Effect of transition metal (M = Co, Ni, Cu) substitution on electronic structure and vacancy formation of Li3N
Shunnian
Wu
ab,
Zhili
Dong
a,
Ping
Wu
*ab and
Freddy
Boey
*a
aSchool of Materials Science and Engineering, Nanyang Technological University, Nanyang Avenue, Singapore 639798. E-mail: mycboey@ntu.edu.sg
bInstitute of High Performance Computing, 1 Fusionopolis Way, #16-16 Connexis, Singapore 138632. E-mail: wuping@ihpc.a-star.edu.sg
First published on 18th October 2010
Abstract
We carried out first principles calculations to investigate the effect of transition metal (M = Co, Ni, Cu) substitution on electronic structure and vacancy formation of Li3N in this study. Transition metals are shown to selectively substitute interplanar Li(1) atoms. Both Co and Ni substitution remarkably reduces the energy band gap to 0.55 eV in comparison with 1.13 eV of Li3N, while Cu substitution insignificantly decreases the energy band gap by 0.07 eV. Covalent bonding between transition metal atom and the coordinated N, which is manifested both visually by the contour plots of valence charge density difference and numerically by bond length variation, results in the formation of Li3−x−yMx□yN with y dependent on the covalency and concentration of transition metal. Ni substitution significantly reduces VLi(2) formation energy, which suggests greatly increased Li vacancy concentration for improved Li ionic mobility and conduction. Therefore, controlling the energy band gap and vacancy concentration by transition metal substitution provides a viable approach to tailor Li3N for variable applications in rechargeable lithium ion batteries.
Introduction
Transition metal substituted lithium nitrides have emerged as promising candidates for anode materials in rechargeable lithium ion batteries.1–5Li3N can react with transition metals at approximately 600–900 °C under nitrogen-rich conditions to form two groups of ternary nitride compounds, one with the anti-fluorite structure and the other with the layered Li3N structure.6–8 The structure of these ternary lithium nitrides follows a predictable trend that the anti-fluorite structure is favored for Ti through Fe and a structure is based on the layered Li3N structure for Co, Ni and Cu.9 Transition metals (M = Co, Ni, Cu) are proposed to substitute Li in Li(1) sites of Li3N as shown in Fig. 1, irrespective of the dopant concentration and the synthesis conditions applied.10–14 The layered morphology is claimed to be beneficial to fast Li-ion conduction and low transition metal oxidation states.9 It is claimed that no ternary lithium nitrides could surpass the high capacity of the Li2.6Co0.4N nitride at low potential, yet substitution of Co by Ni or Cu can dramatically improve the cycling performance with a slightly decreased capacity.1,15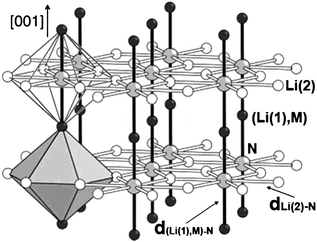 | ||
| Fig. 1 Schematic illustration of transition metal substituted Li3N. | ||
Two disputable models have been proposed to account for the substitution of Li by transition metals in Li3N. Some claimed ostensibly isovalent substitution of Li+ by M+, which leads to the formation of Li3−xMxN. Consequently, the Li vacancy concentration would be about 1–2% generally observed in Li3N. The charge balance of Li2.6Co0.4N was well described with monovalent cobalt.16,17 Negligible lithium vacancies were determined in Li2.6Co0.4N from chemical composition analysis.16 Li2.57Cu0.43N showed 1–2% disordered Li vacancies similar to Li3N,10 which seemed insensitive to reaction temperature. Niewa et al. also claimed that transition metal (M = Ni and Cu) substituted Li3N contains predominately M+ species over the whole range of x in Li3−xMxN based on their X-ray diffraction data, thermal measurements, and chemical analysis.13
Since transition metals rarely exist in the +1 oxidation state as M+ (Cu+ and few Ni+, Co+ oxo-ligands are exceptions18–22), others asserted aliovalent substitution of Li+ by M+2, which gives rise to the formation of Li3−2xMx□xN (□ = Li vacancy). This suggests that high concentration of Li vacancies is required to compensate the charge balance for divalent (or even trivalent) transition metals, and that Li vacancy concentration increases with the concentration of doped transition metal. LiNiN was claimed to serve as an excellent model compound of the stoichiometric LiNi□N with 50% Li vacancies in the [Li2N] plane.23 Ducros et al. declared to synthesize a series of stoichiometric Li3−2xNixN (0.20 ≤ x ≤ 0.60) compounds with the existence of Ni2+ and correspondingly compensated Li vacancies.24 Li1.99Co0.53N synthesized by Gordon et al.,11 Li1.36Ni0.79N and Li2.25Cu0.43N by Gregory et al.,12 and Li2.2Cu0.5N by Weller et al.25 were also proposed to support the predominant aliovalent substitution model.
