Direct evidence of ZnO morphology modification via the selective adsorption of ZnO-binding peptides†
Mei-Keat
Liang
,
Olivier
Deschaume‡
,
Siddharth V.
Patwardhan§
and
Carole C.
Perry
*
Biomolecular and Materials Interface Research Group, School of Science and Technology, Nottingham Trent University, Clifton Lane, Nottingham, NG11 8NS, UK. E-mail: Carole.Perry@ntu.ac.uk
First published on 22nd October 2010
Abstract
Biomolecule-mediated ZnO synthesis has great potential for the tailoring of ZnO morphology for specific application in biosensors, window materials for display and solar cells, dye-sensitized solar cells (DSSCs), biomedical materials, and photocatalysts due to its specificity and multi-functionality. In this contribution, the effect of a ZnO-binding peptide (ZnO-BP, G-12: GLHVMHKVAPPR) and its GGGC-tagged derivative (GT-16: GLHVMHKVAPPRGGGC) on the growth of ZnO crystals expressing morphologies dependent on the relative growth rates of (0001) and (10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0) planes of ZnO have been studied. The amount of peptide adsorbed was determined by a depletion method using oriented ZnO films grown by Atomic Layer Deposition (ALD), while the adsorption behavior of G-12 and GT-16 was investigated using XPS and a computational approach. Direct evidence was obtained to show that (i) both the ZnO-BP identified by phage display and its GGGC derivative (GT-16) are able to bind to ZnO and modify crystal growth in a molecule and concentration dependent fashion, (ii) plane selectivity for interaction with the (0001) versus the (100) crystal planes is greater for GT-16 than G-12; and (iii) specific peptide residues interact with the crystal surface albeit in the presence of charge compensating anions. To our knowledge, this is the first study to provide unambiguous and direct quantitative experimental evidence of the modification of ZnO morphology via (selective and non-selective) adsorption–growth inhibition mechanisms mediated by a ZnO-BP identified from phage display libraries.
0) planes of ZnO have been studied. The amount of peptide adsorbed was determined by a depletion method using oriented ZnO films grown by Atomic Layer Deposition (ALD), while the adsorption behavior of G-12 and GT-16 was investigated using XPS and a computational approach. Direct evidence was obtained to show that (i) both the ZnO-BP identified by phage display and its GGGC derivative (GT-16) are able to bind to ZnO and modify crystal growth in a molecule and concentration dependent fashion, (ii) plane selectivity for interaction with the (0001) versus the (100) crystal planes is greater for GT-16 than G-12; and (iii) specific peptide residues interact with the crystal surface albeit in the presence of charge compensating anions. To our knowledge, this is the first study to provide unambiguous and direct quantitative experimental evidence of the modification of ZnO morphology via (selective and non-selective) adsorption–growth inhibition mechanisms mediated by a ZnO-BP identified from phage display libraries.
Introduction
The realization of the ZnO era in the electronics and optoelectronics industries may be possible if the excellent electrical and optical properties of ZnO,1–3 from sub-nanometre quantum dots to micrometre sized crystals, can be exploited. The challenges ahead can be generally classified into four areas: crystallinity and purity of ZnO; doping effectiveness and efficiency; size, morphology and functional structure assembly; and, last but not least, process feasibility and economics. Among these, morphology control has been given much attention since the successful application of ZnO in many instances relies on its shape- and size-dependent properties.2 Highly crystalline ZnO crystals with diverse nanostructures, from simple to hierarchical, have been produced by vapor route synthesis.1,2,4–12 However, the solution route offers a simpler, low cost scale-up, “green”, and versatile alternative for ZnO synthesis.The morphology of ZnO synthesized in solution (aqueous and non-aqueous) can be tuned via three distinct approaches: (i) the use of structure-directing agents (SDAs); (ii) the retention of ZnO precursor structures, such as layered basic zinc salt (LBZS), with ZnO formation realized by thermal decomposition; and (iii) a localized-reaction approach such as sonochemical and microwave-assisted synthesis. The most commonly used approach is that involving SDAs ranging from simple molecules, such as amines13–16 and anionic species,17–21 to polymers (DBCP,22–29 PVP,30–42 PAM,29 and PAH43) and biomolecules.44–52 The SDAs have been shown to affect the morphology of ZnO by imposition of effects on nucleation and/or growth of the crystals. Among these SDAs, biomolecules have received little attention in ZnO synthesis because of their complexity, high cost, and sensitivity to reaction conditions. Nonetheless, the use of biomolecules to direct ZnO synthesis is attractive for a number of reasons. The first is the potential of biomolecules to direct ZnO formation under mild reaction conditions.53,54 Secondly, some biomolecule-directed syntheses have been shown to impose exquisite control on ZnO morphology/structure;44–46,48,50,51,55 improve ZnO crystallinity;50 template ZnO formation;47,49,52,56 catalyze ZnO formation;57,58 and also suppress ZnO formation.44,45 These diverse effects can possibly be used to tune the morphology/structure-related properties of ZnO and thus its applications. Thirdly, biomolecules show remarkable recognition capabilities, functional specificity and multi-functional characteristics which may contribute to high efficiency and accuracy in ZnO structure tailoring. Finally, there is an infinite reservoir of biomolecules with highly diverse functionalities that can be explored and discovered.53
Some biomolecules have been shown to influence ZnO morphology. As an example, ZnO films have been formed on substrates in the presence of histidine and/or its dipeptides, however, when these biomolecules were absent, discrete ZnO microcrystals were obtained.44,45 In another study, the change of reactant concentrations, including variation of histidine concentration, generated ZnO particles with diverse morphologies including microrods, flowers, and microshells.48 ZnO microshells can also be produced by the use of cysteine59 while the use of a urease enzyme has been shown to produce nanoshells with potentially tunable diameters.47 The transformation of ZnO particles from spheres to hexagonal plates and ellipsoids has been achieved by progressive reduction of the chain length of silk fibroin added.56 Hexagonal plates have also been synthesized using gelatine50,51 and Arabic gum55 from reactions where hexagonal pellets or irregularly shaped crystals were typically formed respectively. In addition, a three-dimensional interwoven mesoporous fibrillar ZnO structure has been obtained using eggshell membrane as a template.49,52 With the exception of studies involving amino acids and dipeptides, biomolecules that have been used in the study of ZnO formation are mainly complex macromolecules and, in some cases, their reactivity is determined by their multiple active sites and secondary/tertiary conformations. Thus many of the “wonders” that occur in their presence are created in a “black box”.
