Dithienopyrrole–quinoxaline/pyridopyrazine donor–acceptor polymers: synthesis and electrochemical, optical, charge-transport, and photovoltaic properties
Xuan
Zhang
a,
Jae Won
Shim
b,
Shree Prakash
Tiwari
b,
Qing
Zhang
a,
Joseph E.
Norton
a,
Pei-Tzu
Wu
c,
Stephen
Barlow
a,
Samson A.
Jenekhe
c,
Bernard
Kippelen
b,
Jean-Luc
Brédas
a and
Seth R.
Marder
*a
aSchool of Chemistry and Biochemistry and Center for Organic Photonics and Electronics, Georgia Institute of Technology, Atlanta, Georgia 30332-0400, USA. E-mail: seth.marder@gatech.edu
bSchool of Electrical and Computer Engineering and Center for Organic Photonics and Electronics, Georgia Institute of Technology, Atlanta, Georgia 30332-0400, USA
cDepartment of Chemical Engineering and Department of Chemistry, University of Washington, Seattle, Washington 98195-1750, USA
First published on 17th February 2011
Abstract
Soluble alternating copolymers of N-(3,4,5-tri-n-dodecyloxyphenyl)dithieno[3,2-b:2′,3′-d]pyrrole donor groups and 2,3-di-n-decylquinoxaline, 2,3-di-n-decylpyrido[3,4-b]pyrazine, 2,3,6,7-tetrakis(n-decyloxy)benzo[a,c]phenazine, or 2,3,6,7-tetrakis(n-decyloxy)dibenzo[f,h]pyrido[4,3-b]quinoxaline acceptors were synthesised using Stille coupling reactions. Experimental absorption maxima in THF range from 645 to 770 nm. These optical data, along with the results of quantum-chemical calculations and electrochemical measurements, show that, as expected, the pyridopyrazine moiety acts as a stronger acceptor than quinoxaline and that the extended species benzophenazine and dibenzopyridoquinoxaline are stronger acceptors than quinoxaline and pyridopyrazine, respectively. Modest average hole mobilities of up to ca. 3.0 × 10−4 cm2 V−1s−1 were obtained in field-effect transistors. Bulk heterojunction photovoltaic devices made from blends of the benzo[a,c]phenazine-based polymer with 3′-phenyl-3′H-cyclopropa[1,9](C60-Ih)[5,6]fullerene-3′-butanoic acid methyl ester (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 weight ratio) exhibited average power conversion efficiencies up to 1.4%.
:3 weight ratio) exhibited average power conversion efficiencies up to 1.4%.
Introduction
Due to their film-forming and charge-transport properties, and to the possibility for tuning optical and electronic properties through appropriate choice of monomer(s), soluble conjugated polymers have been increasingly used in electronic devices including organic light-emitting diodes (OLEDs), organic photovoltaic cells (OPVs), and organic field-effect transistors (OFETs).1–7 The use of copolymers of alternating electron-rich (donor, D) and electron-poor (acceptor, A) repeat units has been of particular interest for OPV applications since they typically exhibit lower energy absorption bands than all-donor homopolymers such as polythiophenes,2 leading to enhanced coverage of the solar spectrum in the low-energy portion of the visible and/or in the near-infrared. Indeed power conversion efficiencies (PCEs) as high as 7.4% have been reported for blends of alternating D–A conjugated polymers with fullerenes such as the widely used 3′-phenyl-3′H-cyclopropa[1,9](C60-Ih)[5,6]fullerene-3′-butanoic acid methyl ester (PCBM) and its C70 analogue.8–20 Moreover, the relatively low ionisation potentials (IPs) and high electron affinities (EAs) imparted by the donor and acceptor groups, respectively, can result in the possibility of both hole and electron injection at moderate potentials and, therefore, in ambipolar OFET behavior.21–28Here we report copolymers of a dithieno[3,2-b:2′,3′-d]pyrrole-2,6-diyl (DTP) donor with quinoxaline-5,8-diyl (Qx), pyrido[3,4-b]pyrazine-5,8-diyl (PPz), benzo[a,c]phenazine-10,13-diyl, and dibenzo[f,h]pyrido[4,3-b]quinoxaline-10,13-diyl derivatives as acceptors. In recent years, N-alkyl and N-aryl DTP building blocks have been incorporated into a wide range of conjugated polymers.26,29–44DTP is a stronger electron donor than bithiophene or the related fused-ring dithieno[3,2-b:2′,3′-d]thiophene.45–48 Accordingly all-donor DTP-based polymers, such as I (Fig. 1), have rather low IPs, which can lead to adventitious oxidative doping in air and, consequently, poor on–off ratios in OFETs, although promising hole mobilities of up to 0.21 cm2 V−1s−1 have been measured.32 Low current on–off ratios have also been observed for some D–A DTP polymers, such as II (Fig. 1), and may also be attributable to their relatively low IPs.39 The low IPs of DTP-based polymers can also potentially limit the open-circuit voltages attainable in OPV cells;40 on the other hand, the electron-richness of DTPs means that their incorporation into D–A copolymers leads to lower-energy absorption bands than those of analogous D–A polymers incorporating the same acceptor in combination with weaker donors such as bithiophene, dithienothiophene, or carbazole.37–39,49 PCEs of up to 2.8% have been reported for solar cells based on a copolymer of DTP with the [2,1,3]-benzothiadiazole-4,7-diyl (BTD) acceptor, III, blended with PCBM.36
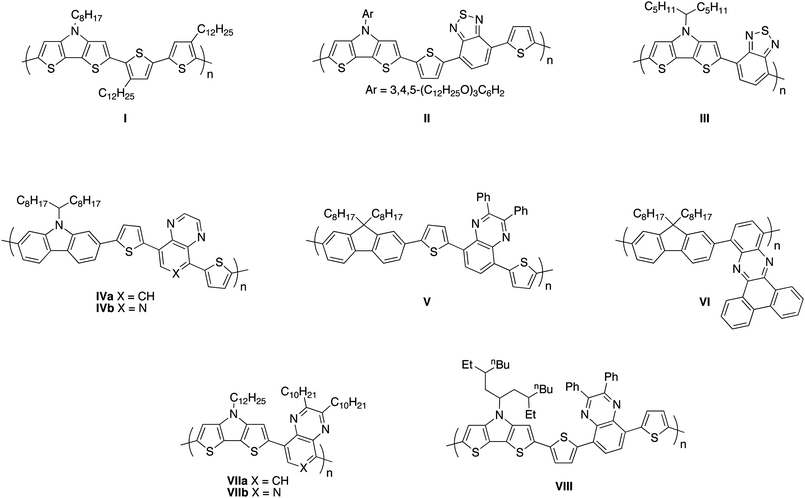 | ||
| Fig. 1 Structures of some previously reported DTP, Qx, and PPz polymers discussed in the text.13,32,36,39,49,56,59,71 | ||
Qx monomers50 have previously been incorporated into conjugated polymers, including “all-acceptor” structures, such as poly(quinoxaline-5,8-diyl)s51 and poly(quinoxaline-5,8-diyl-vinylene)s,52,53 and D–A structures.13,23,54–68PPz monomers have been used much less extensively;52,54,69,70 electrochemical and UV-vis absorption data for D–A copolymers suggest that PPz is a somewhat stronger acceptor than Qx, consistent with the expected effect of the additional electronegative nitrogen atom.59 Some D–A polymers incorporating Qx or PPz acceptors have, when blended with fullerene acceptors, exhibited good OPV performance. For example, blends of IVa and IVb (Fig. 1) with PCBM gave PCEs of 1.8% and 1.1%, respectively,59 while blends of V (Fig. 1) with the C70 analogue of PCBM gave a PCE of 5.5%.13Benzo[a,c]phenazine and dibenzo[f,h]pyrido[4,3-b]quinoxaline can be considered as extended Qx and PPz derivatives, respectively, but we are aware of only one example of a conjugated polymer containing one of these moieties (VI, Fig. 1).56
To date there are only a handful of examples of polymers in which DTP donors are combined with Qx or PPz acceptors. Some of us recently reported the DTP–Qx/PPz polymers VII (Fig. 1) as partner materials for PbS nanocrystals in PV cells,71 while the (dithienyl-DTP)–Qx polymer VIII has been compared to other D–A Qx polymers for use in OPV cells when combined with PCBM or its C70 analogue.49 Here we report in detail on the synthesis, spectroscopy, electrochemistry, and OFET and OPV performance of copolymers of a DTP monomer with Qx, PPz, and their extended analogues, benzo[a,c]phenazine and dibenzo[f,h]pyrido[4,3-b]quinoxaline (P1–P4, Scheme 1).