These two discrepant substitution models bring about distinct transition metal oxidation states and thus Li vacancy concentrations. The ionic conduction of these substituted lithium nitrides originates from hopping of Li ions among the Li vacancies and Li occupied sites. Therefore, an understanding of the nature of Li vacancies in transition metal substituted Li3N is essential for their potential applications. However, to date, there is little relevant theoretic work reported in the literature. In this study, we carried out first principles calculations to study the effect of transition metal substitution on the electronic structure and vacancy formation of Li3N. These results would provide an insight into the dopant selection and vacancy tailoring for improved performance of ternary lithium nitrides.
Methodology
First principles calculations were carried out using the plane-wave based Vienna ab initio simulation package VASP.26–28 Exchange and correlation were treated within the Perdew–Burke–Ernzerhof (PBE) generalized gradient approximation (GGA).29,30 For the electronic configuration, the states treated as bands were: Li (2s, 2p), N (2s, 2p), Co (4s, 3d), Ni (4s, 3d) and Cu (4s, 3d). The cutoff energy for the plane-wave expansion was set to be 500 eV, sufficiently high to give well-converged total energies and structures. 4 × 4 × 4k-point mesh according to the Monkhorst–Pack method31 was adopted in all total energy calculations. With this setup, total energies in our calculations are converged to within 0.01 eV. In the relaxation of Li3N unit cell, both cell parameters and ionic positions were fully optimized so as to obtain the minimum total energies. Our calculated lattice constants are a = 3.64 Å and c = 3.87 Å, which are in good agreement with the experimental values 3.65 Å and 3.87 Å,32 respectively. Li3N supercells were constructed by 3 × 3 × 3 expansion of Li3N unit cell and one Li atom was substituted to model the substituted structure, yielding a dopant concentration of 1.23 at%. To establish clearly the effect of transition metal substitution without much interference from structural changes, the lattice parameters of Li3N supercells with transition metal substitution were fixed as those of the Li3N supercells, as we assumed the conditions at the dilute limit. Our preliminary calculations indicate that substitution brings about negligible lattice variation with observed volume change less than 0.3%. A Li vacancy was generated by removing one Li atom near the transition metal atom to observe its effect more clearly. Spin-polarization was also included where excess spins are expected.Results and discussion
Hexagonal layer-structured Li3N consists of alternating [Li(2)2N] and Li(1) layers, where two types of Li exist, i.e. one Li(1) coordinated with 2 N atoms and two equivalent Li(2) coordinated with 3 N atoms. Therefore, transition metal (M = Co, Ni, Cu) may substitute Li either in Li(1) site or in Li(2) site in Li3N structure. Total energy calculations were first carried out to determine the substitution site. The energy differences ΔE for transition metal substitution in two different Li sites are calculated with:| ΔE=ET(MLi(1)) −ET(MLi(2)) | (1) |
The obtained ΔE values for different transition metals (M = Co, Ni, Cu) are listed in Table 1. The ΔE values being less than −1.2 eV suggests that transition metals selectively replace Li in Li(1) sites. This is consistent with the experimentally observed transition metal substitution for Li at the interplanar site.10–14 Therefore, Li3N with transition metal doped in Li(1) site was selected for the following study.