An alternative approach for the rational design of simpler biomolecules for the generation of materials with novel form and for the study of biomolecule-mediated interactions is application of the combinatorial display method where short peptides (7-mer or 12-mer) that have affinity towards the surface of a metal or metal oxide can be identified. Although there are limitations to this approach,60,61 a number of metal-binding peptides (M-BPs)62–65 and metal oxide-binding peptides (MO-BPs)46,58,66–70 have been identified that have either been shown to have an affinity for the surface they were isolated from or have been shown to influence the growth of their respective metal or metal oxide. The identification of ZnO-binding peptides (ZnO-BPs) and understanding how they adhere to the surface of ZnO is potentially useful for the construction of artificial biomaterials, medical implants, and biosensors where biocompatibility is required. Peptides could also be used to control crystal growth, for example, producing ZnO with high surface to volume ratio for applications as solar cells, catalysts and sensors.1–3,71
Application of the combinatorial phage display method identified a ZnO-BP, EAHVMHKVAPRP (EM-12) and its GGGSC derivative, EAHVMHKVAPRP–GGGSC was found to catalyze ZnO formation from a stable Zn(OH)2 sol.58 This EM12–GGGSC peptide derivative was further attached to a genetically modified collagen-like triple helix template to generate ZnO nanowires.72 EM-12 has also been used to assist in the immobilization of green fluorescent protein (GFP) on ZnO surfaces with a 15 amino acid linker (GGGSAGSAASGSGEF) being used to tether the protein to the ZnO-BP.73 The interaction between the EM-12 peptide and the ZnO surface has been shown to occur via electrostatic interaction with the single lysine present in the peptide contributing significantly and surrounding non-charged residues also being influential. Others have isolated a ZnO-BP with 67% sequence similarity to EM-12, GLHVMHKVAPPR (G-12).46 The authors have shown that a transformation of ZnO from bipyramidal hexagonal rods to hexagonal plates occurs when the concentration of a GGGC tagged derivative of G-12, GLHVMHKVAPRP–GGGC (GT-16) in a zinc nitrate–hexamethylenetetramine (Zn(NO3)2–HMTA) reaction system is increased.46 The authors propose that GT-16 inhibited the growth of ZnO along its c-axis by selectively adsorbing on the (0001) plane of ZnO based on three pieces of indirect evidence: (i) the GT-16 concentration dependent reduction of the aspect ratio of ZnO, (ii) the (0001) plane of ZnO crystal was flat and well-defined, and (iii) addition of GT-16 to the reaction had no effect on the diameter of ZnO.
Hitherto, evidence for the ability of biomolecules to tune ZnO morphology has been gained for single amino acids,44,45,48,59 short peptides,44,45,46,58 through to complex macromolecules such as proteins.47,50,51,57,72,74 However, the focus of research in this area has been generally application-driven, and a mechanistic understanding of biomolecule-mediated ZnO formation remains incomplete. It has been postulated widely that the suppression of ZnO growth along its c-axis by biomolecules (also by many synthetic polymers/molecules) may be due to the adsorption of growth-inhibiting additives on the (0001) plane of ZnO via electrostatic interactions13,18–20,46,50,75 or growth-promoting additives on complementary planes21,76,77 but no direct measurement of the interactions has been made.
Recently a theory of competitive adsorption–nucleation to explain changes of ZnO aspect ratio in the presence of polypeptides has been developed.78 According to this theory, the adsorption of polypeptides on the (0001) plane of ZnO would reduce the surface energy of the plane which in turn reduces the aspect ratio of ZnO crystals formed. The calculated aspect ratio-to-polypeptide concentration relationship fitted extremely well with the experimental data from Tomczak et al.46 However, direct empirical evidence has not been provided to support biomolecule (that when present have altered ZnO morphology) adsorption on crystal plane(s) of ZnO. The interaction of specific amino acids such as lysine and alanine with ZnO single crystal planes has been studied,79,80 but their effect on ZnO morphology/growth has not yet been identified.45 As other examples, the interaction of tryptophan,81 bovine serum albumin,82 and plasma proteins83 with ZnO has also been studied experimentally using ZnO nanoparticles mainly characterized in terms of size,81,83 shape,81,83 and ζ potential.83 Crystallographic information relating to the surfaces of these nanoparticles exposed to biomolecules was not provided.
In this study, we sought to obtain direct evidence for the selective adsorption of a ZnO-BP (G-12) and its GGGC tagged derivative (GT-16) on specific ZnO crystal plane(s) and related these data to changes in crystal morphology. The experiments and molecular dynamics simulations were designed to provide direct evidence and provide answers to the following questions: (i) are the G-12 and GT-16 peptides able to bind to ZnO and/or modify crystal growth? (ii) is peptide binding dependent on ZnO crystal-plane and peptide-sequence? (iii) can we elucidate the molecular level interactions between peptide and ZnO?
Materials and methods
Zinc nitrate hexahydrate (Zn(NO3)2·6H2O), hexamethylenetetramine (HMTA, C6H12N4), sodium monophosphate (NaH2PO4), sodium phosphate dibasic (Na2HPO4), sodium chloride (NaCl), trifluoroacetic acid (TFA, C2HF3O2), thioanisole (C7H8S), 3,6-dioxa-1,8-octanedithiol (DODT, C6H14O2S2), 1 M hydrochloric acid (HCl), and a zinc atomic absorption standard solution (1000 ppm) were purchased from Sigma-Aldrich; diethyl ether was purchased from Fisher Scientific; O-benzotriazole-N,N,N′,N′-tetramethyl-uronium-hexafluoro-phosphate (HBTU) and all Fmoc-protected amino acids required for peptide synthesis were purchased from CEM Corporation while the arginine- and cysteine-preloaded Wang resins were obtained from Novabiochem®. All chemicals were used without further treatment. Distilled, deionized water (ddH2O) with conductivity less than 1 µS cm−1 was used in all experiments.Solid phase peptide synthesis
G-12 and GT-16 were prepared by solid phase methods using a microwave-assisted solid phase peptide synthesizer (CEM Corporation) via Fmoc chemistry.84 Wang resins were used as the solid support. The following side chain protections were used: Pbf for arginine; Trt for cysteine and histidine; and Boc for lysine. The couplings, via a O-benzotriazole-N,N,N′,N′-tetramethyl-uronium-hexafluoro-phosphate (HBTU) condensation agent, were single except for arginine where a double coupling was used. The cleavage of the peptide from the resin and the deprotection of side chains were achieved in one step by treating the peptide resin with cleaving agent containing trifluoroacetic acid, thioanisole, ddH2O, and 3,6-dioxa-1,8-octanedithiol (DODT). The cleaved peptides were re-precipitated in chilled diethyl ether, washed three times with the same solvent prior to lyophilisation at −70 °C using a Virtis-110 freeze-dryer. The purity of the lyophilized peptides was analyzed with a Dionex HPLC equipped with a UV detector (set at 214 nm) and a Jupiter 4u Proteo 90A C12 Reverse Phase column (Phenomenex). The purity of G-12 and GT-16 was determined as 95% and 88% respectively. The molecular weight (Mw) of the peptides was measured using a Bruker UltraflexIII TOF/TOF mass spectrometer and analyzed with Daltonics flexAnalysis software. The Mw of the peptides was confirmed to be on target where the calculated Mw for G-12 and GT-16 were 1341.6 g mol−1 and 1615.9 g mol−1 respectively while their measured Mw were 1341.7 g mol−1 and 1615.8 g mol−1 respectively.ZnO synthesis and kinetic studies
100 mM Zn(NO3)2·6H2O, 100 mM HMTA, 30 mM G-12, and 30 mM GT-16 were prepared as stock solutions for this study. Prior to each reaction, equal volumes of the Zn(NO3)2·6H2O and HMTA stock solutions were mixed and predetermined volumes of G12 or GT-16 stock solutions were added immediately to obtain final G-12 concentration or GT-16 concentration of 0 mM, 0.03 mM, 0.15 mM and 0.30 mM, respectively. The pH of the final solutions prepared was pH 6.9 ± 0.1 regardless of the presence of additives. Prepared solutions were incubated at 20 °C (24 hours ± 1 min) followed by immersion in a 65 °C water bath (72 hours ± 1 min). During the course of reaction, samples were taken at selected times (±1 min). The control reaction was performed in triplicate in a single run. Collected samples were centrifuged at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 3 minutes to separate any precipitate from the supernatant. The supernatant obtained was re-centrifuged using the same conditions before being sampled into acidified water for the determination of Zn2+ concentration, [Zn2+], using ICP-OES (Perkin Elmer Optima 2100DV). The precipitates were washed three times thoroughly with ddH2O. Cleaned precipitates were lyophilized at −70 °C using a Virtis-110 freeze-dryer.
000 rpm for 3 minutes to separate any precipitate from the supernatant. The supernatant obtained was re-centrifuged using the same conditions before being sampled into acidified water for the determination of Zn2+ concentration, [Zn2+], using ICP-OES (Perkin Elmer Optima 2100DV). The precipitates were washed three times thoroughly with ddH2O. Cleaned precipitates were lyophilized at −70 °C using a Virtis-110 freeze-dryer.
Characterization of lyophilized ZnO precipitates
The wurtzite phase of ZnO was confirmed using XRD (PANalytical X'Pert PRO, CuKα radiation with wavelength of 1.54056 Å). Ground samples (if necessary) packed into an aluminium sample holder were scanned for 2θ from 5° to 70° at 45 kV accelerating voltage, 40 mA filament current, using a scan speed of 0.02° s−1 at room temperature. Diffraction patterns were analyzed using X'Pert-HighScore Plus (version 2.0a) program for diffractogram manipulation, background determination and peak identification. The morphology and size of the ZnO crystals of selected precipitates were studied using SEM (JEOL JSM-840A, 20 kV, gold sputter-coated samples). For size analysis, at least 50 crystals were studied and the aspect ratio (L/D = length/average diameter) of each individual crystal was determined. FTIR (Nicolet Magna IR-750, 256 scans, 2 cm−1 resolution) was used to detect ZnO, peptides, and any incorporated species in the precipitates with a pure KBr disk being used for the background. The amount of non-ZnO component (peptide) in the precipitates was determined by TGA (Mettler Toledo TGA/SDTA 851e) where samples were heated at 10° min−1 from 30 °C to 900 °C in air to ensure complete combustion of all organic material.Peptide adsorption studies
The selective adsorption of G-12 and GT-16 on the (0001) and (100) planes of ZnO was studied at 25 °C using oriented ZnO films (∼9 cm2) grown on silicon wafers by the ALD technique. This temperature was chosen to minimize any change of peptide concentration during the experiment arising from experimental factors (only small film sizes could be prepared by ALD such that the concentration of zinc in solution was necessarily very low) other than direct binding. The crystal orientation of ZnO films is controlled by its deposition temperature: 280 °C and 165 °C being used for the (0001) and (100) planes respectively.85 The as-grown ZnO films were annealed at 800 °C (1 hour) prior to the confirmation of crystal orientation by XRD. All substrates studied, ZnO films, silicon wafers, and glass slide, were cleaned with acetone, dried in air, and placed into 12 mL peptide solution prepared in PBS (phosphate buffered saline, 50 mM phosphate and 150 mM NaCl, pH 7.0). Given the calculated planar Zn atom density on (0001) and (100) planes of ZnO crystals are 10.94 atoms per nm2 and 5.91 atoms per nm2 respectively,86 the number of peptide molecules added were 5 times the estimated number of Zn atoms present on the ZnO films (peptide-to-Zn ratio = 5). At predetermined incubation times (t = 0, 5 hours), aliquots were sampled from the peptide solution and assayed using the Micro-BCA (Bicinchoninic acid) method for peptide concentration (Thermo Fisher Scientific). The absorbance of aliquots from the assays was measured at 562 nm using a Unicam UV2 UV-VIS spectrometer and compared against a series of calibration peptide solutions (0–10.5 µM), prepared in the same medium. Adsorption tests were performed in independent triplicates. At the end of the adsorption test, ZnO films were cleaned with ddH2O and air-dried at room temperature before being analyzed by XPS using a Kratos AXIS ULTRA with a monochromated Al-Kα X-ray source (1486.6 eV) operated at a 10 mA emission current and 10 kV anode potential (100 W). The pressure of the analysis chamber was 3 × 10−9 torr. A wide scan (10 minutes) and high resolution scans (10, 5, 20 and 5 minutes for Zn, O, N, and C respectively) were performed on each sample where the latter was charge-corrected to the hydrocarbon C1s peak (285 eV) and then quantified to compare the amounts of the relevant elements present using Kratos sensitivity factors. The pass energies for the wide scan and the high resolution scan were 80 eV and 20 eV respectively. Fresh ZnO films (not exposed to peptide solution), G-12, and GT-16 were also analyzed and used as references. Six areas were analyzed for the selected samples.
Computational calculations
A molecular dynamics (MD) simulation tool, GROMACS,87 was used to computationally determine the stable configurations of G-12 and GT-16 in water. The energy of the peptides was minimized in explicit single point charge (SPC) water at 300 K with a time step of 2 fs and total simulation time of 2 ns using the OPLS-AA-(L)-all atom force field. Using the stable configurations obtained, the interactions of G-12 and GT-16 with the (0001) and (100) planes of ZnO were studied in vacuum using the Materials Studio®4.3 Adsorption Locator module based on the Metropolis Monte Carlo algorithm.88 The adsorption modes and adsorption energies of G-12 and GT-16 on these ZnO planes were studied and the adsorbing segments/moieties were identified.
Results and discussion
Understanding the anisotropic growth of ZnO in the absence of peptides
For comparison purposes, the ZnO-forming reaction used by Tomczak et al.46 was adopted in this study. In brief, solutions of Zn(NO3)2–HMTA were incubated initially at 20 °C for 24 hours followed by raising the temperature of the reaction medium to 65 °C, where samples were taken and the progress of reaction monitored using analytical methods to measure the amount of Zn2+ remaining in solution, XRD to assess any crystalline phases present and SEM to analyse the morphology of the products. In the absence of the peptides, a gel-like intermediate compound was formed at 20 °C, which was found to be neither ZnO nor pure Zn(NO3)2·6H2O (ESI, Fig. S1†). ZnO was first detected by XRD at 1 hour (first sampling at 65 °C) after the reaction temperature was increased to 65 °C (t20 °C = 24 h, t65 °C = 1 h), Fig. 1 inset. The diffraction peaks obtained can be indexed to the hexagonal wurtzite structure of ZnO (JCPDS card no. 36-1451). At 65 °C, the amount of Zn2+ consumed for ZnO formation steadily increased from the stable level of ∼10% at 20 °C until an equilibrium state was reached when ∼60% of Zn2+ was reacted, Fig. 1. The non-ZnO gel-like precipitates collected after 24 hours (t20 °C = 24 h) appeared to be continuous sheets with rough surfaces (Fig. 2a) as opposed to hexagonal ZnO crystals obtained at 65 °C (Fig. 2b–d). The absence of a centre of inversion in the wurtzite structure is known to result in ZnO having inherent asymmetry along its c-axis.1 In this study, the anisotropic growth of ZnO crystals was clearly observed through the increase of crystal aspect ratio (L/D ± standard error) from 2.7 ± 0.1 to 11.4 ± 0.2 for crystals collected after 25 hours (t20 °C = 24 h, t65 °C = 1 h, Fig. 2b) and 48 hours (t20 °C = 24 h, t65 °C = 24 h, Fig. 2c) respectively. Oriented attachment of ZnO rods along their c-axes was observed as the reaction was prolonged, Fig. 2d. This phenomenon, believed to be driven by surface energy reduction, has also been described in other publications, however, its cause is still debated.39,76,89–93 Since ZnO was not detected at 20 °C, for simplicity, the reaction time is quoted as the reaction duration at 65 °C in all following sections, i.e. total reaction time of 25 hours and 48 hours are described as t65 °C = 1 h and t65 °C = 24 h respectively. | ||
| Fig. 1 The average and standard deviation (error bar) of Zn2+ consumed (in %) with time in the absence of additives (n = 3). The inset shows the diffraction peaks from precipitates collected after 24 hours (t20 °C = 24 h) and 25 hours (t20 °C = 24 h, t65 °C = 1 h). The diffraction peak marked with an asterisk arises from the aluminium sample holder. | ||
 | ||
| Fig. 2 SEM images for precipitates collected in the absence of additives after (a) 24 hours (t20 °C = 24 h), (b) 25 hours (t20 °C = 24 h, t65 °C = 1 h), (c) 48 hours (t20 °C = 24 h, t65 °C = 24 h), and (d) 72 hours (t20 °C = 24 h, t65 °C = 48 h). Scale bars are 5 µm. | ||
ZnO aspect ratio reduction by G-12 and GT-16
The addition of G-12 or GT-16 affected neither the Zn2+ consumption rate for ZnO formation nor the [Zn2+] at equilibrium compared to the control reaction (ESI, Fig. S2†), suggesting that G-12 and GT-16 may not interact with Zn2+ species in solution. These data also suggest that the presence of these two peptides, for the concentration ranges used, does not affect nucleation. Similar to the control reactions, ZnO crystals were detected by XRD from precipitates collected at t65 °C = 1 h, produced in the presence of G-12 and GT-16 (ESI, Fig. S3†). As anticipated, a reduction of crystal L/D as a function of peptide concentration was observed in the presence of GT-16 and G-12, Fig. 3. However, the effect was more prominent for GT-16 compared to G-12, Fig. 3a. This effect induced by G-12 and GT-16 started from the early stage of crystal growth (t65 °C = 1 h) where the L/D was 1.0–1.7 compared to 2.7 for the control. Compared to the L/D reduction reported for GT-16,46 the level of L/D reduction in this study was slightly higher at lower [GT-16] but these values converged as [GT-16] was increased. | ||
| Fig. 3 (a) The average and 95% confidence interval of ZnO aspect ratio (L/D) as a function of peptide concentration, and SEM images of ZnO crystals formed after t65 °C = 24 h in the presence of (b) 0.03 mM G-12, (c) 0.30 mM G-12, (d) 0.03 mM GT-16, and (e) 0.30 mM GT-16. Scale bars are 2 µm. | ||
ZnO precipitates (t65 °C = 24 h) obtained in the presence of G-12 and GT-16 contain peptides as shown by the presence of the amide-I (1600–1700 cm−1) and amide-II (1500–1600 cm−1) bands in their FTIR spectra (Fig. 4a for GT-16 and ESI, Fig. S4† for G-12). The area under the two amide bands of these FTIR spectra was determined and normalized against the area under the control spectrum for the same frequency range. As shown in Fig. 4b, the amount of peptide incorporated in ZnO precipitates increased with the initial peptide concentration, supporting the coprecipitation of peptides with ZnO.
 | ||
| Fig. 4 The FTIR (a–d) and TGA (e) analyses of ZnO precipitates collected at t65 °C = 24 h, produced in the presence of G-12 and GT-16. (a) The FTIR spectra of precipitates obtained in the presence of GT-16. The peak area ratio between samples (in the presence of peptides) and the control for peak area under (b) amide-I and amide-II bands (1480–1730 cm−1), and (c) nitrate peak (1384 cm−1). (d) The amide bands-to-nitrate peak area ratio within individual spectra as depicted in (a). (e) The weight loss attributed to the decomposition of the organic component (200–600 °C) where the error bar is the standard deviation for the control reactions (0 mM peptide, n = 3). | ||
The presence of NO3− (1384 cm−1, asymmetric stretch)44,59,94,95 was also detected in all 48 hour precipitates including the control (Fig. 4a), and the gel-like intermediate compound produced after 24 hours (t20 °C = 24 h) from the blank (ESI, Fig. S1†). The normalized area of the nitrate peak increased linearly as a function of peptide concentration, Fig. 4c, suggesting that NO3− ions may have formed a complex with, possibly, the cationic moieties of the peptide and co-precipitated with ZnO when peptides adsorbed on ZnO surfaces. A non-constant peak area ratio of the amide-I and II bands to NO3− (1384 cm−1) calculated from FTIR spectra suggests that not all cationic moieties of each peptide molecule have reacted with NO3−, Fig. 4d. It has been suggested that HMTA may adsorb on the (100) plane of ZnO crystal,92 however, this possibility can be discounted in this study based on the absence of the peak expected for adsorption of a tertiary amine (∼1006 cm−1) in all spectra including those from the control reaction (ESI, Fig. S4†).
The coprecipitation of peptides with ZnO was further supported by substantial weight loss for the ZnO samples prepared in the presence of peptides, with respect to the control reaction, monitored using TGA (ESI, Fig. S5†). The trend of organic component weight loss as a function of peptide concentration (Fig. 4e) was similar to the trend shown by FTIR results in Fig. 4b. The coprecipitation of NO3− with ZnO (suggested by FTIR results) did not alter the trend of the incorporated amount of peptide significantly (ESI, Fig. S6†). Therefore while reducing the aspect ratio of ZnO crystals, these two peptides co-precipitated onto/into the ZnO crystals. Furthermore, GT-16 which was incorporated to a greater extent than G-12, perhaps therefore has a stronger effect on L/D reduction compared to G-12. This may imply a correlation between the amount of peptide incorporated and the level of L/D reduction.
There were two observations which indirectly suggest peptide adsorption on ZnO surfaces. Firstly, a blue-shift of the amide-I band (from ZnO-peptide precipitates) relative to the amide-I band of the pure peptides was observed. The observed amide-I band shift of 20–26 cm−1 from 1671 cm−1 was likely to be due to the adsorption of peptides on crystal surfaces which alters the energy of vibration.96 Secondly, a new peak at 1590–1594 cm−1, assignable to the symmetric stretching of bound COO− was detected.40,97 Since both G-12 and GT-16 have no acidic amino acid residues, the bound COO− most probably arose from the C-terminus of these peptides.
Modification of ZnO growth rate by G-12 and GT-16
In order to understand how ZnO growth has been affected and how the additional GGGC-tag of GT-16 enhanced the aspect ratio reduction, the growth rates of individual crystal dimensions from t65 °C = 1 h to t65 °C = 24 h were determined, namely, ΔL/Δt for length along the c-axis and ΔD/Δt for the average diameter along the a-axis, Fig. 5a and b. | ||
| Fig. 5 The growth rate of (a) length, L, and (b) average diameter, D, of ZnO crystals within 23 hours of reaction at 65 °C, from t65 °C = 1 h to t65 °C = 24 h. (c) Schematic showing the effect of increasing G-12 and GT-16 concentrations on the length and diameter of ZnO crystals from t65 °C = 1 h to t65 °C = 24 h. The images of ZnO crystals are scaled. | ||
In the absence of peptide, ΔL/Δt and ΔD/Δt for the ZnO crystals were 149 nm h−1 and 3.4 nm h−1 respectively. Addition of G-12 reduced ΔL/Δt gradually to ∼10 nm h−1 as the peptide concentration was increased. In contrast, addition of GT-16, even at the lowest concentration (0.03 mg mL−1), drastically reduced ΔL/Δt to ∼7 nm h−1, however, this growth rate was not affected by further increase in the peptide concentration. In the presence of GT-16, reduction in growth along the c-axis was compensated for by an increase in ΔD/Δt. This contrasted with the crystal growth observed in the presence of G-12, where the average diameter of the ZnO crystals remained unchanged (ΔD/Δt ≈ 0) for the 23 hour analysis period regardless of the concentration of G-12 showing that G-12 has little or no influence on the a-axis growth of the crystal. The effect of increasing G-12 and GT-16 concentrations on the length and diameter growth of ZnO crystals from t65 °C = 1 h to t65 °C = 24 h, as described above, is illustrated by a scaled schematic in Fig. 5c. In contrast to the data presented by Tomczak et al. in their study using GT-16 where the observed aspect ratio reduction was reported as being due to crystal length reduction, our studies have shown that by separately analyzing the effect of the two peptides (G-12 and GT-16) on both the length and diameter of the crystals formed, we have discovered that the reduction of the ZnO aspect ratio is a combination of the presence of G-12 or GT-16 on both the length (c-axis) and diameter (a-axis) growth of the ZnO crystals.
Up to this point, the presence of G-12 and GT-16 has been confirmed to reduce the c-axis growth of ZnO crystals while the presence of G-12 has also retarded the diameter growth (a-axis) growth of the crystal. In addition, the coprecipitation of G-12 and GT-16 strongly correlated (quantitatively) with the reduction of crystal dimension growth rate. In many instances the presence of capping agents such as DBCP,22–25 citrate ions,19,20,75 hydroxyl ions,13etc. has been shown to reduce the aspect ratio of ZnO crystals. Since (0001) is a Zn-terminated plane, the capping agents that have reduced the aspect ratio of ZnO crystals and/or were proposed to adsorb on the (0001) plane are generally anionic or contain anionic moiety(ies). Therefore, based on this generally accepted rationalization, the inhibition of diameter growth in the presence of G-12 but not with GT-16 would lead to three possible speculations. Firstly, besides adsorbing on (0001), G-12 molecules were able to adsorb onto the (100) plane of ZnO crystals, possibly via a different adsorption mechanism. Secondly, GT-16 selectively adsorbed onto the (0001) planes of ZnO crystals. Thirdly, the plane selectivity of GT-16 was attributable to the GGGC tag not present in G-12. To unravel the cause(s) of these interesting findings, the adsorption behavior of G-12 and GT-16 was studied.
A comparison of the adsorption of G-12 and GT-16 on ZnO crystal planes
The adsorption behavior of G-12 and GT-16 was studied by exposing ALD grown-ZnO films with either (0001) or (100) plane (ESI, Fig. S7†) to peptide solutions. The use of oriented-ZnO films eliminated any uncertainties of peptide adsorption sites, which may arise from the use of ZnO crystals with more than one surface crystallographic plane. The amount of peptide adsorbed and peptide density on the ZnO films were determined by a depletion method from the amount of peptide retained in solution after 5 hours of substrate exposure. From the amount adsorbed on the different substrates (area ≈ 9 cm2), G-12 and GT-16 both showed material selectivity towards ZnO over silicon wafer and glass, Fig. 6a. Peptide density (Fig. 6b) was typically higher on the basal plane, ZnO(0001), compared to the side planes, Zn(100). The inter-plane density difference for GT-16 was 0.31 nmol cm−2 as compared to 0.08 nmol cm−2 for G-12, suggesting a preferential adsorption of GT-16 on the (0001) plane. Preferential adsorption of GT-16 supports the observation of reduced growth rate on the c-axis (ΔL/Δt) but not on the a-axis (ΔD/Δt) in its presence. The lower G-12 density on the (0001) plane and its weaker plane selectivity were demonstrated by the lower extent of ZnO aspect ratio reduction in its presence.
 | ||
| Fig. 6 The adsorption of G-12 and GT-16 on different substrates after 5 hours of exposure: (a) the amount adsorbed on a substrate with an area ∼9 cm2, and (b) peptide density. The error bars shown are the standard deviation (n = 3). | ||
Evidence for peptide adsorption on ZnO crystal planes and the adsorption process being peptide- and crystal plane-dependent was provided by XPS analysis which confirmed that significant amounts of peptide were present on the ZnO films exposed to the solutions of the peptides, Fig. 7. The presence of peptide was confirmed via the detection of nitrogen (N 1s) with binding energy of ∼400 eV. Fig. 7 shows the N 1s atomic % (high resolution scan) on exposed ZnO films, unexposed ZnO films, G-12, and also GT-16. XPS results obtained did not (quantitatively) coincide entirely with the adsorption results, inset of Fig. 7. However, the important outcomes are summarized as follows: (i) the detection of a substantial amount of nitrogen on the surface of ZnO films (exposed to peptide solution) is direct evidence of peptide adsorption on ZnO crystal planes, (ii) the amount of G-12 adsorbed on the (0001) plane was slightly larger than on the (100) plane, which corroborated the experimental observations (Fig. 6b), and (iii) the amount of GT-16 adsorbed on the (0001) plane was significantly higher than G-12 on the same plane for both ZnO planes.
 | ||
| Fig. 7 The atomic % of N 1s determined using XPS (high resolution scan). GT and G denote GT-16 and G-12 respectively. Inset shows the peptide density on ZnO films reproduced from Fig. 6b. | ||
The presented data show that for ZnO formation in the presence of ZnO-BPs, both G-12 and GT-16 altered the growth rates of ZnO crystal planes and are adsorbed on these crystal planes, with the level of growth reduction being positively correlated with the amount of peptide adsorbed. Therefore the modification of ZnO crystal morphology by G-12 and GT-16 occurs via an adsorption–growth inhibition mechanism as illustrated in Scheme 1. Hitherto, the incorporation/adsorption of SDAs on ZnO has been shown qualitatively by FTIR, TGA,21,23,34 fluorescence microscopy,23,58 and SEM (through the presence and multiplication of growth spirals in the presence of DBCPs).25 To our knowledge, this is the first study providing unambiguous and direct quantitative experimental evidence, by the use of ALD-grown single orientation ZnO films, for the (selective and non-selective) adsorption-driven ZnO morphology modification by biomolecules and specifically a ZnO-BP identified from phage display libraries.
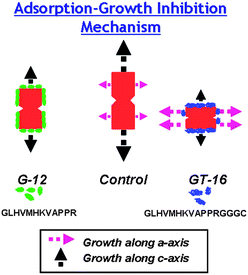 | ||
| Scheme 1 The adsorption–growth-inhibition mechanism. The length and thickness of growth arrows reflects the growth rate of the crystals along their respective axes. | ||
Adsorption and selective adsorption: cause and driving force
To complement the experimental data, a computational approach was employed to provide (calculated) energetic data and the prediction of binding moiety(ies). When exposed to ZnO surfaces in vacuum, the adsorption modes and adsorption energy (Eads) of stable configurations of G-12 and GT-16 (simulated in water by MD) were determined. The adsorption of G-12 and GT-16 on ZnO planes was found to be an energetically favorable process, Fig. 8. The selective adsorption of GT-16 was self-explanatory based on a lower Eads on the (0001) plane and a higher inter-plane Eads of 288 kJ mol−1 as opposed to 100 kJ mol−1 for G-12. The low adsorption selectivity of G-12 can be attributed to competition in binding arising from the smaller difference in its inter-plane Eads, thus an additional limitation of growth along the a-axis was observed. | ||
| Fig. 8 The average adsorption energy and standard deviation of G-12 and GT-16 on (0001) and (100) planes of ZnO. The standard deviation was less than 5% (n = 10). | ||
Much literature has reported/proposed electrostatic interaction/coordination between the Zn2+ of the (0001) plane and additives (DBCP,23,24,98 citrate,19,20 phosphate,17 TEA,15,99etc.) as being responsible for the adsorption of the specific additive on the (0001) plane. The charge and the extent of protonation/de-protonation of amino acid residues in G-12 and GT-16 at pH 6.9 ± 0.1 (based on their pKa values, ESI, Fig. S8†) were determined, and compared with the predicted binding moieties (by simulation as shown in ESI, Fig. S9†) in Table 1.
|
a The side chain charge at pH 6.9 ± 0.1 (based on calculated pKa): green cell for positive, white cell for neutral, and pink cell for negative. The side chain of a residue that binds on different ZnO planes (based on computational prediction): blue circles for (0001) plane and red circles for (100) plane.
b pKb.
|
|---|

|
As shown in Table 1, residues with cationic/neutral side chains were predicted to engage on the (0001) plane of ZnO, namely H3 and K7 for G-12, and G1, H6, and K7 for GT-16. Since neutral imidazole nitrogen atoms could coordinate with Zn2+ using a lone pair on a nitrogen atom,100 the prediction of H3 (of G-12) and H6 (of GT-16) binding on the (0001) plane of ZnO is not surprising. From the FTIR spectra of peptide adsorbed-ZnO precipitates, an absorption peak arising from the asymmetric stretching of NO3− (1384 cm−1) was observed (Fig. 4a). The positive correlation between the amount of NO3− incorporated and the amount of peptide initially present (Fig. 4c) suggests that NO3− interacts with the cationic moieties of G-12 and GT-16. This interaction may reduce the repulsion between the peptide and Zn2+ ions on ZnO crystal planes and will be applicable for any positively charged amino acid residue present in the peptides. The prediction of cationic G1 (of GT-16) and K7 (of G-12 and GT-16) as favorable anchor points on the also cationic Zn-terminated (0001) plane was unexpected although the significance of K7 in binding to ZnO surfaces has recently been shown by other experimentalists using the ZnO-BP EM12.73 It is speculated that the R12 and C16 of G-12 and GT-16 respectively are able to bind to Zn2+ on the (0001) plane. The higher GT-16 density measured on the (0001) plane (adsorption test) may arise due to (i) higher binding capacity of H6 (>50% neutral side chain) from GT-16 as opposed to ∼50% neutral H3 of G-12, and (ii) stronger binding of GT-16 through C16via its C-terminal COO− and its thiol sulfur which is able to bridge two Zn2+ ions.101
In comparison with peptide binding on the (0001) plane, the predicted binding residues on the (100) plane are very different for G-12 and GT-16. The predicted binding moieties from G-12 are polar (H3 and K7) but largely non-polar for GT-16 (M5, R12, and GGGC-tag). Due to the presence of both Zn2+ and O2− on the (100) plane, cationic H3 and K7 are suggested to interact with O2−via electrostatic interactions while neutral H3 could coordinate with Zn2+ ions or hydrogen bond with O2− ions. The adsorption of GT-16 on the (100) plane may occur via hydrogen bonding (neutral R12 with O2−), electrostatic interactions (thiol and COO− of C16 with Zn2+, and M5 sulfur atom lone pair with Zn2+), and hydrophobic interactions (via the GGG-segment). Inferred from these differences, the presence of the GGGC-tag may have lowered the GT-16 density on the (100) plane due to a weaker hydrophobic adsorption contribution. In short, the calculation of adsorption energies and the prediction of binding moieties using a computational approach have provided a plausible explanation for the adsorption phenomenon observed.
Conclusions
A ZnO-BP (G-12) identified by Phage display methods and its -GGGC tagged derivative (GT-16) both affect ZnO crystal morphology in a concentration dependent fashion by altering the c-axis and a-axes growth rate of ZnO crystals. As a consequence, the aspect ratio of ZnO was reduced in their presence with the effect being more prominent in the presence of GT-16. For the first time, the amount of peptide adsorbed on different ZnO crystal planes has been quantified by a depletion method using ALD-grown oriented ZnO films. In conjunction with the confirmation of adsorption by XPS, this study has provided direct evidence for the modification of ZnO morphology via an adsorption–growth inhibition mechanism in the presence of ZnO-BP and its derivative. The correlation of peptide adsorption behavior with the alteration of crystal plane growth rates verified the preferential binding (discriminative behavior) of GT-16 on the (0001) plane over the (100) plane of ZnO crystals. Such behavior was not evident for G-12 which lacked the GGGC-tag, indicating that the GGGC-tag present in GT-16 was not essential for peptide adsorption on the (0001) plane of ZnO but provided GT-16 with plane selectivity characteristics. The calculated adsorption energies of G-12 and GT-16 on different ZnO planes were in broad agreement with the level of plane-selectivity shown experimentally. A prediction of binding moieties by a computational (MD) approach identified GGGC as being the cause of the plane selectivity behavior of GT-16.
The tuning of ZnO morphology via biomolecule adsorption is a complex phenomenon. The solution chemistry of amino acid residues, the nature of counterions, the surface chemistry of ZnO planes, and peptide sequences were all found to play a part and influenced the reaction. The total elucidation of this complex phenomenon would require further extensive studies. However, the results reported in this contribution provide evidence and insight into ZnO morphology-tuning, mediated by the adsorption of biomolecules on specific crystal plane(s) of ZnO crystals. The ultimate goal of these studies is to identify and understand the specific role(s) individual functionalities play within a given biomolecule in ZnO morphology tuning. By doing so, the specificity and multi-functionality of biomolecules in affecting ZnO morphology can be learnt and manipulated to tailor-make ZnO crystals with controllable morphology and properties.
Acknowledgements
This study was supported by funding from US Air Force Office of Scientific Research (AFOSR), FA9550-06-1-0154. We would like to thank Dr David J. Belton and Miss Clare Coveney for carrying out HPLC analysis and molecular weight analysis respectively on peptides studied. We would also like to convey our gratitude to Miss Emily Smith and Mr Ignacio Villar from the Department of Chemistry, University of Nottingham for performing the XPS analysis.Notes and references
- C. F. Klingshirn, ChemPhysChem, 2007, 8, 782 CrossRef CAS.
- Z. L. Wang, J. Phys.: Condens. Matter, 2004, 16, R829 CrossRef CAS.
- S. J. Pearton, D. P. Norton, K. Ip, Y. W. Heo and T. Steiner, Prog. Mater. Sci., 2005, 50, 293 CrossRef CAS.
- C. X. Xu, X. W. Sun, Z. L. Dong and M. B. Yu, Appl. Phys. Lett., 2004, 85, 3878 CrossRef CAS.
- L. Z. Wang, X. Y. Kong and J. M. Zuo, Phys. Rev. Lett., 2003, 91, 185502 CrossRef CAS.
- J. Y. Lao, J. Y. Huang, D. Z. Wang and Z. F. Ren, Nano Lett., 2003, 3, 235 CrossRef CAS.
- X. Y. Kong, Y. Ding, R. Yang and Z. L. Wang, Science, 2004, 303, 1348 CrossRef CAS.
- Z. W. Pan, Z. R. Dai and Z. L. Wang, Science, 2001, 291, 1947 CrossRef CAS.
- P. X. Gao and Z. L. Wang, Science, 2003, 125, 11299 CAS.
- X. Y. Kong and Z. L. Wang, Nano Lett., 2003, 3, 1625 CrossRef CAS.
- W. D. Yu, X. M. Li and X. D. Gao, Appl. Phys. Lett., 2004, 84, 2658 CrossRef CAS.
- Y. Yan, L. Zhou, Q. Xue and Y. Zhang, J. Phys. D: Appl. Phys., 2008, 41, 195402 CrossRef.
- B. Cao and W. Cai, J. Phys. Chem. C, 2008, 112, 680 CrossRef CAS.
- Y. Masuda, N. Kinoshita and K. Koumoto, Electrochim. Acta, 2007, 53, 171 CrossRef CAS.
- R. Xie, D. Li, H. Zhang, D. Yang, M. Jiang, T. Sekiguchi, B. Liu and Y. Bando, J. Phys. Chem. B, 2006, 110, 19147 CrossRef CAS.
- F. Lu, W. Cai and Y. Zhang, Adv. Funct. Mater., 2008, 18, 1047 CrossRef CAS.
- H. Imai, S. Iwai and S. Yamabi, Chem. Lett., 2004, 768 CrossRef CAS.
- Z. R. Tian, J. A. Voigt, J. Liu, B. Mckenzie and M. J. Mcdermott, J. Am. Chem. Soc., 2002, 124, 12954 CrossRef CAS.
- J. Liang, J. Liu, Q. Xie, S. Bai, W. Yu and Y. Qian, J. Phys. Chem. B, 2005, 109, 9463 CrossRef CAS.
- C.-L. Kuo, T.-J. Kuo and M. H. Huang, J. Phys. Chem. B, 2005, 109, 20115 CrossRef CAS.
- T. Zhang, W. Dong, M. Keeter-Brewer, S. Konar, R. N. Njabon and Z. R. Tian, J. Am. Chem. Soc., 2006, 128, 10960 CrossRef CAS.
- M. Öner, J. Norwig, W. H. Meyer and G. Wegner, Chem. Mater., 1998, 10, 460 CrossRef.
- A. Taubert, D. Palms, O. Weiss, T. Piccini and D. N. Batchelder, Chem. Mater., 2002, 14, 2594 CrossRef CAS.
- A. Taubert, G. Glasser and D. Palms, Langmuir, 2002, 18, 4488 CrossRef CAS.
- G. Wegner, P. Baum, M. Müller, J. Norwig and K. Landfester, Macromol. Symp., 2001, 175, 349 CAS.
- A. Taubert, C. Kubel and D. C. Martin, J. Phys. Chem. B, 2003, 107, 2660 CrossRef CAS.
- R. C. Hoffmann, S. Jia, J. C. Bartolomé, T. M. Fuchs, J. Bill, P. C. J. Graat, and F. Aldinger, J. Eur. Ceram. Soc., 2003, 23, 2119 CrossRef CAS.
- R. C. Hoffmann, T. M. Fuchs, T. P. Niesen, J. Bill and F. Aldinger, Surf. Interface Anal., 2002, 34, 708 CrossRef CAS.
- Y. Peng, A.-W. Xu, B. Deng, M. Antonietti and H. Colfen, J. Phys. Chem. B, 2006, 110, 2988 CrossRef CAS.
- J. Zhang, H. Liu, Z. Wang, N. Ming, Z. Li and A. S. Biris, Adv. Funct. Mater., 2007, 17, 3897 CrossRef CAS.
- L. Guo and S. Yang, Chem. Mater., 2000, 12, 2268 CrossRef CAS.
- N. Lepot, M. K. V. Bael, H. V.d. Rul, J. D'Haen, R. Peeters and F. D. Mullens, Mater. Lett., 2007, 61, 2624 CrossRef CAS.
- K. Biswas, B. Das and C. N. R. Rao, J. Phys. Chem. C, 2008, 112, 2404 CrossRef CAS.
- J. Zhang, H. Liu, Z. Wang and N. Ming, J. Cryst. Growth, 2008, 310, 2848 CrossRef CAS.
- M. Bitenc, M. Marinšek and Z. C. Orel, J. Eur. Ceram. Soc., 2008, 28, 2915 CrossRef CAS.
- R. Viswanatha and D. D. Sarma, Chem.–Eur. J., 2006, 12, 180 CrossRef CAS.
- D. Tao, W. Qian, Y. Huang and F. Wei, J. Cryst. Growth, 2004, 273, 353 CrossRef.
- L. Guo, J. X. Cheng, X.-Y. Li, Y. J. Yan, S. H. Yang, C. L. Yang, J. N. Wang and W. K. Ge, Mater. Sci. Eng., C, 2004, 16, 123.
- C. Pacholski, A. Kornowski and H. Weller, Angew. Chem., Int. Ed., 2002, 41, 1188 CrossRef CAS.
- R.-Q. Song, A.-W. Xu, B. Deng, Q. Li and G.-Y. Chen, Adv. Funct. Mater., 2007, 17, 296 CrossRef CAS.
- S. Musić, A. Šarić and S. Popović, J. Alloys Compd., 2008, 448, 277 CrossRef CAS.
- S. Lee, S. Jeong, D. Kim, B. K. Park and J. Moon, Superlattices Microstruct., 2007, 42, 361 CrossRef CAS.
- G. Begum, S. V. Manorama, S. Singh and R. K. Rana, Chem.–Eur. J., 2008, 14, 6421 CrossRef CAS.
- P. Gerstel, R. C. Hoffmann, P. Lipowsky, L. P. H. Jeurgens, J. Bill and F. Aldinger, Chem. Mater., 2006, 18, 179 CrossRef CAS.
- P. Gerstel, P. Lipowsky, O. Durupthy, R. C. Hoffmann, P. Bellina, J. Bill and F. Aldinger, J. Ceram. Soc. Jpn., 2006, 114, 911 CrossRef CAS.
- M. M. Tomczak, M. K. Gupta, L. F. Drummy, S. M. Rozenzhak and R. R. Naik, Acta Biomater., 2009, 5, 876 CrossRef CAS.
- R.d. l. Rica and H. Matsui, Angew. Chem., Int. Ed., 2008, 47, 5415 CrossRef CAS.
- Q. Wu, X. Chen, P. Zhang, Y. Han, X. C. Y. Yan and S. Li, Cryst. Growth Des., 2008, 8, 3010 CrossRef CAS.
- Q. Dong, H. Su, F. Song, D. Zhang and N. Wang, J. Am. Ceram. Soc., 2007, 90, 376 CrossRef CAS.
- L. P. Bauermann, A.d. Campo, J. Bill and F. Aldinger, Chem. Mater., 2006, 18, 2016 CrossRef.
- Y.-H. Tseng, H.-Y. Lin, M.-H. Liu, Y.-F. Chen and C.-Y. Mou, J. Phys. Chem. C, 2009, 113, 18053 CrossRef CAS.
- Q. Dong, H. Su, C. Zhang, D. Zhang, Q. Guo and F. Kiessling, Chem. Eng. J., 2008, 137, 428 CrossRef CAS.
- M. B. Dickerson, K. H. Sandhage and R. R. Naik, Chem. Rev., 2008, 108, 4935 CrossRef CAS.
- L. E. Greene, B. D. Yuhas, M. Law, D. Zitoun and P. Yang, Inorg. Chem., 2006, 45, 7535 CrossRef.
- M. Jitianu and D. V. Goia, J. Colloid Interface Sci., 2007, 309, 78 CrossRef CAS.
- D. Yan, G. Yin, Z. Huang, M. Yang, X. Liao, Y. Kang, Y. Yao, B. Hao and D. Han, J. Phys. Chem. B, 2009, 113, 6047 CrossRef CAS.
- Z. Huang, D. Yan, M. Yang, X. Liao, Y. Kang, G. Yin, Y. Yao and B. Hao, J. Colloid Interface Sci., 2008, 325, 356 CrossRef CAS.
- M. Umetsu, M. Mizuta, K. Tsumoto, S. Ohara, S. Takami, H. Watanabe, I. Kumagai and T. Adschiri, Adv. Mater., 2005, 17, 2571 CrossRef CAS.