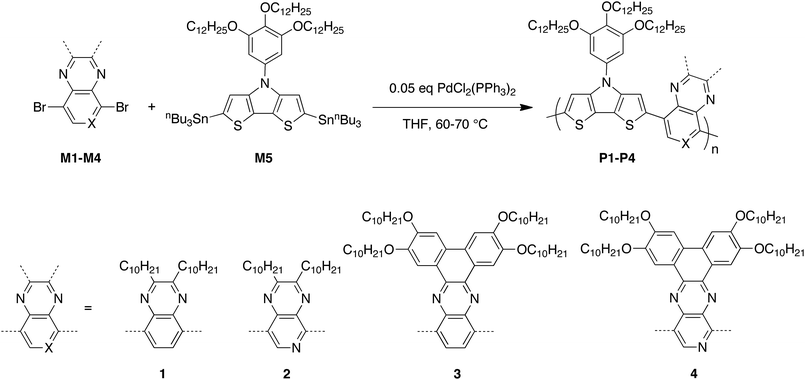 | ||
| Scheme 1 Synthesis of P1–P4. | ||
Results and discussion
Synthesis
Four D–A copolymers P1–P4 incorporating DTP-based donors and Qx or PPz-based acceptors were synthesised using Stille coupling polymerisation of the appropriate dibromo-substituted acceptor derivatives M1–M4 with the 2,6-di(tri-n-butylstannyl) derivative of N-(3,4,5-tris(n-dodecyloxy)phenyl)dithieno[3,2-b:2′,3′-d]pyrrole, M526 (Scheme 1). The acceptor monomers, M1–M4, were synthesised using procedures previously reported for either identical compounds or close analogues.56,59,72,73 Some Qx- and PPz-based polymers have previously been found to be poorly soluble; for example, poly{2,3-di(n-heptyl)pyrido[3,4-b]pyrazine-5,8-diyl-co-thiophene-2,5-diyl} is reported to show good solubility only in acidic media, such as trifluroacetic acid.74 Moreover, our initial polymerisation attempts using N-n-octyl DTP as a donor with M1 resulted in low molecular-weight oligomers or completely insoluble products. Accordingly, M5 was chosen as the DTP monomer based on our previous observations that highly soluble D–A polymers can be obtained using 3,4,5-tris(n-dodecyloxy)phenyl-substituted DTP.26,39 On the other hand, it is worth noting that introduction of this relatively bulky solubilising group will lead to some dilution of the optically and electronically active polymer backbone.The copolymers P1–P4 were obtained in moderate to high yields (Table 1) using optimised Stille coupling conditions, with Pd(PPh3)2Cl2 as a catalyst and THF as the solvent, in pressure vessels. The crude polymers were isolated by reduction of the solvent volume, followed by precipitation into methanol. In each case, multiple precipitations and Soxhlet extractions using several solvents were employed to purify the crude products; in some cases, further purifications were carried out by running flash column chromatography on silica gel, followed by size-exclusion column chromatography.
| Polymer | Yield | M w/kDa | M w/Mna | DP b | T d/°Cc |
|---|---|---|---|---|---|
| a Weight-average molecular weight (Mw) and polydispersity index (Mw/Mn) determined by GPC with toluene as eluentvs.polystyrene standards. b Degree of polymerisation, DP = Mn/Mo, where Mo is the molecular weight of the repeat unit. c The temperature at which 5% weight loss is observed using TGA under N2 at a heating rate of 10 °C min−1. | |||||
| P1 | 93% | 119 | 3.7 | 27 | 364 |
| P2 | 72% | 103 | 1.8 | 47 | 366 |
| P3 | 66% | 70 | 1.8 | 23 | 365 |
| P4 | 75% | 50 | 2.3 | 13 | 376 |
Weight-average molecular weights (Mw) and polydispersities (Mw/Mn) for P1–P4 were estimated using gel-permeation chromatography (GPC, see Experimental section for details); these data, summarised in Table 1, indicate that the polymers have moderate to high molecular weights. The copolymers are readily soluble in solvents including THF, chloroform, and toluene, whereas many previous examples of Qx or PPz-containing copolymers have been reported to exhibit limited solubilities in common organic solvents.55,74Thermogravimetric analysis (TGA) indicated that all four polymers showed good thermal stability with 5% weight loss only occurring at temperatures in excess of 350 °C (see Table 1).
Density functional theory calculations of electronic structure
Optimised geometries and electronic energy levels were calculated for isolated systems using DFT at the B3LYP/6-31G(d,p) level of theory. Excited-state-energy calculations were performed at the same level of theory using the time-dependent method (TD-DFT). These calculations were performed on model donor–acceptor oligomers, H–(D–A)n–H {n = 1, 2, 3}, in which the extended alkyl chains were replaced by methyl groups.As shown for the examples of trimeric models for P1 and P3 (Fig. 2), the LUMOs of the D–A oligomers are somewhat more localised on the acceptor unit, although there are also significant coefficients on the DTP moieties. In contrast, the HOMOs are delocalised along the main conjugation path, with only small coefficients on the pyrazine ring and even smaller coefficients on the additional rings of the acceptors of P3 and P4. This pattern of orbital distribution is broadly similar to that we have previously observed for II and related DTP D–A polymers (Fig. 1);39 however, in these previously studied systems, the LUMOs were more strongly acceptor localised, presumably, at least in part, due to the presence of longer dithienyl-DTP donor-type segments separating adjacent acceptors. The calculated orbital energies for the trimeric models shown in Table 2 indicate that the replacement of a CH group in P1 (or P3) with a N atom in P2 (or P4) stabilises both HOMO and LUMO, consistent with the trend observed in the redox potentials (see below) and with the expected effect of incorporating a more electronegative atom. The HOMO–LUMO separation (Eg) decreases from P1 to P2, consistent with the electronegative atom having a more significant effect on the LUMO, which is more strongly acceptor-localised than the HOMO. The calculated energy of the S0 → S1 optical transition also decreases from Qx to PPz derivatives. These findings are consistent with the electrochemical and optical data (see below), and with previous experimental and computational studies of polymers of type IV (Fig. 1).59 The S0 → S1 optical transitions calculated for the extended systems P3 and P4 are also bathochromically shifted relative to those for P1 and P2, respectively, consistent with the experimental data, although the calculated effects on orbital energies are not consistent with the electrochemical data.
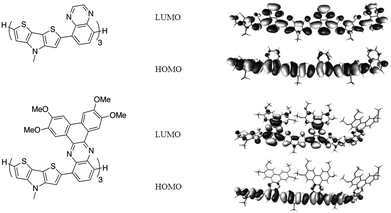 | ||
| Fig. 2 DFT-calculated HOMO and LUMOs for oligomeric models (n = 3) for P1 and P3. | ||
Optical properties
Normalised optical absorption spectra of the polymers in dilute THF solution and in thin films are shown in Fig. 3; absorption maxima and absorption coefficients are summarised in Table 3. The copolymers P1 and P2 show two prominent absorption maxima; the low-energy maximum is bathochromically shifted (by 0.20 eV) on replacing Qx by PPz by considerably more than the high-energy maximum (shifted by 0.06 eV). The low-energy bands presumably have considerable D-to-A charge-transfer character, as is also suggested by the frontier orbitals for the model oligomers (see above). The absorption maximum of P1 in THF is similar to that previously reported for VIIa in chloroform;71 however, the PPz derivative P2 shows a considerably longer wavelength maximum than VIIb and also a rather different spectral profile,71 suggesting that VIIb might contain significant quantities of low molecular weight material. The absorption maxima of both P1 and VIIa71 in both solution and thin films are bathochromically shifted from the values recently reported for the (dithienyl-DTP)–Qx copolymer VIII (617 and 624 nm in CHCl3 and thin films, respectively).49 This is consistent with the bathochromic shift seen between the DTP–BTD polymer III36 and the (dithienyl-DTP)–BTD polymer II.39 The bathochromic shift of the maximum for P2 relative to that of P1 (but not of VIIbvs.VIIa) is consistent with that seen in previous comparisons of analogous Qx- and PPz-based donor–acceptor polymers (IVavs.IVb)59 and with the TD-DFT calculations (vide supra). The low-energy bands are seen at much lower energies than those previously reported for the (dithienyl-carbazole)-based polymers IV (ca. 510 and 550 nm for Qx and PPz examples, respectively, in 1,2,4-trichlorobenzene at 135 °C),59 or for other Qx or PPz-containing conjugated polymers,13,23,49,54–65,67,68 consistent with previous evidence indicating that DTP is a relatively strong electron donor.37–39,45–48 The low-energy bands of P3 and P4 are bathochromically shifted relative to those of P1 and P2, respectively, suggesting that the extended conjugation in the acceptors of these polymers leads to enhanced acceptor strength; this is consistent with previous observations for VI and its 2,3-diphenyl Qx analogue.56,75 Comparison of the absorption maximum of the