| Li3N/Co | Li3N/Ni | Li3N/Cu | |
|---|---|---|---|
| a Li3N/Co, Li3N/Ni and Li3N/Cu denote Li3N supercell with one Li substituted by Co, Ni and Cu separately. | |||
| ΔE/eV | −1.34 | −1.30 | −1.23 |
The densities of states (DOS) and the projected densities of states (PDOS) for Li3N and transition metal substituted Li3N are shown in Fig. 2. It shows that Li3N is an ionic semiconductor with occupied valence band mainly derived from N 2p states and conduction bands from Li 2s and 2p states. The appreciable overlapping of Li 2s states with N 2p states implicates weak covalent bonding in the structure. This is consistent with the electron energy loss spectroscopy study, which discloses that Li3N is not completely ionic but has at least weak covalent characteristics.17 Transition metal substitution significantly changes the electronic structure of Li3N. Spin-polarization is observed in both Li3N/Co and Li3N/Ni, suggesting the existence of unpaired 3d electrons in Co and Ni, while no spin-polarization in Li3N/Cu indicates fully paired 3d electrons in Cu. The valence bands in transition metal substituted Li3N are composed of both M 3d states and N 2p states, and the conduction bands mainly come from Li 2s and 2p states. Substitution of Li by both Co and Ni greatly reduces the energy band gap to 0.55 eV in comparison with 1.13 eV of Li3N, which can be attributed to the formation of tangling bonds at the Fermi surface. This suggests possible electronic conduction via electron hopping in Co or Ni substituted Li3N. It is expected that the substituted Li3N can even become a metallic conductor with formation of infinite N–M–N chains, 1∞[MN2/2], with increasing dopant concentration.33 On the other hand, Cu substitution insignificantly reduces the energy band gap by 0.07 eV without formation of tangling bonds at the Fermi surface. Therefore, different transition metal substitution suggests a viable approach to tailor the electronic conduction of Li3N for various electrochemical applications.
 | ||
| Fig. 2 DOS and PDOS for Li3N (a), and Li3N/Co (b), Li3N/Ni (c) and Li3N/Cu (d) respectively. The PDOS components are s black; px red; py blue; pz green; dxy (overlapped with dx2−y2) yellow; dyz gray; dxz cyan; dz2 pink; and dx2−y2 dark red. The Fermi level is set to zero. | ||
It is seen from Fig. 2 that the five-fold degenerate 3d states of transition metal splits to delocalized dxz state and localized dxy/dx2−y2and dyz/dz2 states. Therefore, there are one unpaired 3d electron in Co of 3d7, two unpaired 3d electrons in Ni of 3d8 and no unpaired electron in Cu of 3d10, which is consistent with the observed spin-polarization. The overlapping of N px/py states with M dxy/dx2−y2 states in the valence band region suggests the occurrence of π-bonding, while the M dyz/dz2 states may interact with N pz state to form σ-bonding. This implicates covalent bonding between N and M in transition metal substituted Li3N. The 3d states of Co and Ni located at approximately 0.6 eV above the original valence band maximum (VBM) of Li3N interact with Li and N states which are partly pushed to higher energies. This upward shift of valence bands decreases the cohesion in Co or Ni substituted Li3N and thus brings about the destabilization. On the contrary, the interaction of 3d states of Cu with N and Li states does not push N and Li states to higher energies, since the 3d states of Cu to build up the valence bands are close to the original VBM of Li3N. Therefore, Cu substitution marginally affects the thermodynamic stability of Li3N. Experiments observed that ternary copper lithium nitrides are more stable than ternary nickel lithium nitrides and ternary cobalt lithium nitrides.14 Destabilization by transition metal substitution was also asserted in lithium borohydride34 and lithium amide–imide structures.35 The destabilization might be beneficial to the intercalation and deintercalation processes in rechargeable lithium ion batteries, which leads to improved charge–discharge cyclability.
