A lab-on-a-chip device for rapid identification of avian influenza viral RNA by solid-phase PCR†
Yi
Sun
a,
Raghuram
Dhumpa
b,
Dang Duong
Bang
b,
Jonas
Høgberg
b,
Kurt
Handberg
c and
Anders
Wolff
*a
aDTU Nanotech, Department of Micro- and Nanotechnology, Technical University of Denmark (DTU), Ørsteds Plads, DK-2800 Kgs, Lyngby, Denmark. E-mail: anders.wolff@nanotech.dtu.dk
bDTU Vet, Laboratory of Applied Micro-Nanotechnology, National Veterinary Institute, Technical University of Denmark, Hangøvej 2, DK-8200, Aarhus, Denmark
cLaboratory of Avian Influenza Virus, National Veterinary Institute, Technical University of Denmark (DTU), Hangøvej 2, DK-8200, Aarhus, Denmark
First published on 2nd March 2011
Abstract
The endemic of Avian Influenza Virus (AIV) in Asia and epizootics in some European regions have caused serious economic losses. Multiplex reverse-transcriptase (RT) PCR has been developed to detect and subtype AIV. However, the number of targets that can be amplified in a single run is limited because of uncontrollable primer–primer interferences. In this paper, we describe a lab-on-a-chip device for fast AIV screening by integrating DNA microarray-based solid-phase PCR on a microfluidic chip. A simple UV cross-linking method was used to immobilize the DNA probes on unmodified glass surface, which makes it convenient to integrate microarray with microfluidics. This solid-phase RT-PCR method combined RT amplification of extracted RNA in the liquid phase and species-specific nested PCR on the solid phase. Using the developed approach, AIV viruses and their subtypes were unambiguously identified by the distinct patterns of amplification products. The whole process was reduced to less than 1 hour and the sample volume used in the microfluidic chip was at least 10 times less than in the literature. By spatially separating the primers, highly multiplexed amplification can be performed in solid-phase PCR. Moreover, multiplex PCR and sequence detection were done in one step, which greatly simplified the assay and reduced the processing time. Furthermore, by incorporating the microarray into a microchamber-based PCR chip, the sample and the reagent consumption were greatly reduced, and the problems of bubble formation and solution evaporation were effectively prevented. This microarray-based PCR microchip can be widely employed for virus detection and effective surveillance in wild avian and in poultry productions.
Introduction
Avian influenza virus (AIV) or “bird flu” is an infectious disease caused by Influenza A virus. Based on antigenic differences in the hemagglutinin (HA) and neuraminidase (NA) proteins, AIV is divided into 16 HA and 9 NA subtypes.1 The AIV is classified as either highly pathogenic avian influenza (HPAI) or low pathogenic avian influenza (LPAI) based on the ability to cause disease and mortality.2Reverse transcriptase PCR (RT-PCR) has been introduced as the most sensitive method for detection and pathotyping of AIV.3 Identification and subtyping of HPAI virus by RT-PCR often involve multi-step amplifications, which make the assay complicated and time-consuming.4 Multiplex RT-PCR was then developed by targeting multiple genes at once.5,6 Detection and subtyping of AIV were gained from a single test run that otherwise would require several tests to perform. However, analysis using multiplex PCR is constrained by two main limitations: the formation of primer–dimers due to the uncontrollable primer–primer interferences and preferential amplification of one target sequence over another.7 Moreover, identification of multiplex PCR amplicons typically requires a secondary method, such as slab gel, melting curve, microarray hybridization or capillary electrophoresis,8 for size separation or sequence verification.To minimize the interferences in multiplex PCR, a technique called solid-phase PCR has been developed by grafting one or both primers on a solid support while keeping other PCR components in the liquid phase.9,10 The primers can be immobilized on microtiter plates,11 flat surfaces,12 or microbeads.13 In each case, enzymatic extension of the primer produces a tethered amplicon. Solid-phase PCR on DNA microarray has become increasingly popular as highly multiplexed amplification could occur in a miniature space with hundreds or thousands of discrete immobilized primers.14–16 The multiplexing capabilities are produced by spatially encoding the array, in which each spot on the array is used to target a specific analyte. As the primers are spatially separated, the interferences between various primers are minimized, and the number of different targets able to be amplified in a single PCR reaction is greatly improved. The technique has been applied for a variety of applications, including diagnosis of infectious diseases16 and single-nucleotide polymorphism (SNP) analysis.17 Solid-phase PCR has shown its advantages of high throughput, ease of operation and specific detection. However, protocols for attaching probes on the glass surface were complicated and the method has never been applied for a fragile material like viral RNA.
In conventional solid-phase PCR, a self-adhesive frame7 or a glass coverslip18 is mounted on the oligonucleotide array to seal the reaction mixture. In both cases, a large amount of PCR samples are required. Moreover, air bubbles are easily trapped in the aqueous phase and the solutions are subject to evaporation, which could adversely affect the efficiency of solid-phase PCR. The problems can be addressed by using lab-on-a-chip technology. Since the concept of lab-on-a-chip was proposed, PCR microchips have been rapidly developed.19–21 The microfabricated chips have characteristic dimensions in the order of µm, and PCR samples could be processed in a much shorter time with a high degree of fluid control. With the advances of microfabrication techniques, DNA microarrays have been incorporated into PCR microchips to detect PCR products by hybridization where unique DNA sequences are recognized through base pairing.22–24 However, PCR microchips with microarrays for solid-phase PCR have seldom been reported.
In this paper, we describe a lab-on-a-chip device for rapid AIV detection by integrating DNA microarray-based solid-phase PCR on microchip. A 2 µl hybrid PDMS–glass chamber-based PCR microchip was fabricated and a DNA microarray for interrogating different influenza types was integrated in the chip. A very simple UV cross-linking procedure was used to immobilize TC-tagged oligonucleotide probes on the glass surface without any surface modification. The immobilization method is fast and thermally stable, making it a convenient to integrate microarrays into microfluidic systems. The fragile viral RNA was used as templates. The on-chip solid-phase PCR combined reverse-transcription amplification of RNA extract in the liquid phase and the simultaneous sequence-specific enzymatic extension of probes on the solid phase. The amplicons remained covalently bound to the glass and could be directly detected after PCR by fluorescence scanning. Using the developed approach, AIV viruses and their subtypes were unambiguously identified by their distinct patterns. Parameters such as the ratio of aqueous primers and solid probe densities were optimized for the system to improve efficiency of solid-phase amplification. This is the first demonstration of incorporation of solid-phase PCR into PCR microchip. Highly multiplex PCR can be performed on chip for viral classification attributed to the high-throughput capabilities of microarrays. Amplification and sequence detection are done during solid-phase PCR in a single, valveless chamber, which eliminates the need for post-PCR step and greatly simplifies the assay. Moreover, despite the sample volume being at least ten times less than that in the literature, ten-fold increase in detection sensitivity has been achieved when compared to both solution-based multiplex PCR and standard microarray method. The device can be considered as a versatile tool for identification of other genetic targets or it can also be integrated with other functionalities such as sample preparation to develop an automated and fully integrated sample-in–answer-out system.
Experimental
Design of on-chip solid-phase PCR
Previous studies revealed that solid-phase PCR is generally less efficient than conventional solution-based reactions.9,10 This inefficiency may be attributed to a low primer concentration, its inability to diffuse through solution or inefficient enzymatic extension. Huber et al. proposed a method to allow the reaction to proceed in the liquid phase and on the surface of the solid phase simultaneously, which dramatically increased the product yield on the solid support.25 The liquid-phase amplification produced DNA templates to initiate amplification on the solid phase and the accumulation of targets in a solution also served to accelerate the solid-phase amplification. In this study, we extended this strategy and developed an approach where RT-PCR occurs in the liquid solution and nested amplification occurs on the microarray elements with specific oligonucleotide probes. For the liquid-phase PCR, three pairs of primers were designed to amplify AIV conserved region on the matrix (M) gene and H5 and H7 regions on the HA gene. For the solid-phase amplification, three nested probes were spotted on the glass surface to target the M, H5 and H7 templates generated in the liquid. Though a multitude of unbound primers were added to the liquid phase, the potential for primer interference is counteracted by the superior specificity of nested amplification on the solid surface.The working model for on-chip solid-phase PCR is illustrated in Fig. 1. The viral RNA and RT-PCR mixture is pumped into the chamber. Subsequently, the chamber is sealed and RT-PCR is performed by temperature cycling (Fig. 1a). The RNA is first reverse transcribed to complementary DNA (cDNA) in the liquid (Fig. 1b). cDNA is then amplified with freely moving PCR primers (Fig. 1c). Simultaneously, the newly amplified PCR amplicons in the liquid phase interact with the nested probes immobilized on the solid phase (Fig. 1d). If the probes match the bases in the template DNA, they are extended by the polymerase (Fig. 1e). In the next cycle, the extended probe served as templates for the second strand elongation primed by the forward primers in the liquid phase (Fig. 1f), thus generating new templates for the solid-phase amplification (Fig. 1g). After the reaction, PCR products remain attached to the glass slide through covalent binding and could be directly visualized as the forward primers in the liquid phase were labelled with Cy5 dyes. Detection of influenza virus A and simultaneous identification of two highly pathogenic AIV subtypes H5 and H7 are achieved by examining the specific patterns of the microarray.
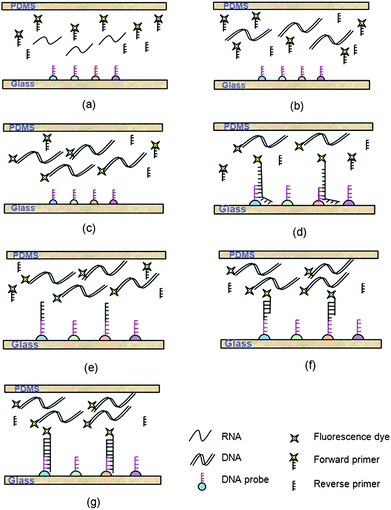 | ||
| Fig. 1 The concept of on-chip solid-phase PCR for rapid AIV detection. (a) The viral RNA and RT-PCR mixture is pumped into the chamber. (b) The RNA is reverse transcribed to cDNA in the liquid. (c) cDNA is amplified with freely moving PCR primers. (d) The newly amplified PCR amplicons in the liquid phase interact with the nested probes immobilized on the solid support. (e) The matched probes are extended by the polymerase. (f) In the next cycle, the forward primers in the liquid phase are annealed to the extended probes. (g) Complementary strands are generated and serve as new templates for the solid-phase amplification. After the reaction, PCR products remain attached to the glass slide through covalent binding and could be directly visualized as the forward primers are labeled with Cy5 dyes. | ||
Viral strains
Four different inactivated AIV strains and one strain of Newcastle diseases virus (NDV) were used in this study. The phenotypes, sources of isolation and HA titres of the viral strains are listed in Table 1. They were provided by the National Veterinary Institute of Denmark.| No. | Viral strains and sources | Titre (HA) |
|---|---|---|
| 1 | H1N1 A/DK/ALB 35/76 | 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :16 :16 |
| 2 | H5N1 A/CK/Scotland/ 59 06.04.67 | 1:256 |
| 3 | H7N5-2 A/Chick/Nether/2993—17/03 AV 506/03 | 1:128 |
| 4 | Newcastle diseases, Ulster strain | 1:128 |
| 5 | H16N3—A/Gull/Denmark/68110/02 | 1:128 |
RNA extraction
Isolation of RNAs from virus strains was performed using the RNeasy Kit (Qiagen, Germany) according to the manufacturer's instruction. The RNAs were eluted using the RNase free water. The isolated RNAs were used as templates for evaluation of the solid-phase PCR.Primer and probes
Table 2 shows a list of primers and probes used for AIV identification and subtyping. The M gene-specific primers and probes were designed for AIV screening as it has been found that the region on the M gene is conserved across all type A influenza viruses.4 Primers and probes for H5 and H7 subtypes were designed to target the H5 and H7 regions on HA gene of Influenza virus with avian origin. Each forward primer was Cy5-labeled at the 5′ end so that the solid-phase PCR results could be directly visualized on the microarray. The probes (immobilized primers) were modified at the 5′ end with a poly(T)10–poly(C)10 tail to facilitate the attachment on the solid substrate. All the oligonucleotide primers and probes were synthesized at DNA Technology A/S, Denmark.| Type or subtype | Target gene | Primers and probes | Sequences (5′–3′) | Amplicon (bp) |
|---|---|---|---|---|
| A | Matrix | Forward DB-MF | CY5-AGA TGA GTC TTC TAA CCG AGG TCG | 98 |
| Reverse DB-MR | TGC AAA AAC ATC TTC AAG TCT CTG | |||
| M gene probe | TTTTTTTTTTCCCCCCCCCC TCA GGC CCC CTC AAA GCC GA | |||
| H5 | HA | Forward DB-H5LH1 | CY5-ACA TAT GAC TAC CCA CAR TAT TCA G | 151 |
| Reverse DB-H5RH1 | AGA CCA GCT AYC ATG ATT GC | |||
| H5 Probe | TTTTTTTTTTCCCCCCCCCC TCW ACA GTG GCG AGT TCC CTA GCA | |||
| H7 | HA | Forward DB-LH6H7 | CY5-GGC CAG TAT TAG AAA CAA CAC CTA TGA | 131 |
| Reverse DB-R4H7 | GCC CCG AAG CTA AAC CAA AGT AT | |||
| H7 Probe | TTTTTTTTTTCCCCCCCCCC CCG CTG CTT AGT TTG ACT GGG TCA ATC T |
Preparation of DNA microarrays on glass substrate