DTP–BTD copolymer III (764 nm in CHCl3)36 with the data in Table 3 suggests that the widely used [2,1,3]-benzothiadiazole-4,7-diyl (BTD) moiety has an acceptor strength intermediate between that of PPz and dibenzo[f,h]pyrido[4,3-b]quinoxaline-10,13-diyl acceptors; this is broadly consistent with data for donor–acceptor polymers with dithienyl carbazole donors that show similar absorption maxima for BTD and PPz derivatives.59 As is typical for conjugated polymers, the thin-film absorption maxima of all the copolymers are bathochromically shifted compared to those in solution. Polymers P1 and P2 show considerably larger shifts between solution and the solid state than P3 and P4, perhaps because the bulkier acceptor groups in the latter species lead to reduced planarisation of the polymer chain in the solid state and/or reduce close approach of neighboring chains; on the other hand, increased π stacking might be anticipated in P3 and P4 since small molecules based on the two extended moieties76 and related extended structures77 readily π stack in solution. The peak absorptivities and solid-state absorption coefficients for P1–P4 are comparable to those for other DTP D–A polymers such as II;78 the peak absorption coefficients are much lower than those for polythiophene donors, such as regioregular poly(3-n-hexylthiophene) (αmax = ca. 1.5 × 105 cm−1 at ca. 520 nm).79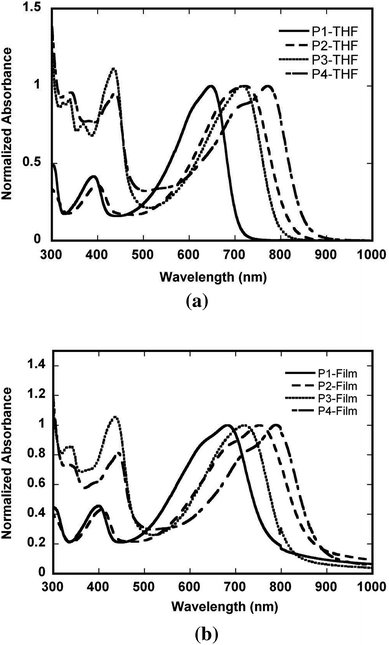 | ||
| Fig. 3 UV-vis-NIR absorption spectra of P1–P4 (a) in dilute THF solution and (b) as thin films. | ||
| Polymer | λ max (THF)/nm (εmax/104 M−1 cm−1) | λ max (film)/nm (αmax/104 cm−1) | E g (opt)a/eV | E ox (onset)b/V | IP (est)c/eV | E red (onset)b/V | EA (est)c/eV | E g (echem)d/eV |
|---|---|---|---|---|---|---|---|---|
| a Optical gap estimated from the onset of thin-film absorption (Fig. 3b). b Onset redox potentials vs.SCE estimated from cyclic voltammograms of polymer films in 0.1 M [nBu4N]+[PF6]−/acetonitrile solution measured at 50 mV s−1. c Estimated from IP (est) = eEox (onset) + 4.4 eV and EA (est) = eEred (onset) + 4.4 eV,80,81 defining EA here as −ΔG for the process P + e− → P˙−. d Electrochemical (“transport”) gap obtained from Eg (echem) = e(Eox (onset) − Ered (onset)). | ||||||||
| P1 | 392 (1.70), 645 (4.68) | 400 (2.1), 683 (4.6) | 1.58 | +0.65 | 5.1 | –1.55 | 2.9 | 2.2 |
| P2 | 401 (1.43), 720 (3.87) | 408 (1.8), 753 (4.1) | 1.43 | +0.80 | 5.2 | –1.30 | 3.1 | 2.1 |
| P3 | 327 (3.65), 369 (3.06), 435 (4.50), 713 (4.10) | 333 (1.9), 374 (1.6), 436 (2.4), 722 (2.3) | 1.52 | +0.69 | 5.1 | –1.36 | 3.0 | 2.1 |
| P4 | 337 (2.77), 438 (2.88), 710 (2.61), 771 (3.02) | 343 (2.7), 443 (3.1), 718 (3.1), 785 (3.8) | 1.39 | +0.82 | 5.2 | –1.10 | 3.3 | 1.8 |
Electrochemistry
Oxidative and reductive cyclic voltammograms of P4, drop cast from solution onto a platinum working electrode and measured in 0.1 M nBu4NPF6 in acetonitrile, are shown as a representative example in Fig. 4. The electrochemical oxidation and reduction onsets (Eoxonset and Eredonset) are summarised in Table 3 and were used to estimate the solid-state IP and EA according to IP = eEox (onset) + 4.4 eV, EA = eEred (onset) + 4.4 eV (defining EA as −ΔG for the reaction P + e− → P˙−), where the onset potentials are quoted vs.SCE;80,81 the “transport gaps”, Eg (echem), were obtained from the difference between Eox (onset) and Ered (onset) values. The estimated IPs of the polymers fall in the range 5.1–5.2 eV, and the estimated EAs are in the range of 2.9–3.3 eV. Both the IPs and EAs of P2 and P4 containing pyridopyrazine-based acceptors are somewhat higher compared to the values in their quinoxaline analogues, P1 and P3, respectively; this is consistent with the similar observation in other copolymers containing PPz or Qx,59,74 as well as the trend from the quantum-chemical calculations described earlier. Extension of the acceptor moieties in P3 and P4 leads to EA values increased by ca. 0.1–0.2 eV relative to P1 or P2, respectively.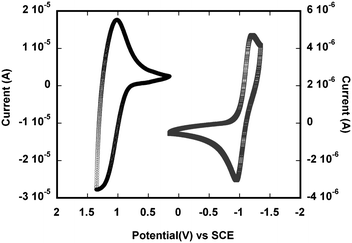 | ||
| Fig. 4 Oxidative (left) and reductive (right) cyclic voltammograms of a P4 film on a platinum working electrode in 0.1 M nBu4NPF6 at a scan rate of 50 mV s−1. | ||
Comparison of the estimated IPs and EAs to those for other polymers is somewhat hampered by the variety of electrochemical techniques (cyclic, differential pulse, and square-wave voltammetries), conventions (onsets or peak potentials at various scan rates), and even values of the vacuum-level offset that have been used in the literature. Nevertheless, it is worth noting that these IPs are slightly higher than those of either “all-donor” DTP-containing polymers (4.7–5.0 eV; 4.9 eV for I) and regioregular poly(3-n-hexylthiophene) (5.0 eV) estimated using essentially the same approach.32 The IPs are also a little higher than those reported for a (dithienyl-DTP)–Qx polymer VIII (5.0 eV),49 and those for some other DTP D–A polymers such as the BTD derivatives II and III (4.8–4.9 eV).36,39 As expected from the presence of the relatively strong DTP donor, the IPs are lower than those obtained in comparable ways for some other examples of Qx or PPz polymers; for example, values of 5.4–5.5 eV have been reported for Qx-containing copolymers with dialkoxylphenylene, dithienylfluorene, and dithienylcarbazole donors,49,60,62 while values of 5.7 and 5.4 eV have been reported for PPz-containing copolymers with fluorene or dithienylfluorene donors, respectively.70 The EAs fall in the range of previous estimates made in a similar way for Qx and PPz polymers (2.6–3.5 eV)49,60,62,63,70 and are close to those obtained for DTP–BTD polymers (3.1–3.2 eV for II and III36,39), consistent with other studies showing that the EA of the PPz compound IVb is similar to that of its BTD analogue.59 The electrochemical gaps, estimated from the difference between the onset oxidation and reduction potentials (or between IP and EA), follow the same variation with chemical structure as the optical gaps obtained from the onset of absorption in thin films; however, the former values are ca. 0.5–0.6 eV larger, demonstrating the influence of exciton-binding energies on the excited-state energies.
Field-effect transistor characteristics
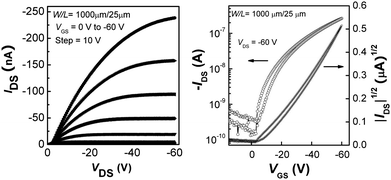
Charge transport in P1–P4 was investigated by fabricating OFETs with polymer active layers, top-contact gold source and drain electrodes, and a trichloroalkylsilane-treated SiO2 gate dielectric layer. Field-effect mobilities, µ, and threshold voltages, VT, were measured in the saturation regime from the highest slopes of plots of |IDS|1/2vs.VGS according to the saturation-region current equation for a standard MOSFET:where Ci is the capacitance per unit area of the gate dielectric, and W and L are respectively the width and length of the semiconductor channel defined by the source and drain electrodes. In all four cases p-channel behavior was observed. In contrast to one example of a dithienylfluorene–Qx copolymer,23 no n-channel behavior was observed for any of the present polymers. Representative output and transfer characteristics of an OFET (W/L = 1000 µm/25 µm) based on P3 are shown in Fig. 5, while Table 4 summarises values of µh, VT, and the current on/off ratios (Ion/Ioff). The hole mobilities fall in the range previously reported for Qx or PPz-based D–A copolymers,55,59,70 those of P1–P3 being around an order of magnitude lower than the highest values reported for these types of material (3.6 × 10−3 and 4.4 × 10−3 cm2 V−1s−1 for Qx and PPz D–A polymers55,70). The mobility in P4 is significantly lower than for P1–P3; this could be related to the lower degree of polymerisation achieved for this material (Table 1). The hole mobilities in P1–P3 are also comparable to those previously reported for other DTP-based D–A copolymers.23,39,40 The low on/off ratios are also reminiscent of those previously observed for other DTP polymers;32,39 these are possibly due to some limited doping by air, although the estimated IPs of the current polymers are higher than those for the previously reported DTP polymers.
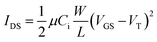
 | ||
| Fig. 5 Output (left) and transfer (right) characteristics of a P3OFET. | ||
| Polymer | µ h/cm2 V−1s−1 | V T/V | I on/Ioff |
|---|---|---|---|
| P1 | 1.20 (±0.18) × 10−4 | –9.34 (±0.78) | 103 |
| P2 | 2.30 (±0.30) × 10−4 | –8.20 (±3.60) | 5 × 102 |
| P3 | 2.94 (±0.08) × 10−4 | –8.36 (±0.85) | 103 |
| P4 | 1.0 (±0.1) × 10−5 | –13.6 (±3.9) | 1 × 102 |
Photovoltaic cell characteristics
The hole-transporting properties and low-energy visible-near-IR absorption bands of P1–P4 suggest their possible use as donors in bulk-heterojunction solar cells. The soluble fullerene-3′-phenyl-3′H-cyclopropa[1,9](C60-Ih)[5,6]fullerene-3′-butanoic acid methyl ester (PCBM) was chosen as an acceptor due to its widespread use, allowing comparison of our device data to a wide range of literature. The solid-state EA of PCBM is estimated to be 3.7 eV82 using a similar approach to that used for estimated polymer IPs and EAs, close to values of 3.8–3.9 eV obtained directly from inverse photoelectron spectroscopy,83,84 indicating that PCBM is indeed an acceptor relative to the present polymers (Fig. 6a). Moreover, the free-energy changes, ΔG, for the formation of separated polymer positive polarons and PCBM radical anions from polymer excitons, estimated according to:| ΔG = IPpolymer − EAPCBM − Eg(opt)polymer |
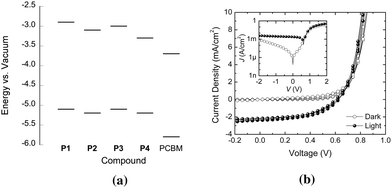 | ||
| Fig. 6 (a) Energy level diagram for P1–P4 and PCBM based on the electrochemically estimated IPs and EAs (IP of PCBM from UPS data in ref. 84). (b) Representative J–V characteristics measured in the dark (open circles) and under illumination (filled circles) for multiple cells based on a 1:3 (w/w) P3/PCBM blend. (Inset shows the same data in a semilogarithmic plot.) | ||
OPV devices were fabricated by spin-coating 80–90 nm films of polymer/PCBM blends from chlorobenzene solutions. Representative J–V characteristics are shown in Fig. 6b for an example of a P3-based device. Moderate PCEs ranging from 0.08–1.40% were obtained; as noted in the Introduction, considerably higher values have been reported for other D–A polymer/fullerene blends. For example, a 1:3 blend of the DTP–BTD polymer III with PCBM gave a PCE of 2.8%,36 a 1:4 blend of Qx D–A polymer V with the C70 analogue of PCBM gave a PCE of 5.5%,13 and 1:2 blends of the previously reported (dithienyl-DTP)–Qx polymer, VIII, with PCBM and its C70 analogue exhibited PCEs of 1.2% and 1.8%, respectively.49,87 For the best P3 and P4 devices, the open-circuit voltages, Voc, are quite high (0.6–0.7 V); however, Voc varies with the polymer/PCBM ratio (see below), indicating that, at least in these systems, Voc is not solely determined by the difference between the donor IP and polymer EA.2,4 Nevertheless, the Voc values obtained for the better P3 and P4 devices are larger than the values of 0.4–0.5 V reported for blends of the DTP–BTD polymers II or III with PCBM,36,39 consistent with the higher IPs estimated for the current polymers (see above), while values >0.9 V have been obtained for blends of PCBM with high-IP materials, such as the carbazole-based D–A polymers, IV.59 External quantum efficiency (EQE) spectra for P3 blends (Fig. 7) show that light is harvested throughout the visible, extending out to beyond ca. 800 nm, but that the EQE is low (<20%) over the wavelength range studied, accounting for the overall moderate PCEs, despite the relatively high values of Voc and fill factor, FF, obtained in the best devices. The differences between the EQE spectra and the thin-film spectrum of P3 suggest that PCBM absorption contributes a significant portion of the photocurrent.88 In part, the low EQE values can be attributed to the modest absorption coefficients of the polymers (for example, the αmax value shown in Table 3 for the low-energy absorption of P3 means that the polymer in an 85 nm film of a 1:1 blend would only absorb ca. 20% of the incident light at a wavelength of 720 nm). The low driving forces estimated above for charge separation may also limit the efficiency of exciton generation.
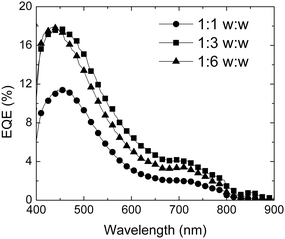 | ||
| Fig. 7 EQE spectra of OPV devices fabricated for different P3/PCBM ratios. | ||
The PCEs vary considerably with the polymer/fullerene ratio used in the devices, as has been found for other bulk heterojunction devices based on blends, the details of the variation depending on the choice of polymer. Thus, the PCE of P3-based devices is higher at a 1:3 than at a 1:6 ratio, while for P4, a 1:6 ratio performs better than 1:3. This is consistent with previous literature reports indicating that the optimum polymer/fullerene ratios depend on the specific polymer in question;89,90 for example, the optimum P3HT/PCBM ratio is ca. 1:1,91 while for poly[2-methoxy-5-(3′,7′-dimethyloctyloxy)-1,4-phenylene vinylene]/PCBM blends, the optimum ratio is closer to 1:4.92
AFM images of some of the P3/PCBM films were obtained; as shown in Fig. 8, the 1:3 blend shows a similar surface morphology to the 1:1 system, but with larger scale features, while the 1:6 blend shows a qualitatively different pattern. The increase in feature size with increasing PCBM content from the 1:1 to 1:3 blends is similar to what has been found in other conjugated polymer blends, such as poly[2-methoxy-5-(3′,7′-dimethyloctyloxy)-1,4-phenylene vinylene]/PCBM.93–95 As has been suggested for these other blends, the larger features in the 1:3 blend may be associated with larger polymer and fullerene domains leading to better phase percolation and, hence, a higher short-circuit current density, JSC. Accordingly, the 1:3 blend also shows a higher external quantum efficiency (EQE) than the 1:1 blend (Fig. 7). However, as shown in Table 5, other parameters contributing to the PCE—VOC and FF—also vary with blend composition.
 | ||
| Fig. 8
AFM height (left) and phase (right) images of films of (a) 1:1, (b) 1:3, and (c) 1:6 P3/PCBM blends (vertical grey scale is 50 nm for all three height images; vertical grey scale for phase images are 70, 80, and 45° for a, b and c, respectively, while each image shows a 1 × 1 µm square). RMS roughness values for (a), (b), and (c) are 7.2, 9.2 and 8.6 nm, respectively. | ||
| Polymer | Polymer/PCBM wt ratio | V OC/mV | J SC/mA cm−2 | FF | PCE (%) |
|---|---|---|---|---|---|
| P1 | 1:1 |
439 ± 3 | 2.20 ± 0.10 | 0.44 ± 0.01 | 0.57 ± 0.02 |
| P2 | 1:1 |
468 ± 5 | 1.71 ± 0.29 | 0.33 ± 0.01 | 0.37 ± 0.07 |
| P3 | 2:1 |
544 ± 3 | 1.23 ± 0.02 | 0.35 ± 0.01 | 0.31 ± 0.01 |
| 1:1 |
579 ± 4 | 2.12 ± 0.08 | 0.42 ± 0.01 | 0.76 ± 0.02 | |
| 1:2 |
594 ± 9 | 2.68 ± 0.06 | 0.49 ± 0.01 | 1.00 ± 0.10 | |
| 1:3 |
626 ± 7 | 2.69 ± 0.03 | 0.61 ± 0.02 | 1.40 ± 0.07 | |
| 1:4 |
624 ± 5 | 1.87 ± 0.13 | 0.66 ± 0.03 | 1.00 ± 0.10 | |
| 1:5 |
624 ± 5 | 1.67 ± 0.03 | 0.64 ± 0.03 | 0.87 ± 0.05 | |
| 1:6 |
624 ± 26 | 2.27 ± 0.02 | 0.57 ± 0.12 | 1.10 ± 0.28 | |
| P4 | 3:1 |
634 ± 37 | 0.38 ± 0.01 | 0.24 ± 0.01 | 0.08 ± 0.01 |
| 1:1 |
496 ± 18 | 0.88 ± 0.02 | 0.25 ± 0.01 | 0.16 ± 0.06 | |
| 1:3 |
527 ± 42 | 1.82 ± 0.03 | 0.35 ± 0.01 | 0.48 ± 0.03 | |
| 1:6 |
705 ± 14 | 2.14 ± 0.04 | 0.33 ± 0.02 | 0.71 ± 0.05 |
Conclusions
D–A copolymers, P1–P4, based on DTP donors coupled to acceptors based on Qx, PPz, and extended analogues have been synthesised and characterised. These polymers exhibit absorption bands with maxima in either the long-wavelength part of the visible or in the near-IR; comparison with other Qx and PPz polymers indicates that DTP is a strong donor, consistent with previous work, while the acceptors are found to be roughly comparable in strength to the widely used BTD acceptor (with acceptor strengths in the order dibenzopyridoquinoxaline > BTD > PPz > benzophenazine > Qx). These trends are further supported by electrochemical data. Electrochemically estimated IPs are higher than for all-donor DTP-based polymers and for some DTP-based D–A polymers, such as those containing BTD, although still much lower than those of many other D–A polymers that have been used in solar cells.All four copolymers function as hole-transport materials in OFETs with mobilities in the range 10−5 to 10−4 cm2 V−1s−1, although on/off current ratios were relatively low, in common with other DTP polymers, for which inadvertent doping by air has been postulated, despite the somewhat higher IPs of the current materials. In view of their optical and charge-transport characteristics, the polymers were used as donors in bulk heterojunction cells in combination with PCBM. Consistent with the estimated IPs, the best values of VOC obtained in solar cells fabricated from blends with PCBM are larger than those found for blends based on other DTP polymers, but fall short of those obtained with, for example, D–A polymers with carbazole-based donors. Despite the relatively good VOC and FF values found for some blends, PCEs are only moderate due to poor values of Jsc, arising from rather low EQEs. Further work is needed to determine the role of the morphologies obtained in the cells, the details of exciton diffusion within the polymer, and the rather small driving forces estimated for charge transfer from a polymer exciton to the fullerene in giving these low efficiencies. However, the relatively small absorption coefficients of the polymers undoubtedly play a major role in limiting the EQE. Thicker active layers could partially circumvent this problem, although fabrication of such layers from solution is not trivial and this would have to be done without adversely affecting charge transport. It is also worth noting that in the current polymers solubilising trialkoxylphenyl and alkyl or alkoxy groups account for a significant portion of the molecular weight (for example, 73% in the case of P3) and so, in principle, much higher absorption coefficients could be achieved by greatly reducing this proportion; a future challenge for materials of the present type, and for D–A polymers more generally, will be to reduce the dilution effects of solubilising groups while retaining acceptable solubility for device fabrication.
Experimental section
General
1H NMR and 13C{1H} NMR spectra were measured on a Varian Mercury 300 MHz or a Bruker 400 MHz spectrometer. The microwave used was a CEM Discover Labmate. Mass spectra were measured on a VG Instruments 70-SE using the electron impact (EI) mode or on an Applied Biosystems 4700 Proteomics Analyzer using MALDI mode. Elemental analyses were carried out by Atlantic Microlabs using a LECO 932 CHNS elemental analyzer. Gel permeation chromatography (GPC) analysis for P1–P4 was performed at 25 °C on Waters styragel HR 4, 3, and 1 columns coupled with a Waters 2410 Refractive Index detector and 2690 separations module, using toluene as eluent, against polystyrene standards, and a flow rate of 1 mL min−1. UV-vis absorption spectra were recorded on Varian Cary 500 spectrophotometer. Thicknesses of films were measured using a profilometer (Dektak 6M Stylus, Veeco, Plainview, NY). Cyclic voltammetry was carried out using a BAS 100B electrochemical analyzer, 0.1 M [nBu4N]+[PF6]− in acetonitrile as electrolyte, a polymer film drop cast onto the platinum working electrode, a platinum wire auxiliary electrode, and a Ag+/Ag pseudo-reference electrode consisting of a silver wire immersed into a solution of electrolyte containing AgNO3. The pseudo-reference electrode was calibrated using the ferrocenium/ferrocene couple; values reported relative to the saturated calomel electrode (SCE) were derived using a value of +0.40 V vs.SCE for the ferrocenium/ferrocene couple in 0.1 M [nBu4N]+[PF6]−acetonitrile solution.96Thermogravimetric analysis (TGA) analysis was conducted with a NETZSCH thermogravimetric analyzer (model STA 449C) under a nitrogen flow at a heating rate of 10 °C min−1. AFM images were taken on a Digital Instruments NanoScope™ Scanning Probe Microscope.5,8-Dibromo-2,3-di-n-decylquinoxaline (M1)
The title compound was synthesised as a white solid (0.50 g, 55%) in a similar fashion to the analogous diphenyl compound.721H NMR (300 MHz, CDCl3): δ 7.79 (s, 1H), 3.05 (t, J = 7.5 Hz, 4H), 1.91 (quint, J = 7.5 Hz, 4H), 1.46–1.24 (m, 28H), 0.86 (t, J = 6.8 Hz, 6H). 13C{1H} NMR (100 MHz, CDCl3): δ 158.3, 139.3, 131.9, 123.3, 34.8, 31.9, 29.6 (two peaks separated by 0.04 ppm), 29.5 (two peaks separated by 0.04 ppm), 29.3, 27.8, 22.7, 14.1. HRMS (EI) calcd for C28H44N2Br2 (M+): 566.1895; found: 566.1871. Anal. Calcd for C28H44N2Br2: C, 59.16; H, 7.80; N, 4.93. Found: C, 59.26; H, 7.99; N, 4.84%.5,8-Dibromo-2,3-di-n-decyl-pyrido[3,4-b]pyrazine (M2)
The title compound was synthesised as a white solid (0.40 g, 50%) in a similar fashion to the analogous diphenyl compound.70,741H NMR (300 MHz, CDCl3): δ 8.66 (s, 1H), 3.08 (t, J = 7.5 Hz, 2H), 3.07 (t, J = 7.5 Hz, 2H), 1.89 (m, 4H), 1.46–1.24 (m, 28H), 0.86 (t, J = 6.8 Hz, 6H). 13C{1H} NMR (75 MHz, CDCl3): δ 163.2, 160.5, 146.4, 145.7, 142.5, 136.0, 120.1, 35.1, 34.9, 31.9, 29.6, 29.5 (two peaks separated by 0.08 ppm), 29.4 (two peaks separated by 0.04 ppm), 29.3, 27.5, 22.7, 14.1 (8 alkyl carbon resonances not observed, presumably due to overlap). HRMS (EI) calcd. for C27H43N3Br2: 567.1824; found: 567.1848. Anal. Calcd for C27H43N3Br2: C, 56.95; H, 7.61; N, 7.38. Found: C, 57.05; H, 7.46; N, 7.36%.10,13-Dibromo-2,3,6,7-tetrakis(n-decyloxy)dibenzo[a,c]phenazine (M3)
2,3,6,7-Tetrakis(n-decyloxy)phenanthrene-9,10-dione 73 (0.35 g, 0.42 mmol), 3,6-dibromobenzene-1,2-diamine (0.30 g, 1.1 mmol),72 and acetic acid (30 mL) were heated to reflux overnight in a 100 mL three-necked flask. After the reaction mixture was allowed to cool to room temperature, water (50 mL) was added and the resulting yellow precipitate was collected by filtration. The crude product was recrystallised from a mixture dichloromethane and acetone to give a yellow solid (0.35 g, 78%). 1H NMR (300 MHz, CDCl3): δ 8.75 (s, 2H), 7.92 (s, 2H), 7.62 (s, 2H), 4.32 (t, J = 6.6 Hz, 4H), 4.26 (t, J = 6.6 Hz, 4H), 1.98 (m, 8H), 1.65–1.24 (m, 56H), 0.86 (t, J = 6.8 Hz, 12H). 13C{1H} NMR (100 MHz, CDCl3): δ 152.5, 149.4, 142.6, 139.2, 131.8, 127.1, 123.8, 122.9, 109.3, 106.2, 69.6, 69.0, 31.9, 29.7 (two peaks separated by 0.03 ppm), 29.6 (two peaks separated by 0.09 ppm), 29.4, 29.2, 26.2 (two peaks separated by 0.04 ppm), 22.7, 14.1 (7 alkyl carbon resonances not observed, presumably due to overlap). MS (MALDI): m/z 1063 (M+). Anal. Calcd for C60H90N2BrO4: C, 67.78; H, 8.53; N, 2.63; Found: C, 67.91; H, 8.61; N, 2.56%.10,13-Dibromo-2,3,6,7-tetrakis(n-decyloxy)dibenzo[f,h]pyrido[4,3-b]quinoxaline (M4)
2,3,6,7-Tetrakis(n-decyloxy)phenanthrene-9,10-dione 73 (0.40 g, 0.50 mmol), 2,5-dibromopyridine-3,4-diamine (0.20 g, 0.75 mmol),59acetic acid (0.5 mL), and ethanol (3 mL) were sealed in a 10 mL vial, heated to 150 °C for ca. 25 min (standard mode, run time of 5 min, hold time of 20 min, maximum pressure of 110 psi, maximum power of 100 W). The reaction mixture was allowed to cool to room temperature, and then irradiated for another ca. 25 min using the same conditions. The same reaction was carried out for another batch with the same amounts of starting materials and solvents. The reaction mixture from two vials was combined, and washed with aqueous NaHCO3 solution. The organic phase was separated and evaporated under reduced pressure. The crude product was purified by column chromatography (silica gel, CH2Cl2 as eluent) to give a orange solid (0.66 g, 65%). 1H NMR (300 MHz, CDCl3, 0.01 M):97δ 8.70 (s, 1H), 8.54 (s, 1H), 8.51 (s, IH), 7.47 (s, 1H), 7.45 (s, 1H), 4.25 (m, 8H), 1.99 (m, 8H), 1.63–1.30 (m, 56H), 0.90 (t, J = 7.8 Hz, 12H). 13C{1H} NMR (75 MHz, CDCl3): δ 153.4, 152.9, 149.5, 149.3, 146.5, 155.5, 145.2, 143.9, 141.6, 135.5, 128.2, 127.1, 122.2, 121.8, 120.2, 109.3, 109.0, 105.7, 105.5, 69.4, 69.3, 68.9, 31.9, 29.8, 29.7 (two peaks separated by 0.06 ppm), 29.6, 29.4, 29.3 (two peaks separated by 0.04 ppm), 29.2, 26.2 (two peaks separated by 0.03 ppm), 22.7, 14.1 (24 alkyl carbon resonances not observed, presumably due to overlap). MS (MALDI): m/z 1064 (M+). Anal. Calcd for C59H89N3BrO4: C, 66.59; H, 8.43; N, 3.95. Found: C, 66.74; H, 8.43; N, 3.93%.Polymer P1
Monomers M1 (0.40 g, 0.70 mmol) and 2,6-bis(tri-n-butylstannyl)-N-(3,4,5-tris(n-dodecyloxy)phenyl)-dithieno[3,2-b:2′,3′-d]pyrrole (M5)26 (0.98 g, 0.70 mmol), dry THF (30 mL), and PdCl2(PPh3)2 (0.025 g, 0.04 mmol) were sealed in a pressure vessel under nitrogen. The resulting mixture was stirred at 60–70 °C for 4 d, after which time the vessel was opened to air; the organics were washed with aqueous KF solution, and extracted with chloroform. The organic layer was separated, concentrated under reduced pressure, and dropped into methanol (ca. 500 mL). The resulting solid was filtered and purified by successive Soxhlet extractions with methanol, acetone and hexanes, each for 1 d. The remaining solid was redissolved into chloroform and was precipitated into methanol again to give a black solid (0.80 g, 93%). 1H NMR (300 MHz, CD2Cl2): δ 8.16 (br, 2H), 7.02–6.76 (br, 4H), 4.05 (br, 6H), 3.12 (br, 4H), 2.10–1.12 (br, 92H), 0.91 (br, 15H). Anal. Calcd for (C78H123N3O3S2)n: C, 77.11; H, 10.20; N, 3.46. Found: C, 76.57; H, 10.07; N, 3.42%.Polymer P2
Monomers M2 (0.38 g, 0.67 mmol) and M526 (0.93 g, 0.67 mmol), dry THF (30 mL), and PdCl2(PPh3)2 (0.024 g, 0.04 mmol) were sealed in a pressure vessel under nitrogen. The vessel was sealed after the additions and taken out of the glove box. The resulting mixture was stirred at 60–70 °C for 4 d, after which time the vessel was opened to air; the organics were washed with aqueous KF solution, and extracted with toluene. The organic layer was separated and evaporated under reduced pressure. The resulting solid was dissolved into THF (ca. 20 mL), and dropped into methanol (ca. 500 mL), and the solid was filtered. The resulting solid was filtered and purified by successive Soxhlet extractions with methanol, acetone, and hexanes, each for 1 day. The remaining solid was dissolved into chloroform and was precipitated into methanol. The precipitate was filtered and dissolved in THF and passed through a silica gel plug, eluting with THF, and then subjected to size-exclusion column chromatography (SX-1 bio-beads, THF as eluent). The solution was concentrated, and precipitated again into methanol to give a black solid (0.58 g, 72%). 1H NMR (300 MHz, CD2Cl2): δ 8.90–9.10 (br, 1H), 7.62 (br, 1H), 7.28 (br, 1H), 6.80–7.02 (br, 2H), 4.10 (br, 6H), 3.16 (br, 4H), 2.10–1.12 (br, 92H), 0.91 (br, 15H). Anal. Calcd for (C77H122N4O3S2)n: C, 76.06; H, 10.11; N, 4.61. Found: C, 75.11; H, 10.02; N, 4.34%.Polymer P3
Monomers M3 (0.50 g, 0.47 mmol) and M526 (0.65 g, 0.47 mmol), dry THF (25 mL), and PdCl2(PPh3)2 (0.017 g, 0.03 mmol) were sealed in a pressure vessel under nitrogen. The resulting mixture was stirred at 60–70 °C for 4 d, after which time the vessel was opened to air; the organics were washed with aqueous KF solution, and extracted with toluene. The organic layer was separated and concentrated under reduced pressure. The concentrated solution was dropped into methanol (ca. 500 mL); the resulting solid was collected and purified by successive Soxhlet extractions with methanol, acetone, and hexanes, each for 1 d. The hexane extracts were evaporated under reduced pressure, dissolved into chloroform, and precipitated into methanol to give a black solid (0.53 g, 66%). 1H NMR (300 MHz, THF-d8): δ 9.36 (br, 2H), 8.50–7.80 (br, 6H), 7.15 (br, 2H), 4.62–4.02 (br, 14H), 2.00–1.02 (br, 124H), 0.90 (br, 21H). Anal. Calcd for (C110H169N3O7S2)n: C, 77.28; H, 10.20; N, 2.46. Found: C, 76.83; H, 10.06; N, 2.49%.Polymer P4
Monomers M4 (0.44 g, 0.42 mmol) and M526 (0.59 g, 0.42 mmol), dry THF (30 mL), and PdCl2(PPh3)2 (0.015 g, 0.02 mmol) were sealed in a pressure vessel under nitrogen. The resulting mixture was stirred at 60–70 °C for 4 d, after which time the vessel was opened to air; the organics were washed with aqueous KF solution, and extracted with chloroform. The organic extracts were concentrated under reduced pressure and dropped into methanol (ca. 500 mL); the resulting solid was collected and purified by successive Soxhlet extraction with methanol, acetone and hexanes, each for 1 d. The hexane extracts were concentrated under reduced pressure and were precipitated into methanol. The precipitate was filtered and dissolved in THF and passed through a silica gel plug, eluting with THF, and then subjected to size-exclusion column chromatography (SX-1 bio-beads, THF as eluent). The solution was concentrated and precipitated from methanol to give a black solid (0.53 g, 75%). 1H NMR (300 MHz, THF-d8): δ 9.42 (br, 1H), 8.80 (br, 2H), 8.40–6.80 (br, 4H), 6.70 (br, 2H), 4.62–3.86 (br, 14H), 2.00–1.10 (br, 124H), 0.90 (br, 21H). Anal. Calcd for (C109H168N4O7S2)n: C, 76.53; H, 9.90; N, 3.28. Found: C, 75.69; H, 9.90; N, 3.25%.Fabrication and characterisation of thin film transistors
OFETs were fabricated on heavily doped n-type silicon substrates (which also serve as gate electrodes) with 200 nm thick thermally grown SiO2 as the gate dielectric, in a top-contact configuration. Ti/Au (10 nm/100 nm) metallisation on the backside of the substrate was performed to enhance the gate electrical contact. Firstly, the substrates were cleaned using an O2 plasma for 3 min. Surface treatment (with 5 mM solutions in toluene) was done by soaking the substrates in n-butyl-trichlorosilane (BTS, used for P1 and P3) or n-octyl-trichlorosilane (OTS, used for P2 and P4) solution for 17 h in a N2-filled dry box. The substrates were rinsed with toluene, and annealed at 60 °C for 5 min. The capacitance of the BTS or OTS treated SiO2 was about 16.2 nF cm−2. A thin layer of organic semiconductor (measured at 60 nm for P2 and 40 nm for P4) was formed on the substrates by spin coating with a solution (10 mg mL−1 for P1–P3, 15 mg mL−1 for P4) in chlorobenzene (60 s at 700 rpm for P1–P3 and 1000 rpm for P4). To remove solvent, the films were annealed at 90 °C for 30 min. 50 nm thick Au was deposited through a shadow mask to act as top source and drain electrodes. The prepared devices were annealed at 130 °C for 35 min inside a N2 glove box.Fabrication and characterisation of photovoltaic cells
Devices were fabricated using ITO-coated glass (Colorado Concept Coating LLC) substrates with resistivity of ca. 15 Ω sq−1. The substrate was cleaned by immersing in deionised water with detergent and rinsing with de-ionised water in an ultrasonic bath for 20 min, followed by successive ultrasonic cleaning using acetone and isopropanol, each for 20 min. Nitrogen was used to dry the substrates after each of the last three baths. Next, SiOx (300 nm) was deposited through a first shadow mask on half of the cleaned ITO surface by electron-beam deposition (AXXIS, Kurt J. Lesker) at a rate of 5 Å s−1 at 3.3 × 10−7 Torr and room temperature. Prior to deposition of the hole-conducting layer, the ITO/glass substrate with SiOx was cleaned with isopropanol through ultrasonication for 10 min and the surface was prepared by exposure to an air plasma for 3 min. PEDOT:PSS (Baytron P VP AI 4083 PE FL) was filtered though 0.45 µm pore-PVDF filters and spin-coated onto the substrate at the speed of 5000 rpm for 1 min, followed by annealing at 140 °C for 10 min in the atmosphere. Mixtures of polymers and PCBM with different weight ratios were dissolved in chlorobenzene at the concentration of 20 mg mL−1 without any filtration and were then spin-coated onto the active region at 1000 rpm for 1 min to give a film of 80–90 nm thickness. Each film deposition step was performed in the M-Braun nitrogen glove box. The substrate was loaded on a second shadow mask for cathode deposition and taken into a thermal evaporation system (SPECTROS, Kurt J. Lesker) connected with the glove box. And 200 nm Al electrode was deposited on the top of the active region at a rate of 10–20 nm s−1 under the 5.0 × 10−8 Torr at the room temperature. The sample was then annealed at 150 °C for 25 min. Electrical properties were measured with a source meter (2400 Keithley) controlled by a LabVIEW program under nitrogen environment. For testing solar cell properties under illumination, filtered light from a 175 W Xenon lamp (ASB-XE-175EX, CVI) was used for broad light source with an irradiance of ∼71 to 74 mW cm−2.
Quantum-chemical calculations
Optimised geometries and electronic energy levels were calculated in the gas phase using DFT at the B3LYP/6-31G(d,p) level of theory. Excited-state-energy calculations were performed at the same level of theory using the time-dependent method (TD-DFT).Acknowledgements
We thank Séverine Coppée for acquiring the AFM images and Raghunath Dasari for synthesising one of the diketone starting materials. This work was funded in part by DARPA (N00014-06-1-0897), the Office of Naval Research (N00014-08-1-0928), the AFOSR (FA9550-09-1-0320) and the STC Program of the National Science Foundation under agreement No. DMR-0120967. The solid-state device work was performed in part at the Microelectronics Research Center at Georgia Institute of Technology, a member of the National Nanotechnology Infrastructure Network, which is supported by NSF (Grant No. ECS-03-35765).Notes and references
- R. H. Friend, R. W. Gymer, A. B. Holmes, J. H. Burroughes, R. N. Marks, C. Taliani, D. D. C. Bradley, D. A. D. Santos, J. L. Brédas, M. Lögdlund and W. R. Salaneck, Nature, 1999, 397, 121 CrossRef CAS
.
- C. Winder and N. S. Sariciftci, J. Mater. Chem., 2004, 14, 1077 RSC
- B. de Boer and A. Facchetti, Polym. Rev., 2008, 48, 423 Search PubMed
- G. Dennler, M. C. Scharber and C. J. Brabec, Adv. Mater., 2009, 21, 1323 CrossRef CAS
- Y.-J. Cheng, S.-H. Yang and C.-S. Hsu, Chem. Rev., 2009, 109, 5868 CrossRef CAS
-
R. Hamilton, M. Heeney, T. Anthopoulos and I. McCulloch, in Organic Electronics, ed. F. So, Boca Raton, FL, 2010 Search PubMed
-
M. Tamilvanan and S.-H. Jin, in Organic Electronics, ed. F. So, Boca Raton, FL, 2010 Search PubMed
- J. Peet, J. Y. Kim, N. E. Coates, W. L. Ma, D. Moses, A. J. Heeger and G. C. Bazan, Nat. Mater., 2007, 6, 497 CrossRef CAS
- N. Blouin, A. Michaud and M. Leclerc, Adv. Mater., 2007, 19, 2295 CrossRef CAS
- E. G. Wang, L. Wang, L. F. Lan, C. Luo, W. L. Zhuang, J. B. Peng and Y. Cao, Appl. Phys. Lett., 2008, 92, 033307 CrossRef
- S. H. Park, A. Roy, S. Beaupre, S. Cho, N. Coates, J. S. Moon, D. Moses, M. Leclerc, K. Lee and A. J. Heeger, Nat. Photonics, 2009, 3, 297 CrossRef CAS
- H.-Y. Chen, J. Hou, S. Zhang, Y. Liang, G. Yang, Y. Yang, L. Yu, Y. Wu and G. Li, Nat. Photonics, 2009, 3, 649 CrossRef CAS
- D. Kitazawa, N. Watanabe, S. Yamamoto and J. Tsukamoto, Appl. Phys. Lett., 2009, 95, 053701 CrossRef
- Y. Liang, D. Feng, Y. Wu, S.-T. Tsai, G. Li, C. Ray and L. Yu, J. Am. Chem. Soc., 2009, 131, 7792 CrossRef CAS
- P.-T. Wu, T. Bull, F. S. Kim, C. K. Luscombe and S. A. Jenekhe, Macromolecules, 2009, 42, 671 CrossRef CAS
- Y. Zou, A. Najari, P. Berrouard, S. Beaupré, B. R. Aïch, Y. Tao and M. Leclerc, J. Am. Chem. Soc., 2010, 132, 5330 CrossRef CAS
- Y. Zhang, S. K. Hau, H.-L. Yip, Y. Sun, O. Acton and A. K.-Y. Jen, Chem. Mater., 2010, 22, 2696 CrossRef CAS
- Y. Liang, Z. Xu, J. Xia, S.-T. Tsai, G. L. Yue Wu, C. Ray and L. Yu, Adv. Mater., 2010, 22, E135 CrossRef CAS
- C. Piliego, T. W. Holcombe, J. D. Douglas, C. H. Woo, P. M. Beaujuge and J. M. J. Fréchet, J. Am. Chem. Soc., 2010, 132, 7595 CrossRef CAS
- J.-Y. Wang, S. K. Hau, H.-L. Yip, J. A. Davies, K.-S. Chen, Y. Zhang, Y. Sun and A. K.-Y. Jen, Chem. Mater., 2011, 23, 765 CrossRef CAS
- J. Zaumseil, C. L. Donley, J. S. Kim, R. H. Friend and H. Sirringhaus, Adv. Mater., 2006, 18, 2708 CrossRef CAS
- L. Bürgi, M. Turbiez, R. Pfeiffer, F. Bienewald, H.-J. Kirner and C. Winnewisser, Adv. Mater., 2008, 20, 2217 CrossRef CAS
- A. Gadisa, W. Mammo, L. M. Andersson, S. Admassie, F. Zhang, M. R. Andersson and O. Inganäs, Adv. Funct. Mater., 2007, 17, 3836 CrossRef CAS
- H. Usta, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2008, 130, 8580 CrossRef CAS
- M. C. Gwinner, S. Khodabakhsh, H. Giessen and H. Sirringhaus, Chem. Mater., 2009, 21, 4425 CrossRef CAS
- T. T. Steckler, X. Zhang, J. Hwang, R. Honeyager, S. Ohira, X. H. Zhang, A. Grant, S. Ellinger, S. A. Odom, D. Sweat, D. B. Tanner, A. G. Rinzler, S. Barlow, J. L. Bredas, B. Kippelen, S. R. Marder and J. R. Reynolds, J. Am. Chem. Soc., 2009, 131, 2824 CrossRef CAS
- A. P. Zoombelt, S. G. J. Mathijssen, M. G. R. Turbiez, M. M. Wienk and R. A. J. Janssen, J. Mater. Chem., 2010, 20, 2240 RSC
- F. S. Kim, X. Guo, M. D. Watson and S. A. Jenekhe, Adv. Mater., 2010, 22, 478 CrossRef CAS
- Although most reports of DTP polymers have appeared in the last few years, the electropolymerisation of DTP derivatives was first reported in the early 1990s ( A. Berlin, G. Pagani, G. Zotti and G. Schiavon, Makromol. Chem., 1992, 193, 399 Search PubMed
- K. Ogawa and S. C. Rasmussen, Macromolecules, 2006, 39, 1771 CrossRef CAS
- G. Koeckelberghs, L. De Cremer, A. Persoons and T. Verbiest, Macromolecules, 2007, 40, 4173 CrossRef CAS
- J. Y. Liu, R. Zhang, G. Sauve, T. Kowalewski and R. D. McCullough, J. Am. Chem. Soc., 2008, 130, 13167 CrossRef CAS
- E. J. Zhou, M. Nakamura, T. Nishizawa, Y. Zhang, Q. S. Wei, K. Tajima, C. H. Yang and K. Hashimoto, Macromolecules, 2008, 41, 8302 CrossRef CAS
- W. Zhang, J. Li, B. Zhang and J. Qin, Macromol. Rapid Commun., 2008, 29, 1603 CrossRef CAS
- W. Zhang, J. Li, L. Zou, B. Zhang, J. Qin, Z. Lu, Y. F. Poon, M. B. Chan-Park and C. M. Liw, Macromolecules, 2008, 41, 8953 CrossRef CAS
- W. Yue, Y. Zhao, S. Y. Shao, H. K. Tian, Z. Y. Xie, Y. H. Geng and F. S. Wang, J. Mater. Chem., 2009, 19, 2199 RSC
- X. W. Zhan, Z. A. Tan, E. J. Zhou, Y. F. Li, R. Misra, A. Grant, B. Domercq, X. H. Zhang, Z. S. An, X. Zhang, S. Barlow, B. Kippelen and S. R. Marder, J. Mater. Chem., 2009, 19, 5794 RSC
- S. Zhang, Y. Guo, H. Fan, Y. Liu, H.-Y. Chen, G. Yang, X. Zhan, Y. Liu, Y. Li and Y. Yang, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5498 CrossRef CAS
- X. Zhang, T. T. Steckler, R. R. Dasari, S. Ohira, W. J. Potscavage, S. P. Tiwari, S. Coppee, S. Ellinger, S. Barlow, J. L. Bredas, B. Kippelen, J. R. Reynolds and S. R. Marder, J. Mater. Chem., 2010, 20, 123 RSC
- M. Zhang, H. Fan, X. Guo, Y. He, Z. Zhang, J. Min, J. Zhang, G. Zhao, X. Zhan and Y. Li, Macromolecules, 2010, 43, 5706 CrossRef CAS
- R. S. Ashraf, J. Gilot and R. A. J. Janssen, Sol. Energy Mater. Sol. Cells, 2010, 94, 1759 CrossRef CAS
- W. Vanormelingen, L. Pandey, M. Van der Auweraer, T. Verbiest and G. Koeckelberghs, Macromolecules, 2010, 43, 2157 CrossRef CAS
- E. Zhou, K. Tajima, C. Yang and K. Hashimoto, J. Mater. Chem., 2010, 20, 2362 RSC
- T. L. Nelson, T. M. Young, J. Liu, S. P. Mishra, J. A. Belot, C. L. Balliet, A. E. Javier, T. Kowalewski and R. D. McCullough, Adv. Mater., 2010, 22, 4617 CrossRef CAS
- M. Fujitsuka, T. Sato, F. Sezaki, K. Tanaka, A. Watanabea and O. Ito, J. Chem. Soc., Faraday Trans., 1998, 94, 3331 RSC
- K. Ogawa and S. Rasmussen, J. Org. Chem., 2003, 68, 2921 CrossRef CAS
- S. A. Odom, K. Lancaster, L. Beverina, K. M. Lefler, N. J. Thompson, V. Coropceanu, J.-L. Brédas, S. R. Marder and S. Barlow, Chem.–Eur. J., 2007, 13, 9637 CrossRef CAS
- S. Barlow, S. A. Odom, K. Lancaster, Y. A. Getmanenko, R. Mason, V. Coropceanu, J.-L. Brédas and S. R. Marder, J. Phys. Chem. B, 2010, 114, 14397 CrossRef CAS
- E. Zhou, J. Cong, K. Tajima and K. Hashimoto, Chem. Mater., 2010, 22, 4890 CrossRef CAS
-
Conjugated polymers incorporating quinoxaline-2,6-diyl and -2,3-diyl groups have also been reported ( N. Saito, T. Kanbara, T. Kushida, K. Kubota and T. Yamamoto, Chem. Lett., 1993, 1775 Search PubMed
- Y. Yang and Q. Pei, Appl. Phys. Lett., 1997, 70, 1926 CrossRef CAS
- M. Jonforsen, T. Johansson, O. Inganäs and M. R. Andersson, Macromolecules, 2002, 35, 1638 CrossRef CAS
- L. Huo, Z. Tan, Y. Zhou, E. Zhou, M. Han and Y. Li, Macromol. Chem. Phys., 2007, 208, 1294 CrossRef CAS
- T. Yamamoto, Z. Zhou, T. Kanbara, M. Shimura, K. Kizu, T. Maruyama, Y. Nakamura, T. Fukuda, B.-L. Lee, N. Ooba, S. Tomaru, T. Kurihara, T. Kaino, K. Kubota and S. Sasaki, J. Am. Chem. Soc., 1996, 118, 10389 CrossRef CAS
- R. D. Champion, K. F. Cheng, C. L. Pai, W. C. Chen and S. A. Jenekhe, Macromol. Rapid Commun., 2005, 26, 1835 CrossRef CAS
- R. S. Ashraf, H. Hoppe, M. Shahid, G. Gobsch, S. Sensfuss and E. Klemm, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 6952 CrossRef CAS
- W.-C. Wu, W.-Y. Lee, C.-L. Pai, W.-C. Chen, C.-S. Tuan and J.-L. Lin, J. Polym. Sci., Part B: Polym. Phys., 2006, 45, 67
- L. J. Huo, Z. A. Tan, X. Wang, Y. Zhou, M. F. Han and Y. F. Li, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 4038 CrossRef CAS
- N. Blouin, A. Michaud, D. Gendron, S. Wakim, E. Blair, R. Neagu-Plesu, M. Belletete, G. Durocher, Y. Tao and M. Leclerc, J. Am. Chem. Soc., 2008, 130, 732 CrossRef CAS
- C. L. Liu, J. H. Tsai, W. Y. Lee, W. C. Chen and S. A. Jenekhe, Macromolecules, 2008, 41, 6952 CrossRef CAS
- Z. Chen, J. Bouffard, S. E. Kooi and T. M. Swager, Macromolecules, 2008, 41, 6672 CrossRef CAS
- M. H. Lai, C. C. Chueh, W. C. Chen, J. L. Wu and F. C. Chen, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 973 CrossRef CAS
- J. H. Tsai, C. C. Chueh, M. H. Lai, C. F. Wang, W. C. Chen, B. T. Ko and C. Ting, Macromolecules, 2009, 42, 1897 CrossRef CAS
- J. W. Chen and Y. Cao, Acc. Chem. Res., 2009, 42, 1709 CrossRef CAS
- J.-Y. Lee, W.-S. Shin, J.-R. Haw and D.-K. Moon, J. Mater. Chem., 2009, 19, 4938 RSC
- S. Tarkuc, Y. A. Udum and L. Toppare, Polymer, 2009, 50, 3458 CrossRef CAS
- G. Zhang, Y. Fu, Q. Zhang and Z. Xie, Polymer, 2010, 51, 2313 CrossRef CAS
- J.-H. Tsai, C.-C. Chueh, W.-C. Chen, C.-Y. Yu, G.-W. Hwang, C. Ting, E.-C. Chen and H.-F. Meng, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2351 CrossRef CAS
- N. Jonforsen, I. Ahmad, T. Johansson, J. Larsson, L. S. Roman, M. Svensson, O. Inganäs and M. R. Andersson, Synth. Met., 2001, 119, 185 CrossRef
- P. T. Wu, F. S. Kim, R. D. Champion and S. A. Jenekhe, Macromolecules, 2008, 41, 7021 CrossRef CAS
- K. M. Noone, E. Strein, N. C. Anderson, P.-T. Wu, S. A. Jenekhe and D. S. Ginger, Nano Lett., 2010, 10, 2635 CrossRef CAS
- M. J. Edelmann, J. M. Raimundo, N. F. Utesch, F. Diederich, C. Boudon, J. P. Gisselbrecht and M. Gross, Helv. Chim. Acta, 2002, 85, 2195 CrossRef CAS
- B. Mohr, V. Enkelmann and G. Wegner, J. Org. Chem., 1994, 59, 635 CrossRef CAS
- B. L. Lee and T. Yamamoto, Macromolecules, 1999, 32, 1375 CrossRef CAS
- Note that the literature comparison is between an unsubstituted benzo[a,c]phenazine-10,13-diyl and a 2,3-diphenyl Qx derivative, while the current comparison is between a 2,3,6,7-tetraalkoxybenzo[a,c]phenazine-10,13-diyl and a 2,3-dialkyl Qx.
- C. Lavigueur, E. J. Foster and V. E. Williams, J. Am. Chem. Soc., 2008, 130, 11791 CrossRef CAS
- B. X. Gao, M. Wang, Y. X. Cheng, L. X. Wang, X. B. Jing and F. S. Wang, J. Am. Chem. Soc., 2008, 130, 8297 CrossRef CAS
- For example, for II (P1 of ref. 39): THF, λmax (αmax) 472 (2.31), 674 (3.92) nm (M−1 cm−1); film, λmax (αmax) 470 (1.1), 686 (2.1) nm (cm−1). For its 6,7-di(n-hexyl)thiadiazolo[3,4-g]quinoxaline analogue (P2 of ref. 39): 508 (3.11), 931 (2.93) nm (M−1 cm−1); film, λmax (εmax) 514 (2.0), 941 (2.0) nm (cm−1). For its benzo[1,2-c;4,5-c′]bis[1,2,5]thiadiazole analogue (P3 of ref. 39): THF, λmax (εmax) 516 (2.69), 1154 (2.49) nm (M−1 cm−1); film, λmax (εmax) 526 (3.5), 1206 (3.5) nm (cm−1).
- D. E. Motaung, G. F. Malgas and C. J. Arendse, J. Mater. Sci., 2010, 45, 3276 CrossRef CAS
- L. Micaroni, F. C. Nart and I. A. Hummelgen, J. Solid State Electrochem., 2002, 7, 55 CrossRef CAS
- Ref. 80 specifically derives an approximate offset of 4.4 eV for potentials measured vs. the AgCl/Ag electrode, but due to the similarity of AgCl/Ag and SCE potentials (0.222 and 0.242 V vs.NHE, respectively; Handbook of Chemistry and Physics, CRC Press, Boca Raton, FL, 63rd edn, 1982), the same relation can be applied to potentials vs.SCE. The 4.4 eV offset is also equivalent to the offset of 4.8 eV commonly used for potentials vs.FeCp2+/0.
- This value is estimated using a value of −1.08 V vs.FeCp2+/0 reported for E1/2(PCBM0/–) in o-dichlorobenzene/acetonitrile ( F. B. Kooistra, J. Knol, F. Kastenberg, L. M. Popescu, W. J. H. Verhees, J. M. Kroon and J. C. Hummelen, Org. Lett., 2007, 9, 551 Search PubMed
- K. Akaike, K. Kanai, H. Yoshida, J. Tsutsumi, T. Nishi, N. Sato, Y. Ouchi and K. Seki, J. Appl. Phys., 2008, 104, 023710 CrossRef
- Z.-L. Guan, J. B. Kim, H. Wang, C. Jaye, D. A. Fischer, Y.-L. Loo and A. Kahn, Org. Electron., 2010, 11, 1779 CrossRef CAS
- H. Ohkita, S. Cook, Y. Astuti, W. Duffy, S. Tierney, W. Zhang, M. Heeny, I. McCulloch, J. Nelson, D. D. C. Bradley and J. R. Durrant, J. Am. Chem. Soc., 2008, 130, 3030 CrossRef CAS
- T. Clarke, A. Ballantyne, F. Jamieson, C. Brabec, J. Nelson and J. Durrant, Chem. Commun., 2009, 89 RSC
- A low PCE of 0.005% was reported for a 1:2 blend of the only previous report of a polymer incorporating a benzo[a,c]phenazine acceptor—polymer VI—with PCBM (ref. 56).
- Solid PCBM exhibits strong absorption in the 400–500 nm range ( S. Cook, H. Ohkita, Y. Kim, J. J. Benson-Smith, D. D. C. Bradley and J. R. Durrant, Chem. Phys. Lett., 2007, 445, 276 Search PubMed
- H. Hoppe, M. Niggemann, C. Winder, J. Kraut, R. Hiesgen, A. Hinsch, D. Meissner and N. S. Sariciftci, Adv. Funct. Mater., 2004, 14, 1005 CrossRef CAS
- The ability or inability of PCBM to intercalate between polymer side chains is one of the factors that have been shown to influence the optimum polymer/fullerene ratio in OPV devices ( A. C. Mayer, M. F. Toney, S. R. Scully, J. Rivnay, C. J. Brabec, M. Scharber, M. Kopppe, M. Heeney, I. McCulloch and M. D. McGehee, Adv. Funct. Mater., 2009, 19, 1173 Search PubMed
- M. Reyes-Reyes, K. Kim and D. L. Carroll, Appl. Phys. Lett., 2005, 87, 083506 CrossRef
- B. C. Thompson and J. M. J. Fréchet, Angew. Chem., Int. Ed., 2008, 47, 58 CrossRef
- J. K. J. van Duren, X. N. Yang, J. Loos, C. W. T. Bulle-Lieuwma, A. B. Sieval, J. C. Hummelen and R. A. J. Janssen, Adv. Funct. Mater., 2004, 14, 425 CrossRef CAS
- X. N. Yang, J. K. J. van Duren, R. A. J. Janssen, M. A. J. Michels and J. Loos, Macromolecules, 2004, 37, 2151 CrossRef CAS
- T. Bull, L. S. C. Pingree, S. A. Jenekhe, D. S. Ginger and C. K. Luscombe, ACS Nano, 2009, 3, 627 CrossRef CAS
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877 CrossRef CAS
- The 1H chemical shifts of M4 vary with concentration; presumably, as with observations for structurally similar compounds (ref. 77) this is due to aggregation.
| This journal is © The Royal Society of Chemistry 2011 |