The different bonding in Li3N and transition metal substituted Li3N is illustrated by the contour plots of valence charge density difference of the plane through substitution site in [001] direction shown in Fig. 3. The difference is defined as Δρ = ρsolid − ∑ρatom, i.e. the difference between the total charge density of the solid and the superposition of independent atomic charge densities placed at the atomic sites of the same solid, which is instructive to evaluate valence charge density distribution. The weak interactions between Li(1) and M in the (Li(1),M) layer are not shown here. It can be seen that conspicuous ionic bonding between Li and N predominates in Li3N. Electrons accumulate around the N atoms while deplete around the Li atoms, indicating that the charges transfer from Li to N. On the other hand, electrons accumulate between N and M in transition metal substituted Li3N, forming a marked directional bonding. This remarkable charge polarization indicates formation of both ionic and covalent bonding between N and M, significantly different from the predominant ionic bonding in Li3N. A distinct difference is seen between Co/Ni and Cu in the binding with nitrogen. Nitrogen back donates electrons to Co and Ni while does not back donate electrons to Cu. This can be attributed to the unpaired electrons in 3d orbits in Co and Ni and totally paired electrons in the fully filled 3d orbits in Cu. The 3d orbits with unpaired electrons in Co and Ni can accept electrons back donated by nitrogen, while the fully filled 3d orbits in Cu cannot accept back donated electrons. The covalent binding between transition metal and N reduces the oxidation state of transition metal and leads to an effective oxidation state below the nominal +2 depending on the covalency. Consequently, this decreases the Li vacancies required to compensate the charge balance derived from the nominal oxidation state of transition metal. This indicates that nonstoichiometric Li vacancies would form in the substituted structure due to the covalent bonding between transition mental and nitrogen.
![Valence charge density difference of the plane through substitution site in [001] direction for Li3N (a), and Li3N/Co (b), Li3N/Ni (c) and Li3N/Cu (d) respectively. The contour interval is 0.004 e Å−3. The electron accumulation is depicted by pink lines, while the electron depletion is represented by green lines.](/image/article/2011/JM/c0jm01883j/c0jm01883j-f3.gif) | ||
| Fig. 3 Valence charge density difference of the plane through substitution site in [001] direction for Li3N (a), and Li3N/Co (b), Li3N/Ni (c) and Li3N/Cu (d) respectively. The contour interval is 0.004 e Å−3. The electron accumulation is depicted by pink lines, while the electron depletion is represented by green lines. | ||
Therefore, both ostensibly isovalent substitution model and aliovalent substitution model could not capture the apparent oxidation state of transition metal and Li vacancy concentration due to occurrence of the marked covalent bonding between transition metal and N, particularly the apparent charge transfer from nitrogen to Co and Ni. The reported transition metal substituted Li3N rarely approaches the supposed composition Li3−xMxN or Li3−2xMx□xN. LiNiN can be regarded as an exception owing to alteration of crystal structure and formation of ordered structure. It is concluded that transition metal substitution results in the formation of Li3−x−yMx□yN, in which y is dependent on the covalency and concentration of transition metal in the structure. The preparation conditions may also affect the covalency of transition metal, consequently the Li vacancy concentration. For example, a series of Li3−x−yNix□yN with x = 0.339, y = 0.126; x = 0.360, y = 0.162; x = 0.362, y = 0.320; x = 0.565, y = 0.412; x = 0.577, y = 0.188; x = 0.743, y = 0.626 reported by Stoeva et al.14 show that Li vacancy concentrations are higher than 1–2% of Li3N but lower than the corresponding substitution concentrations with ambiguous correlation.
The covalent bonding between transition metal and N may be further manifested by variation of the bond lengths d(Li(1),M)–N and dLi(2)–N as listed in Table 2. The calculated dLi(1)–N and dLi(2)–N in Li3N are 1.944 Å and 2.106 Å respectively, which is well consistent with the experimental values.32 Substitution for Li by Co, Ni and Cu reduces the interplanar d(Li(1),M)–N in the order Li3N/Co < Li3N/Ni < Li3N/Cu, while increases the dLi(2)–N in [Li2N] layer in the order Li3N/Co > Li3N/Ni > Li3N/Cu. This trend is in agreement with experimental observations.11,12,23,25,36 However, it is difficult to make strict comparison of the calculated bond lengths with the available experimental values, since these values are dependent on transition metal substitution concentration and Li vacancy concentration.25 The ionic radii for Co, Ni, Cu and Li in their low oxidation states from Shannon37 are 0.65, 0.69, 0.77 and 0.76 Å, respectively. The six-coordination values are used here to examine the trend since 2-coordination values are not available for all the ions considered. The apparent decrease of d(Li(1),M)–N with substitution for Li by Cu of similar size indicates the covalent binding between Cu and N. However, the markedly decreased d(Li(1),M)–N with substitution for Li by Co/Ni indicates that the dopant size also plays an important role. Covalent bonds between transition metal and N strengthen the interplanar binding, thus the d(Li(1),M)–N is reduced with the reduction dependent on the covalent binding strength. The interplanar covalent binding decreases the charge of N, thus weakens the N–Li(2) binding in the [Li2N] layer. This results in the extension of dLi(2)–N.