- D. Weinzierl, D. Touraud, A. Lecker, A. Pfitzner and W. Kunz, Mater. Res. Bull., 2008, 43, 62 CrossRef CAS.
- S. V. Patwardhan, G. Patwardhan and C. C. Perry, J. Mater. Chem., 2007, 17, 2875 RSC.
- M. Sarikaya, C. Tamerler, A. K.-Y. Jen, K. Schulten and F. Baneyx, Nat. Mater., 2003, 2, 577 CrossRef CAS.
- S. Brown, M. Sarikaya and E. Johnson, J. Mol. Biol., 2000, 299, 725 CrossRef CAS.
- S. Brown, Nat. Biotechnol., 1997, 15, 269 CAS.
- S. Brown, Nano Lett., 2001, 1, 391 CrossRef CAS.
- R. R. Naik, S. J. Stringer, G. Agarwal, S. E. Jones and M. O. Stone, Nat. Mater., 2002, 1, 169 CrossRef CAS.
- C. K. Thai, H. Dai, M. S. R. Sastry, M. Sarikaya, D. T. Schwartz and F. Baneyx, Biotechnol. Bioeng., 2004, 87, 129 CrossRef CAS.
- K. Kjærgaard, J. K. Sørensen, M. A. Schembri and P. Klemm, Appl. Environ. Microbiol., 2000, 66, 10 CrossRef.
- R. R. Naik, L. L. Brott, S. J. Clarson and M. O. Stone, J. Nanosci. Nanotechnol., 2002, 2, 95 CrossRef CAS.
- E. Eteshola, L. J. Brillson and S. C. Lee, Biomol. Eng., 2005, 22, 201 CAS.
- H. Chen, X. Su, K.-G. Neoh and W.-S. Choe, Langmuir, 2008, 24, 6852 CrossRef CAS.
- T. P. Chou, Q. Zhang, G. E. Fryxell and G. Cao, Adv. Mater., 2007, 19, 2588 CrossRef CAS.
- H. Bai, F. Xu, L. Anjia and H. Matsui, Soft Matter, 2009, 5, 966 RSC.
- N. Yokoo, T. Togashi, M. Umetsu, K. Tsumoto, T. Hattori, T. Nakanishi, S. Ohara, S. Takami, T. Naka, H. Abe, I. Kumagai and T. Adschiri, J. Phys. Chem. B, 2010, 114, 480 CrossRef CAS.
- L. Shi, J. Zhou and S. Gunasekaran, Mater. Lett., 2008, 62, 4383 CrossRef CAS.
- Z. R. Tian, J. A. Voigt, J. Liu, B. Mckenzie, M. J. Mcdermott, M. A. Rodriguez, H. Konishi and H. Xu, Nat. Mater., 2003, 2, 821 CrossRef CAS.
- B. Liu and H. C. Zeng, J. Am. Chem. Soc., 2003, 125, 4430 CrossRef CAS.
- B. Liu and H. C. Zeng, Langmuir, 2004, 20, 4196 CrossRef CAS.
- M. Muthukumar, J. Chem. Phys., 2009, 130, 161101 CrossRef CAS.
- Y. K. Gao, F. Traeger, O. Shekhah, H. Idriss and C. Wöll, J. Colloid Interface Sci., 2009, 338, 16 CrossRef CAS.
- S. Irrera, D. Costa and P. Marcus, J. Mol. Struct., 2009, 903, 49 CrossRef CAS.
- G. Mandal, S. Bhattacharya and T. Ganguly, Chem. Phys. Lett., 2009, 472, 128 CrossRef CAS.
- M. Bardhan, G. Mandal and T. Ganguly, J. Appl. Phys., 2009, 106, 034701 CrossRef.
- Z. J. Deng, G. Mortimer, T. Schiller, A. Musumeci, D. Martin and R. F. Minchin, Nanotechnology, 2009, 20, 455101 CrossRef.
- W. C. Chan and P. D. White, Fmoc Solid Phase Peptide Synthesis, Oxford university Press, New York, 2000 Search PubMed.
- S.-Y. Pung, K.-L. Choy, X. Hou and C. Shan, Nanotechnology, 2008, 19, 435609 CrossRef.
- C. H. Liu, M. Yan, X. Liu, E. Seelig and R. P. H. Chang, Chem. Phys. Lett., 2002, 355, 43 CrossRef CAS.
- GROMACS is a free software available under the GNU General Public License.
- N. Metropolis, A. W. Rosenbluth, M. N. Rosenbluth, A. H. Teller and E. Teller, J. Chem. Phys., 1953, 21, 1087 CrossRef CAS.
- Z. Zhang, M. Lu, H. Xu and W.-S. Chin, Chem.–Eur. J., 2007, 13, 632 CrossRef CAS.
- Q. Li, V. Kumar, Y. Li, H. Zhang, T. J. Marks and R. P. H. Chang, Chem. Mater., 2005, 17, 1001 CrossRef CAS.
- S.-C. Liou, C.-S. Hsiao and S.-Y. Chen, J. Cryst. Growth, 2005, 274, 438 CrossRef CAS.
- A. Sugunan, H. C. Warad, M. Boman and J. Dutta, J. Sol-Gel Sci. Technol., 2006, 39, 49 CrossRef CAS.
- H. Wang, C. Xie, D. Zeng and Z. Yang, J. Colloid Interface Sci., 2006, 297, 570 CrossRef CAS.
- S. Aisawa, E. Ishida, S. Takahashi, H. Hirahara and E. Narita, Chem. Lett., 2005, 630 CrossRef CAS.
- S. Aisawa, S. Takahashi, W. Ogasawara, Y. Umetsu and E. Narita, J. Solid State Chem., 2001, 162, 52 CrossRef CAS.
- A. Barth, Prog. Biophys. Mol. Biol., 2000, 74, 141 CrossRef CAS.
- O. Deschaume, A. Fournier, K. Shafran and C. C. Perry, New J. Chem., 2008, 32, 1346 RSC.
- K. Gorna, R. Muñoz-Espí, F. Gröhn and G. Wegner, Macromol. Biosci., 2007, 7, 163 CrossRef CAS.
- Y.-S. Fu, X.-W. Du, J. Sun, Y.-F. Song and J. Liu, J. Alloys Compd., 2008, 461, 527 CrossRef CAS.
- I. Bertini, H. B. Gray, E. I. Stiefel and J. S. Valentine, Biological Inorganic Chemistry: Structure and Reactivity, University Science Book, California, 2007 Search PubMed.
- U. Brand, M. Rombach, J. Seebacher and H. Vahrenkamp, Inorg. Chem., 2001, 40, 6151 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: FTIR spectra, XRD diffractograms, and TGA curves for ZnO precipitates formed; Zn2+ (%) reacted as a function of reaction time for reactions in the presence of peptide; crystal orientation of ALD-grown ZnO films; pKa values and structure of G-12 and GT-16 obtained using Marvin software; predicted adsorbing moieties of G-12 and GT-16 on (0001) and (100) planes of ZnO, FTIR peak area analysis for nitrate ion amide bands, and the estimation of weight loss between 200 and 900 °C region due to NO3−. See DOI: 10.1039/c0jm02124e |
| ‡ Present address: Unité POLY, Université Catholique de Louvain, Boltzmann A + 2, Croix du Sud, 1, B-1348, Louvain-la-Neuve, Belgium. |
| § Present address: Department of Chemical and Process Engineering, University of Strathclyde, 75 Montrose St, Glasgow G1 1XJ, UK. |
| This journal is © The Royal Society of Chemistry 2011 |