Microarray was produced on an unmodified glass substrate by simple UV cross-linking according to a method developed in our lab.26 Briefly, a glass chip of 10 mm × 10 mm was diced from a 0.5 mm glass wafer using a Dicing Saw (Disco, Japan) and was used without any pre-treatment or modification. The three oligonucleotide probes with poly(T)10–poly(C)10 tails were diluted in 150 mM sodium phosphate buffer (pH 8.5) to a final concentration from 5 to 50 µM and spotted using a non-contact array nano-plotter 2.1 (GeSim, Dresden, Germany). The layout is shown in Fig. 3a. The array was spotted at the centre of the glass chip and each probe was repeated four times for easy identification of the reaction products. The spot had a diameter of 150 µm and the spot-to-spot distance was 300 µm. The spots were allowed to dry and then exposed to UV irradiation at 254 nm with an energy of 0.3 J cm−2 for 10 min (Stratalinker 2400, Stratagene, CA, USA). Subsequently, the glass chip was washed under agitation in 0.1× standard saline citrate (SSC) with 0.1% (w/v) sodium dodecyl sulfate (SDS) (Promega, WI, USA) solution for 10 min, then rinsed in deionized water and dried under nitrogen.Microfabrication
The PCR microchamber was fabricated in polydimethylsiloxane (PDMS) by rapid prototyping. The mask layout was designed using a CAD program. The master for molding was fabricated in SU-8 50 (MicroChem, MA, USA) by the standard photolithography. A 10:1 mixture of PDMS pre-polymer and curing agent (Sylgard 184, Dow Cornig, MI, USA) was stirred thoroughly and then poured onto the master and cured for 1 h at 65 °C. After curing, the 1 mm thick PDMS replica was peeled from the master. Inlet and outlet holes were punched by a needle. The PDMS substrate and the glass chip with DNA microarray were sealed by plasma bonding. Both PDMS and glass substrates were exposed to oxygen plasma for 30 s at 100 W with an oxygen flow rate of 240 ml min−1 (Plasma Processor 300, PVA TePla, Germany). Right after the removal from the plasma chamber, the substrates were brought into conformal contact where an irreversible seal formed spontaneously. Though the probe array was also exposed to oxygen plasma, test showed that the functionality of DNA as probes for PCR was not affected at such low power exposure. As shown in Fig. 2, the outer dimension of the microchip was 10 mm × 10 mm and the volume of the microchamber was 2 µl.
 | ||
| Fig. 2 Photograph of the hybrid PDMS–glass microchip. DNA microarray was spotted on the glass substrate. The outer dimension of the microchip was 10 mm × 10 mm and the volume of the microchamber was 2 µl. | ||
On-chip solid-phase PCR
25 µl PCR reaction mix was prepared which consisted of 10 µl of 5 x RT-PCR buffer, 1 µl of 10 mM DNTP mix, 1 µl of 2 µl /reaction enzyme mix, 2.5 µl of 2.5 µg µl−1 BSA (Onestep RT-PCR kit, Qiagen, Germany), three pairs of primers with a final concentration of 1 µM for forward primers and 0.5 µM for reverse primers, and 5 µl RNA sample. 2 µl of the mixture was loaded into the microchamber. The whole chip was placed in a homemade chip holder and the inlet and outlet were then sealed by pressing rubber plugs down on the holes. The chip holder was put on a flatbed thermocycler (MJ Research Inc., MA, USA). PCR was carried out according to the following program: 15 min at 50 °C for reverse transcription, followed by 40 cycles of 1 s at 95 °C, 5 s at 54 °C and 3 s at 72 °C, and finally 3 min at 72 °C for extension. After the cycling, the microchamber was washed with 0.1× SSC per 0.1% SDS and deionized water.The microarray in the microchip was scanned by an array scanner (LaVision BioTec, Germany). Fluorescence imaging and processing software (LaVision BioTec, Germany) was used to quantify the spots by calculating the average pixel intensity inside the defined spots.