The alteration of Li3N electronic structure with transition metal substitution significantly influences Li vacancy formation. Li vacancies are crucial to the fast Li ionic conduction of lithium nitrides for the hopping of Li ions between occupied Li sites and Li vacancies. Theoretical study has disclosed that Frenkel defect is not possible to be formed in lithium nitride,38 therefore, Li vacancy is generated from removal of one Li(1) or Li(2) atom in the parent and doped Li3N supercells. Neutral Li vacancy formation energy EF is evaluated as:
| EF = ET(V) − ET(bulk) + µLi | (2) |
| lµLi + mµM + nµN = µbulk | (3) |
![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) and bulk Cu (Fmm) were chosen as the upper limits of chemical potential of nitrogen, cobalt, nickel and copper, respectively. The calculated total energies per unit formula were applied as the chemical potentials of the studied systems. We consider the Li-poor limitation employed in practical synthetic conditions.
m) and bulk Cu (Fmm) were chosen as the upper limits of chemical potential of nitrogen, cobalt, nickel and copper, respectively. The calculated total energies per unit formula were applied as the chemical potentials of the studied systems. We consider the Li-poor limitation employed in practical synthetic conditions.
The calculated formation energies EF for Li vacancy in Li(1) site VLi(1) and in Li(2) site VLi(2) are listed in Table 3. It shows that the formation energy of VLi(1) is much larger than that of VLi(2) in transition metal substituted Li3N with the same trend as in Li3N. Thus, Li vacancies mainly form in [Li2N] layers owing to the energetically favorable process. This is in agreement with the reported Li vacancies in [Li2N] layers.11–14
| Li3N | Li3N/Co | Li3N/Ni | Li3N/Cu | |
|---|---|---|---|---|
| V Li(1) | 2.23 | 1.99 | 1.88 | 2.13 |
| V Li(2) | 0.52 | 0.24 | −0.14 | 0.46 |
It is found from Table 3 that Co and Ni substitution reduces the VLi(2) formation energy by 0.28 eV and 0.66 eV respectively, while Cu substitution slightly reduces the VLi(2) formation energy by 0.06 eV. The effective decrease of VLi(2) formation energy with Co and Ni substitution can bring about significant increase of VLi(2) concentration, which remarkably improves the Li diffusion, Li ionic conduction and thus the charge–discharge capacity.8,33 This makes lithium cobalt nitride and particularly lithium nickel nitride outstanding candidates for use as anode materials in rechargeable lithium ion batteries. The slight decrease of VLi(2) formation energy with Cu substitution indicates unsubstantial increase of Li vacancy concentration. The distinct discrepancy in VLi(2) formation energy between Co, Ni and Cu substituted Li3N is consistent with the observed back donation of nitrogen electrons in Fig. 3. It seems that N back donates charge into the dxy orbit of Co in Li3N/Co while back donates charge into both dxy and dx2−y2 orbits of Ni in Li3N/Ni, which correlates with the unpaired electrons in 3d orbits. Back donation of nitrogen electrons to Co and Ni reduces the charge of nitrogen, and thus weakens the binding between N and Li(2), which facilitates the VLi(2) formation. Since Ni has more unpaired 3d electrons in the splitted 3d orbits than Co, thus nitrogen may back donate more electrons in Ni substituted Li3N, which is beneficial to stabilize the VLi(2) containing structure. This can explain the reported better stability on cycling of Li3−xNix□yN due to its tolerance to low Li concentration during a wider range of Li deintercalation and intercalation.32 The low VLi(2) formation energy may also lead to the formation of the exceptionally ordered compound LiNiN. The ordering reduces the Li ionic mobility but improves the structural stability during charge–discharge cycling. The different effect of Co, Ni and Cu substitution on Li vacancy formation suggests that transition metal substitution could be employed to control the ionic mobility and conduction properties of Li3N for various electrochemical applications.
Back donated nitrogen electrons seems to correlate with the unpaired electrons in transition metals, therefore, it is possible to predict the effects of other transition metals assuming that their ternary lithium nitrides can maintain the layered Li3N structures at diluted doping concentrations. Sc, V, Cr and Mn are expected to have unpaired electrons in the splitted 3d orbits, thus can accept the electrons back donated by nitrogen. Therefore, their substitution for Li may also significantly reduce Li vacancy formation energy, which brings about improved Li ion mobility and conduction. The ionic radii for Sc, V, Cr and Mn in their low oxidation states are 0.75, 0.79, 0.80 and 0.67 Å, respectively.37 Compared with Sc, V and Cr of similar size to Li or larger than Li, the much smaller Mn can form interplanar covalent binding with N with shortened bond length, which suggests that Mn be a promising candidate for Li substitution for further investigation.
Conclusions
Transition metal (M = Co, Ni, Cu) substitution significantly influences the electronic structures and the Li vacancy formation of Li3N. First principles calculations indicate that transition metal substitution takes place mainly at Li(1) sites, which agrees well with the experimental observations. Co or Ni substitution can reduce the energy band gap and even change Li3N from a semiconductor to a metallic-like conductor, which has the advantage of both electronic and ionic conduction. Transition metal substitution reduces the thermodynamic stability of Li3N, which might benefit the intercalation and deintercalation of lithium during charge–discharge cycling in rechargeable lithium batteries. Covalent bonding is formed between transition metal atom and the coordinated N, which results in the weakening of the intraplanar binding and the strengthening of the interplanar binding. This reduces the nominal oxidation state of transition metal, and decreases the Li vacancies required for charge compensation, leading to the formation of Li2−x−yMx□yN with y dependent on the transition metal nature and the substitution concentration. Co or Ni substitution brings about remarkable reduction of Li vacancy formation energy in comparison with Li3N, which is attributed to their 3d orbits with unpaired electrons to accept the electrons back donated by nitrogen. This leads to significant improvement in ionic mobility and conduction. Therefore, transition metal substitution provides a viable approach to control the electronic properties and electrochemical performance of Li3N in anode applications. It may render appreciable electronic conduction to the original material and generate a vacancy-controlled charge compensation system for active oxidation–reduction reaction during charge–discharge cycling.References
- D. H. Gregory, Chem. Rec., 2008, 8, 229–239 CrossRef CAS.
- M. Nishijima, T. Kagohashi, M. Imanishi, Y. Takeda, O. Yamamoto and S. Kondo, Solid State Ionics, 1996, 83, 107–111 CrossRef CAS.
- M. Nishijima, T. Kagohashi, Y. Takeda, M. Imanishi and O. Yamamoto, J. Power Sources, 1997, 68, 510–514 CrossRef CAS.
- T. Shodai, S. Okada, S. Tobishima and J. Yamaki, Solid State Ionics, 1996, 86–88, 785–789 CrossRef CAS.
- T. Shodai, S. Okada, S. Tobishima and J. Yamaki, J. Power Sources, 1997, 68, 515–518 CrossRef CAS.
- R. Juza, K. Langer and K. V. Benda, Angew. Chem., Int. Ed. Engl., 1968, 7, 321–406 CrossRef CAS.
- R. Niewa, D. Zherebtsov and Z. Hu, Inorg. Chem., 2003, 42, 2479–2816 CrossRef.
- Z. Stoeva, R. Gomez, D. H. Gregory, G. B. Hix and J. J. Titman, Dalton Trans., 2004, 3093–3097 RSC.
- J. L. C. Rowsell, V. Pralong and L. F. Nazar, J. Am. Chem. Soc., 2001, 123, 8598–8599 CrossRef.
- A. G. Gordon, D. H. Gregory, A. J. Blake, D. P. Weston and M. O. Jones, Int. J. Inorg. Mater., 2001, 3, 973–981 CrossRef CAS.
- A. G. Gordon, R. I. Smith, C. Wilson, Z. Stoeva and D. H. Gregory, Chem. Commun., 2004, 2812–2813 RSC.
- D. H. Gregory, P. M. O'Meara, A. G. Gordon, J. P. Hodges, S. Short and J. D. Jorgensen, Chem. Mater., 2002, 14, 2063–2070 CrossRef CAS.
- R. Niewa, Z.-L. