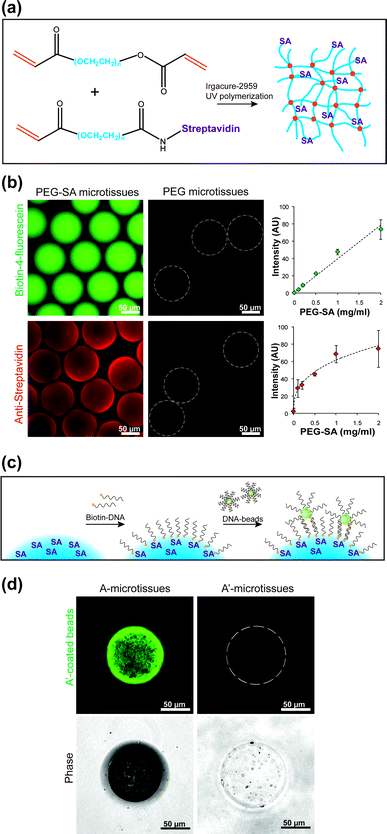 | ||
| Fig. 3 Microtissue functionalization. (a) The primary hydrogel component, acrylate–PEG20k–acrylate macromonomer, was mixed with conjugated acrylate–PEG–streptavidin (0–2 mg ml−1) before photo-initiated free radical polymerization, forming a hydrogel network that is decorated with pendant streptavidin proteins. (b) PEG–streptavidin microtissues stained with biotin-4-fluorescein, which can freely diffuse through the hydrogel network, and anti-streptavidin IgG, which is restricted to the surface of the microtissues. The intensity of biotin-4-fluorescein staining increased linearly with the bulk concentration of covalently bound streptavidin, while antibody stains for surface concentration increased only as a power of bulk concentration. (c) PEG–SA microtissues are further functionalized with biotin-ssDNA. The availability of this ssDNA to hybridize with a templating surface was tested using 1 μm fluorescent beads coated with DNA. (d) Microtissues with the appropriate complementary sequence were coated with hybridized beads. No beads hybridized to control-sequence microtissues, which remained dark in the green channel and showed only encapsulated marker beads in the phase image. | ||
With streptavidin incorporated into the hydrogel network, we were able to encode the microtissues post-polymerization with 5′-biotin terminated oligonucleotides (Fig. 3c). Streptavidin–biotin based DNA-functionalization of microtissues is simple, modular, and cytocompatible. Post-polymerization encoding of microtissues with biotin-DNA avoids UV damage that would occur by pre-mixing acrylated-DNA into the prepolymer,38,39 and allows the same batch of microtissues to be labeled after culture in various conditions. Other bioconjugation methods exist to modify hydrogel networks post-encapsulation, such as maleimide or NHS chemistries40 but often require reaction conditions that are incompatible with maintaining the viability of encapsulated cells. To ensure that DNA bound to microtissues using the streptavidin–biotin interaction was available to hybridize with DNA displayed on a surface, we incubated DNA-encoded microtissues with 1 μm polystyrene beads coated with the complementary oligonucleotide (Fig. 3c). After washing to remove non-specifically bound material, microtissues encoded with the complementary sequence were thoroughly coated with beads visible as bright, punctate spots (Fig. 3d). Conversely, beads did not specifically hybridize to control microtissues (Fig. 3d). In order to maximize bead-microtissue hybridization, we investigated conjugating acrylate–PEG–SVA to streptavidin at several molar ratios (Fig. S1†). As expected, microtissues incorporating streptavidin with few acrylate pendants (10:1 molar ratio, mobility shift assay) did not promote bead hybridization as effectively as streptavidin modified with a higher number of acrylate groups (25:1 to 50:1 molar ratio), which was used for all further studies. Gels incorporating over-decorated streptavidin (1000:1 molar ratio) were also not as efficient in mediating bead-microtissue hybridization, suggesting that overmodification and/or steric hindrance plays an important role in DNA-binding capacity.
Binding efficiency and specificity of DNA-templated assembly
Having shown that cell-free microtissues can be coated with DNA and hybridize specifically to complementary beads, we next investigated the potential of microtissue assembly into mesoscale patterns determined by an encoded template. To create such a template, we spotted increasing concentrations of DNA (sequence A′) onto a functionalized glass slide using conventional microarray technology. DNA-functionalized microtissues (A; containing green marker beads) were allowed to settle onto microarray slides from suspension, at which time non-hybridized microtissues were gently washed off the slide. The number of microtissues bound to templating array spots increased with higher spotting concentrations of templating ssDNA (Fig. 4a), plateauing at 250 μM, an order of magnitude higher than typical epoxy-silane based microarray spotting concentrations. Spots were fully covered by microtissues at this highest DNA density. To determine the capture efficiency, we seeded microtissues at varying densities (microtissues per mm2, Fig. 4b). At contact-limited (hexagonally close-packed) seeding concentrations, we achieved 100% capture efficiency, indicating that if a microtissue settled onto a complementary spot, hybridization and binding would occur.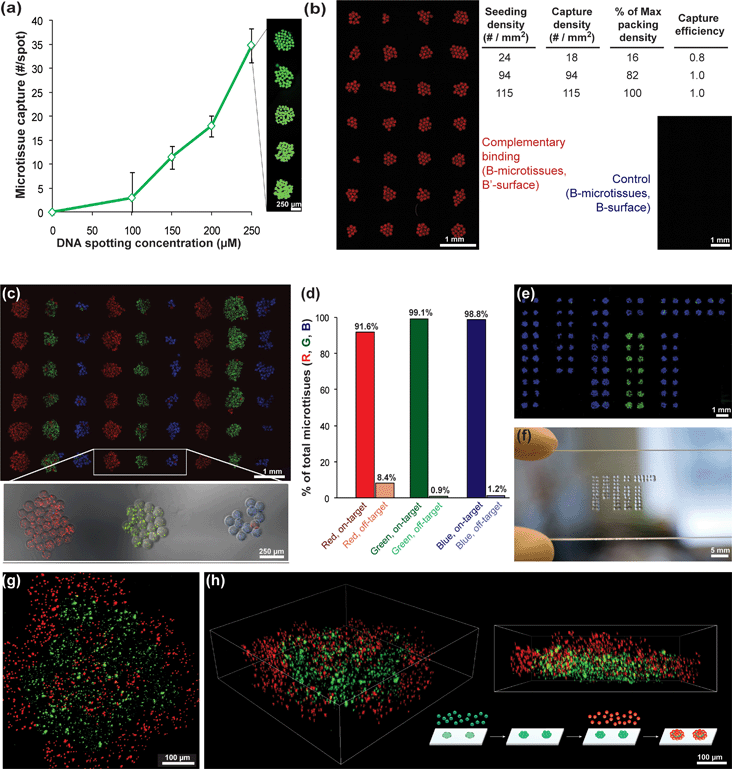 | ||
| Fig. 4 Capture efficiency and specificity of DNA-directed microtissue assembly. (a) The number of DNA-functionalized microtissues containing fluorescent beads as markers captured on microarray spots with increasing spotting concentration of complementary oligonucleotide. (b) Quantified assembly results from microtissues seeded over an array of complementary spots at low, medium (shown on the left), and high (close-packed) % surface coverage. Control arrays of non-complementary spots remained blank. (c) Three-color (RGB) microtissue assembly using a set of orthogonal oligonucleotide sequences: B (red), C (green), and D (blue). Microtissues contain encapsulated marker beads. (d) Quantified percentages of microtissues on target spots (1 column) vs. off-target spots (2 columns). (e) MIT logo assembled in microtissues of C (green) and D (blue), and (f) photograph of templating slide illustrating scale of assembled microtissue patterns. (g) Maximum intensity projection and (h) volume reconstructions from multi-photon scans of the 3D microtissue structure formed by templating a first layer of microtissues (B, green) and then assembling a second layer of complementary microtissues (B′, red). | ||
Similar efficiencies have been observed during the DNA-templated assembly of materials ranging in scale from molecules to nanoparticles to single cells.23,41–46 Until now, DNA-templated assembly has not been extended to larger units such as microtissues (100 μm), which present unique challenges in mass transport.47 At these mesoscopic scales, gravity and friction become important factors in the ability of DNA-coated surfaces to sufficiently interact. During washing steps, stronger viscous drag forces on the microtissues necessitate a large number of hybridization bonds between the microtissues and templating surface to overcome microtissue removal. Here, to compensate for microtissue size, we optimize microtissue DNA functionalization and template spotting to achieve high DNA surface densities, enabling the first demonstration of large structure DNA-templated assembly.
During our assembly process, minimal microtissue binding was observed between spots and on non-complementary templating spots (Fig. 4b), which was largely made possible by our control over microtissue shape. This low background binding allowed us to sequentially pattern multiple microtissue types, each encoded with an orthogonal oligonucleotide sequence, with over 90% specificity (Fig. 4c and d) and across large areas in under 15 minutes (Fig. 4e and f). Furthermore, we were able to build 3D structures (Fig. 4g and h) by filling template spots (B′) with a layer of microtissues (B), and then seeding a second layer of complementary microtissues (B′) that bind on and around microtissues in the first layer. Together, these experiments demonstrate the ease of achieving organizational control at macroscopic length scales by microtissue assembly.
DNA-templated assembly of multicellular tissue constructs
In order to apply DNA-templated patterning to the assembly of multicellular constructs, we next focused on encapsulating cells into uniform and highly viable cell-laden microtissues. To improve the consistency of cell encapsulation (Fig. S2†), we increased the specific gravity of our cell suspensions to prevent cell settling during injection. We chose a density gradient medium (OptiPrep), based on an iodinated small molecule, that increases specific gravity without affecting viscosity or cross-linked hydrogel network density, and easily diffuses out of the polymerized microtissues. With these changes, we attained cell encapsulation matching a Poisson distribution (Fig. 5b). In addition, we replaced the hydrocarbon oil phase with an oxygen-permeable fluorocarbon oil (Fomblin) to allow immediate quenching of excess free radicals post-UV exposure.48 Notably, using fluorocarbon oil, cells were able to tolerate a wide range of total UV exposures (mJ cm−2) while maintaining >90% viability (Fig. 5c). As a result of these changes, several adhesive and suspension cell lines, including adherent mesenchymal (fibroblasts), nonadherent mesenchymal (lymphoblasts) and adherent epithelial (adenocarcinoma), were uniformly encapsulated into microtissues with consistently high viability (Fig. 5a). Variations in average viability between cell types (e.g.J2-3T3vs.TK6) could be due to a number of cell type differences including susceptibility to DNA damage.49 For cell lines sensitive to UV, photoinitiators in the visible-light range could be substituted into our material system.50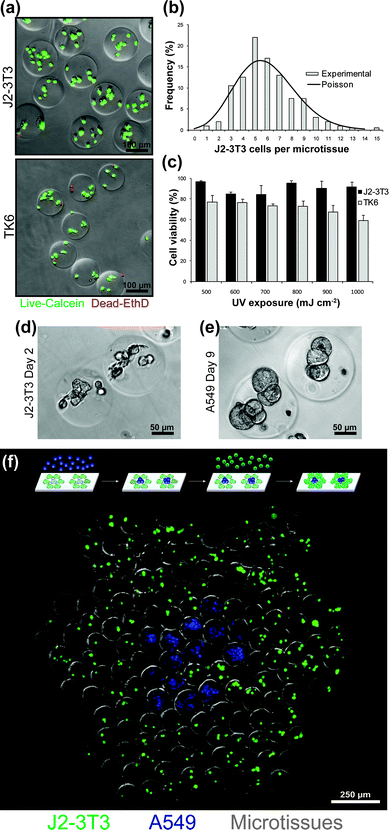 | ||
| Fig. 5 Cell encapsulation and microtissue culture. (a) Rat fibroblast (J2-3T3) and human lymphoblast (TK6) cell lines uniformly encapsulated within microtissues and stained for viability. (b) Histogram of J2-3T3 distribution within microtissues and comparison to optimal Poisson statistics. (c) Viability of J2-3T3 and TK6 cells three hours post-encapsulation at increasing UV overexposure past the minimum intensity required to fully polymerize microtissues. (d) J2-3T3 cells attached and spread within microtissues decorated with RGDS peptides. (e) Human lung adenocarcinoma (A549) cells aggregated to form multicellular tumor spheroids within microtissues. (f) Microtissues encapsulating either J2-3T3 (CellTracker Green) or A549 cells (CellTracker Blue) were self-assembled into composite hexagonal clusters. | ||
These are many advantages associated with patterning cellular microtissues rather than single cells.43,44 Firstly, cells can be encapsulated in a modular scaffold with customized ECM molecules (e.g. RGDS) to promote certain phenotypes. As an example, we added acrylated RGDS peptide to the prepolymer during fibroblast encapsulation. By day 2 post-encapsulation, fibroblasts began spreading within these adhesive microtissues (Fig. 5d and S3†). Secondly, microtissues containing one cell type can be first cultured separately to stabilize homotypic interactions before they are self-assembled with other microtissues to activate heterotypic interactions. For instance, when cultured for several days, adenocarcinoma cells encapsulated from a single-cell suspension formed multicellular spheroids (Fig. 5e). In addition, encoding DNA is bound to the hydrogel scaffold rather than directly onto the cell membrane,43,44 where covalently bound ligands may be susceptible to recycling or may potentially modify cell function. Encoded microtissues can remain in assembled patterns for an extended period of time without additional measures for immobilization (e.g. embedding in agarose23), and then removed for further culture, isolation, and biochemical analysis.27DNA provides a way for programmed detachment via dehybridization (e.g. competitive binding with free ssDNA) or cleavage (e.g. restriction enzymes).43 Alternatively, patterned microtissues could be stabilized into a contiguous tissue by a secondary hydrogel polymerization29 or cell adhesion between microtissues to form 3D sheets for implantation (Fig. S4†).
Finally, to demonstrate DNA-templated positioning of microtissues containing distinct cell types into pre-defined patterns, we encapsulated adenocarcinoma cells (blue) and fibroblasts (green) into separate microtissues and encoded them with orthogonal DNA sequences (C and D respectively). These microtissues were then seeded onto an array printed with hexagonal clusters of complementary DNA (C′ centered within 6 spots of D′), forming co-cultures of the two cell types representative of a tumor nodule surrounded by stromal cells (Fig. 5f). Multicellular constructs patterned using this method could be relevant model systems for studying cancer–stroma interactions in 3D. Notably, although DNA-templated microtissues are patterned on a 2D template, cells are encapsulated and respond to a locally 3D microenvironment, e.g. developing into tumor spheroids (Fig. 5e) rather than growing as a 2D monolayer.16 Heterotypic signaling from stromal cells has been shown to contribute to tumor invasion and metastasis.9 The combination of precise spatial control, similar to that achieved in 2D,10 but with a 3D environment, will be critical toward elucidating such cell signaling mechanisms.
Conclusions
We have presented a method to organize multiple cell types within a 3D microenvironment that integrates the top-down patterning of a DNA microarray template with the bottom-up assembly of DNA-encoded, cell-laden microtissues. This is the first demonstration of microtissue assembly that is directed by specific biomolecular interactions. The speed and scalability of the assembly process are compatible with DNA templates that can be fabricated by other top-down techniques, such as microfabrication and micro-contact printing, for a diverse range of features and patterning resolution. The programmable molecular interaction of DNA to direct assembly has the potential to be extended to even larger sets of encoding sequences to create more complex heterogeneous structures. The ability to precisely control cell–cell interactions (e.g. cancer–stromal cell, hepatic–nonparenchymal cell) via microfluidic cell encapsulation and DNA-templated microtissue assembly provides a unique opportunity to increase our fundamental understanding of complex diseases or to construct highly functional tissue-engineered implants.Acknowledgements
We thank Dr Alice A. Chen and Dr Gabriel A. Kwong for helpful discussions and editing the manuscript, and Dr Bevin P. Engelward (MIT) for providing the TK6 cell line. We acknowledge the Swanson Biotechnology Center (SBC) of the Koch Institute at MIT for the use of microscopy facilities. This work was supported by the NIH (grants R01-DK56966, R01-EB008396) and Stand Up To Cancer (SU2C). C.Y.L. acknowledges support from the NDSEG and NSF Graduate Fellowships. D.K.W. acknowledges support from the Kirschstein NRSA Fellowship. S.N.B. is an HHMI Investigator.Notes and references
- L. G. Griffith and M. A. Swartz, Nat. Rev. Mol. Cell Biol., 2006, 7, 211–224 CrossRef CAS.
- A. Khademhosseini, R. Langer, J. Borenstein and J. P. Vacanti, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 2480–2487 CrossRef CAS.
- J. Jung, M. Zheng, M. Goldfarb and K. S. Zaret, Science, 1999, 284, 1998–2003 CrossRef CAS.
- O. Cleaver and D. A. Melton, Nat. Med., 2003, 9, 661–668 CrossRef CAS.
- S. N. Bhatia, U. J. Balis, M. L. Yarmush and M. Toner, FASEB J., 1999, 13, 1883–1900 CAS.
- S. J. Morrison and A. C. Spradling, Cell, 2008, 132, 598–611 CrossRef CAS.
- N. A. Bhowmick, E. G. Neilson and H. L. Moses, Nature, 2004, 432, 332–337 CrossRef CAS.
- S. L. Friedman, J. Biol. Chem., 2000, 275, 2247–2250 CrossRef CAS.
- D. Hanahan and R. A. Weinberg, Cell, 2011, 144, 646–674 CrossRef CAS.
- E. E. Hui and S. N. Bhatia, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 5722–5726 CrossRef CAS.
- S. March, E. E. Hui, G. H. Underhill, S. Khetani and S. N. Bhatia, Hepatology, 2009, 50, 920–928 CrossRef CAS.
- D. Huh, B. D. Matthews, A. Mammoto, M. Montoya-Zavala, H. Y. Hsin and D. E. Ingber, Science, 2010, 328, 1662–1668 CrossRef CAS.
- M. A. LaBarge, C. M. Nelson, R. Villadsen, A. Fridriksdottir, J. R. Ruth, M. R. Stampfer, O. W. Petersen and M. J. Bissell, Integr. Biol., 2009, 1, 70–79 RSC.
- G. H. Underhill, A. A. Chen, D. R. Albrecht and S. N. Bhatia, Biomaterials, 2007, 28, 256–270 CrossRef CAS.
- K. R. Stevens, K. L. Kreutziger, S. K. Dupras, F. S. Korte, M. Regnier, V. Muskheli, M. B. Nourse, K. Bendixen, H. Reinecke and C. E. Murry, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 16568–16573 CrossRef CAS.
- C. Fischbach, R. Chen, T. Matsumoto, T. Schmelzle, J. S. Brugge, P. J. Polverini and D. J. Mooney, Nat. Methods, 2007, 4, 855–860 CrossRef CAS.
- D. R. Albrecht, V. L. Tsang, R. L. Sah and S. N. Bhatia, Lab Chip, 2005, 5, 111–118 RSC.
- D. R. Albrecht, G. H. Underhill, T. B. Wassermann, R. L. Sah and S. N. Bhatia, Nat. Methods, 2006, 3, 369–375 CrossRef CAS.
- V. Liu Tsang, A. A. Chen, L. M. Cho, K. D. Jadin, R. L. Sah, S. DeLong, J. L. West and S. N. Bhatia, FASEB J., 2007, 21, 790–801 CrossRef.
- V. Chan, P. Zorlutuna, J. H. Jeong, H. Kong and R. Bashir, Lab Chip, 2010, 10, 2062–2070 RSC.
- W. Tan and T. A. Desai, Biomaterials, 2004, 25, 1355–1364 CrossRef CAS.
- A. P. McGuigan and M. V. Sefton, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11461–11466 CrossRef CAS.
- Z. J. Gartner and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4606–4610 CrossRef CAS.
- Y. Du, E. Lo, S. Ali and A. Khademhosseini, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9522–9527 CrossRef CAS.
- B. Guillotin and F. Guillemot, Trends Biotechnol., 2011, 29, 183–190 CrossRef CAS.
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS.
- A. A. Chen, G. H. Underhill and S. N. Bhatia, Integr. Biol., 2010, 2, 517–527 RSC.
- D. A. Bruzewicz, A. P. McGuigan and G. M. Whitesides, Lab Chip, 2008, 8, 663–671 RSC.
- Y. Du, M. Ghodousi, E. Lo, M. K. Vidula, O. Emiroglu and A. Khademhosseini, Biotechnol. Bioeng., 2010, 105, 655–662 CrossRef CAS.
- A. Khademhosseini and R. Langer, Biomaterials, 2007, 28, 5087–5092 CrossRef CAS.
- S. Y. Teh, R. Lin, L. H. Hung and A. P. Lee, Lab Chip, 2008, 8, 198–220 RSC.
- A. Liau, R. Karnik, A. Majumdar and J. H. Cate, Anal. Chem., 2005, 77, 7618–7625 CrossRef CAS.
- P. Panda, S. Ali, E. Lo, B. G. Chung, T. A. Hatton, A. Khademhosseini and P. S. Doyle, Lab Chip, 2008, 8, 1056–1061 RSC.
- G. M. Cruise, D. S. Scharp and J. A. Hubbell, Biomaterials, 1998, 19, 1287–1294 CrossRef CAS.
- R. Novak, Y. Zeng, J. Shuga, G. Venugopalan, D. A. Fletcher, M. T. Smith and R. A. Mathies, Angew. Chem., Int. Ed., 2011, 50, 390–395 CrossRef CAS.
- L. F. Yu, M. C. W. Chen and K. C. Cheung, Lab Chip, 2010, 10, 2424–2432 RSC.
- E. Jabbari, Curr. Opin. Biotechnol., 2011 DOI:10.1016/j.copbio.2011.01.003.
- D. J. Quick and K. S. Anseth, J. Controlled Release, 2004, 96, 341–351 CrossRef CAS.
- D. C. Pregibon, M. Toner and P. S. Doyle, Science, 2007, 315, 1393–1396 CrossRef CAS.
- G. T. Hermanson, Bioconjugate Techniques, Academic Press, San Diego, 1996 Search PubMed.
- Z. J. Gartner and D. R. Liu, J. Am. Chem. Soc., 2001, 123, 6961–6963 CrossRef CAS.
- C. A. Mirkin, R. L. Letsinger, R. C. Mucic and J. J. Storhoff, Nature, 1996, 382, 607–609 CrossRef CAS.
- G. A. Kwong, C. G. Radu, K. Hwang, C. J. Shu, C. Ma, R. C. Koya, B. Comin-Anduix, S. R. Hadrup, R. C. Bailey, O. N. Witte, T. N. Schumacher, A. Ribas and J. R. Heath, J. Am. Chem. Soc., 2009, 131, 9695–9703 CrossRef CAS.
- E. S. Douglas, R. A. Chandra, C. R. Bertozzi, R. A. Mathies and M. B. Francis, Lab Chip, 2007, 7, 1442–1448 RSC.
- M. P. Valignat, O. Theodoly, J. C. Crocker, W. B. Russel and P. M. Chaikin, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 4225–4229 CrossRef CAS.
- D. Nykypanchuk, M. M. Maye, D. van der Lelie and O. Gang, Nature, 2008, 451, 549–552 CrossRef CAS.
- G. M. Whitesides and M. Boncheva, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4769–4774 CrossRef CAS.
- D. Dendukuri, P. Panda, R. Haghgooie, J. M. Kim, T. A. Hatton and P. S. Doyle, Macromolecules, 2008, 41, 8547–8556 CrossRef CAS.
- R. C. Fry, J. P. Svensson, C. Valiathan, E. Wang, B. J. Hogan, S. Bhattacharya, J. M. Bugni, C. A. Whittaker and L. D. Samson, Genes Dev., 2008, 22, 2621–2626 CrossRef CAS.
- S. J. Bryant, C. R. Nuttelman and K. S. Anseth, J. Biomater. Sci., Polym. Ed., 2000, 11, 439–457 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1lc20318e |
| This journal is © The Royal Society of Chemistry 2011 |