Sensitivity of solid-phase PCR
The sensitivity of the solid-phase PCR was evaluated. Ten-fold serial dilution of an AIV strain H16N3 ranging from 100 to 10−9 was made and the RNA was isolated from the virus dilution series according to the method described above. The hemagglutination titre (HA titre) value for the AIV strain was measured to be 1:64. The fifty percent egg infectious dose (EID50) calculated for H16N3 strain was 6.7 log10EID50 per ml. 5 µl of RNA was used as template and added to 20 µl PCR master mix.
Results and discussion
DNA immobilization on unmodified glass surface
Conventionally, fabrication of DNA microarrays on glass slides requires tedious and time-consuming procedures for surface treatment and DNA immobilization,14,25 which seriously hinders the integration of microarrays into microfluidic systems. In this study, a much faster and easier immobilization method was developed by us where DNA probes with poly(T)10–poly(C)10 tails were immobilized on the unmodified glass surface by simple UV cross-linking. During UV exposure, covalent bonds are formed between the TC tag and the glass surface. The probes modified with TC tails offer up to 70% cost reduction when compared to the commonly used amino group modifications of the probes and make solid-phase PCR a convenient possibility for AIV screening. Another advantage of our immobilization method is that the probes are thermally stable. For other immobilisation methods it can be a problem that the primer detaches during thermo-cycling. A 60% loss of grafted primers during thermo-cycling was reported.27 This is, however, not a problem with our method. No significant decrease in fluorescence signal was observed after 20 min incubation in water at 100 °C.26Specificity of on-chip solid-phase PCR
On-chip solid-phase amplifications were conducted with RNA extracts from four viral strains: H1N1, H5N1, H7N5, and NDV respectively. As shown in Fig. 3, with the developed microchip, three AIV viral strains were accurately identified by the distinct patterns of amplified products. No positive signal was obtained for the NDV. In particular, non-specific amplification was almost negligible as the probes in solid-phase PCR were spatially separated. The simple visual inspection of the fluorescence scan images suggested that the multiplex solid-phase amplification is an efficient and specific procedure. But it was also noticed that significant amounts of spot intensity variations were produced for different kinds of probes. As suggested by Huber et al.,28 this was due to some inherent factors such as microarray substrate quality, template quality, and sequence properties of the analyzed DNA fragments. Nevertheless, all strains tested in this study were unambiguously distinguished from each other based on their specific combinations of fluorescence products. The number of solid-phase probes can be continually extended to include sequences for additional AIV subtypes.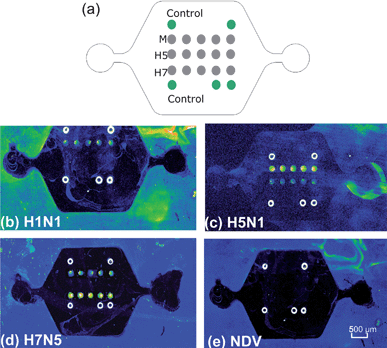 | ||
| Fig. 3 Specificity of on-chip solid-phase PCR. (a) Microarray layout. The DNA probes for M gene and HA genes specific for H5 and H7 are represented as gray circles. Each dot is 150 µm in diameter with a center-to-center distance of 300 µm. Green circles are guiding dots used for orientation. Fluorescent images after 40 cycle amplification of four different viral RNA strains: (b) AIV H1N1, (c) AIV H5N1, (d) AIV H7N5 and (e) Newcastle disease virus (NDV). The strong fluorescence outside of the chip is due to the autofluorescence of PDMS residues. | ||
Optimization of solid-phase amplification
In this study, parameters such as the ratio of aqueous primers and solid-phase probe density that are likely to affect solid-phase PCR were optimized to ensure efficient amplification. RNA extracted from 10−1AIV H16N3 strain was used as template for the amplification.As the aqueous forward primers were designed to bind to both templates in the solution and amplicons tethered on the solid support, the reverse primers were included at a limiting concentration to lower the competition with solid probes. The optimum primer ratios were identified by keeping the concentration of reverse primers at 0.5 µM while increasing the concentration of forward primers from 0.5 µM to 5 µM. When the forward primers in the liquid were doubled from 0.5 µM to 1 µM, 20% increase in the amount of immobilized amplicons was observed. Further increase in the concentration had no effect on the yield of solid-phase amplifications. Therefore, 1 µM forward primers were used to ensure amplification was not limited by the amount of aqueous primers in the reaction.
The probe density is another important factor that could limit the amount of amplicons. The density of probes immobilized on the glass substrate was changed by varying the spotting concentrations from 5 µM to 50 µM. As shown in Fig. 4, the amount of amplicons increased linearly when the probe concentration was raised from 5 µM to 20 µM. But increasing the concentration from 20 µM to 40 µM had no significant effect on the yield of the immobilized amplicons, while an excess number of probes reduced the yield of solid-phase amplifications, indicating that too high probe density negated amplification. The low amplification efficiency at high probe concentration may have several explanations. One explanation could be screening effect: when the probes are immobilized at high density, the high negative charges in close proximity to a solid support disturb polymerase functioning on the surface and repel of target DNA in solution, thus reducing the extension efficiency. Another explanation could be that abnormal target–probe and probe–probe interactions on planar microarrays are intensified at higher probe density, therefore decreasing the amount of accessible probes on the chip and reducing priming and amplification efficiency. For this system, the maximum amplification efficiency was achieved at the combined condition of 1 µM forward primers and 20 µM immobilized probes.
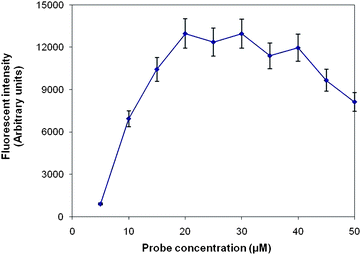 | ||
| Fig. 4 Effect of probe concentration on solid-phase RT-PCR. Solid-phase amplification of 10−1H16N3 viral RNA was performed in replicate reactions with probe concentration varied from 5 µM to 50 µM. 1 µM forward primers and 0.5 µM reverse primers were contained in the PCR solution. Fluorescence intensities of probes specific to M gene were measured. Values represent the mean fluorescence and the standard deviation of triplicate reactions for each probe concentration. | ||
Detection limit of on-chip solid-phase PCR
The sensitivity of the on-chip solid-phase PCR was determined and compared analytically with solution-based multiplex RT-PCR. 10-Fold serial dilution (from 100 to 10−9) of RNA extracted from AIV H16N3 strain was amplified in triplicate reactions. As shown in Fig. 5, the lowest concentration of template at which positive fluorescence signals could be detected for solid-phase PCR was 10−6, or equivalent to 0.7 log10EID50 per ml. The fluorescence increased with template concentration, suggesting that more amplification occurred on the surface at higher template concentration. The signal reached a plateau when the RNA template was present at 10−1, indicating the surface became saturated with the PCR products. The coefficient of variation of the replicate reactions was 8% on average. The detection limit of the solution-based multiplex PCR using the same biological model was 10−5, or equivalent to 1 log10EID50 per ml, similar to that published in the literature29 (see ESI†). Compared to liquid-based amplification, 10-fold improvement was achieved by on-chip solid-phase PCR.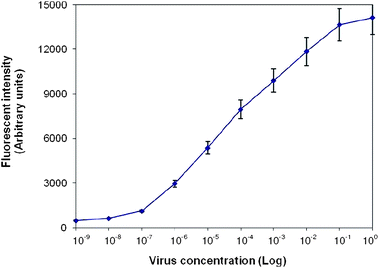 | ||
| Fig. 5 Sensitivity of solid-phase RT-PCR. Serial 10-fold dilutions of viral RNA extracted from H16N3, ranging from 100 to 10−9, were used as templates. Replicate solid-phase PCR was prepared each containing 1 µM forward primers and 0.5 µM reverse primers in solution, and 20 µM probes immobilized on glass surface. Fluorescence intensities of probes specific to M gene were measured. Values represent the mean fluorescence and the standard deviation of triplicate reactions for each template concentration. The detection limit was 10−6, or equivalent to 0.7 log10EID50 per ml. | ||
Compared to solid-phase PCR using a coverslip or a self-adhesive frame
In conventional solid-phase PCR, a glass coverslip or a self-adhesive frame is mounted onto the oligonucleotide array to seal the reaction mixture. In both cases, a large amount of PCR samples (25 µl for standard Gene frame) are required.7 The large sample mass requires lengthy time for denaturation and annealing, and total reaction times are typically in excess of 2 hours. In addition, air bubbles are easily trapped under the coverslip or in the frame even with very careful handling, which could adversely affect the efficiency of solid-phase PCR. Moreover, self-seal reagent is usually contained in the reaction mixture to prevent evaporation during PCR, which polymerizes upon contact with air at high temperature and thereby seals the reaction at the edges. While the effect of polymerization on solid-phase PCR is still under investigation, MJ Research Inc. has stopped the supply of the self-seal reagent and a substitute product has not appeared in the market. This poses a great challenge to solid-phase amplification.In our approach, a 2 µl chamber-based PCR microchip was fabricated to replace the conventional coverslip or frame. As the sample volume was greatly reduced, the denaturation and annealing steps occurred as soon as the correct temperature was reached, and the process was only limited by the extension step. Consequently, the total reaction time was shortened by half. Furthermore, due to the micron sized feature dimension and the closed fluidic format of the microchip, the small amount of samples could be manipulated and processed with a high degree of control. Solution evaporation and bubble formation were effectively prevented. Compared to solid-phase PCR carried out using the coverslip or frame, the amplification in PCR microchip provided better performance as the detection limit achieved on-chip (10−6) was 10-fold better than that obtained using the coverslip (10−5)30 (see ESI†). PCR microchip and DNA microarray have been shown to be a perfect combination, which affords gains in terms of control, speed, efficiency and functionality.
Conclusions
In conclusion, we have developed a simple lab-on-a-chip device for rapid screening and subtyping AIV viral RNA based on on-chip solid-phase PCR. The platform combines RT-PCR in the liquid phase and simultaneous nested amplification with species-specific oligonucleotide probes on the solid surface. Different AIV viral strains were accurately identified by the distinct patterns of reaction products. By immobilizing probes on the glass surface, primer interference effects are minimized, such that the number of AIV subtypes to be identified can easily be extended by adding more species-specific oligonucleotides to the microarray. Moreover, no post-PCR step is needed apart from array scanning, which prevents potential cross-contamination and facilitates the process for AIV detection. By combining the superior specific multiplex format of solid-phase PCR with low reagent consumption and fast cycling provided by the PCR microchip, the platform can be widely employed by veterinarians for on-site rapid screening of AIV in wild and industrial domestic poultries.Acknowledgements
This work is financially supported by Technical University of Denmark (DTU), Food Pathogen Project No. 8, Grant No. 150627, Forskningsrådet Teknologi og Produktion (FTP), Sagsnr. 09066477, and EU FP-7 IP LABONFOIL project, Grant No. 224306.References
- D. J. Alexander, Vaccine, 2007, 25, 5637–5644 CrossRef CAS.
- D. J. Alexander, Vet. Microbiol., 2000, 74, 3–13 CrossRef CAS.
- M. S. Lee, P. C. Chang, J. H. Shien, M. C. Cheng and H. K. Shieh, J. Virol. Methods, 2001, 97, 13–22 CrossRef CAS.
- E. Starick, A. Romer-Oberdorfer and O. Werner, J. Vet. Med., Ser. B, 2000, 47, 295–301 Search PubMed.
- B. Chaharaein, A. R. Omar, I. Aini, K. Yusoff and S. S. Hassan, Microbiol. Res., 2009, 164, 174–179 CrossRef CAS.
- Z. X. Xie, Y. S. Pang, H. B. Uu, X. W. Deng, X. F. Tang, H. H. Sun and M. I. Khan, Mol. Cell. Probes, 2006, 20, 245–249 CrossRef CAS.
- A. Pemov, H. Modi, D. P. Chandler and S. Bavykin, Nucleic Acids Res., 2005, 33, e11 CrossRef CAS.
- K. A. Hagan, C. R. Reedy, M. L. Uchimoto, D. Basu, D. A. Engel and J. P. Landers, Lab Chip, 2011, 11, 957–961 RSC.
- C. Adessi, G. Matton, G. Ayala, G. Turcatti, J. J. Mermod, P. Mayer and E. Kawashima, Nucleic Acids Res., 2000, 28, 87e CrossRef.
- M. H. Shapero, K. K. Leuther, A. Nguyen, M. Scott and K. W. Jones, Genome Res., 2001, 11, 1926–1934 CAS.
- C. M. Niemeyer, M. Adler and R. Wacker, Nat. Protoc., 2007, 2, 1918–1930 Search PubMed.
- M.v. Nickisch-Rosenegk, X. Marschan, D. Andresen and F. F. Bier, Anal. Bioanal. Chem., 2008, 391, 1671–1678 CrossRef CAS.
- R. Palanisamy, A. R. Connolly and M. Trau, Bioconjugate Chem., 2010, 21, 690–695 CrossRef CAS.
- G. Mitterer, M. Huber, E. Leidinger, C. Kirisits, W. Lubitz, M. W. Mueller and W. M. Schmidt, J. Clin. Microbiol., 2004, 42, 1048–1057 CrossRef CAS.
- A. Suomalainen and A. C. Syvanen, Mol. Biotechnol., 2000, 15, 121–131 Search PubMed.
- D. A. Khodakov, N. V. Zakharova, D. A. Gryadunov, F. P. Filatov, A. S. Zasedatelev and V. M. mikhailovich, BioTechniques, 2008, 44, 241–248 CrossRef CAS.
- H. N. Liu, S. Li, L. Liu, L. Tian and N. Y. He, Anal. Biochem., 2009, 386, 126–128 CrossRef CAS.
- G. Mitterer, O. Bodamer, C. Harwanegg, W. Maurer, M. W. Mueller and W. M. Schmidt, Genet. Test., 2005, 9, 6–13 CrossRef CAS.
- Y. Sun, M. V. D. Satya, Y. C. Kwok and N. T. Nguyen, Sens. Actuators, B, 2008, 130, 836–841 CrossRef.
- Y. Sun, Y. C. Kwok, F. P. Lee and N. T. Nguyen, Anal. Bioanal. Chem., 2009, 394, 1505–1508 CrossRef CAS.
- Y. H. Zhang and P. Ozdemir, Anal. Chim. Acta, 2009, 638, 115–125 CrossRef CAS.
- Z. Guttenberg, H. Muller, H. Habermuller, A. Geisbauer, J. Pipper, J. Felbel, M. Kielpinski, J. Scriba and A. Wixforth, Lab Chip, 2005, 5, 308–317 RSC.
- M. Hashimoto, F. Barany and S. A. Soper, Biosens. Bioelectron., 2006, 21, 1915–1923 CrossRef CAS.
- D. Sabourin, J. Petersen, D. Snakenborg, M. Brivio, H. Gudnadson, A. Wolff and M. Dufva, Biomed. Microdevices, 2010, 12, 673–681 CrossRef CAS.
- M. Huber, D. Losert, R. Hiller, C. Harwanegg, M. W. Mueller and W. M. Schmidt, Anal. Biochem., 2001, 299, 24–30 CrossRef CAS.
- H. Gudnason, M. Dufva, D. D. Bang and A. Wolff, BioTechniques, 2008, 45, 261–271 CrossRef CAS.
- M. Fedurco, A. Romieu, S. Williams, I. Lawrence and G. Turcatti, Nucleic Acids Res., 2006, 34, e22 CrossRef.
- M. Huber, A. Mundlein, E. Dornstauder, C. Schneeberger, C. B. Tempfer, M. W. Mueller and W. M. Schmidt, Anal. Biochem., 2002, 303, 25–33 CrossRef CAS.
- C. W. Lee and D. L. Suarez, J. Virol. Methods, 2004, 119, 151–158 CrossRef CAS.
- Y. Sun, R. Dhumpab, D. D. Bang, K. Handberg and A. Wolff, Diagn. Microbiol. Infect. Dis., 2011, 69, 432–439.
Footnote |
| † Electronic supplementary information (ESI) available: Information on sensitivity of two other AIV identification methods—multiplex solution-based RT-PCR and conventional solid-phase PCR with coverslip. See DOI: 10.1039/c0lc00528b |
| This journal is © The Royal Society of Chemistry 2011 |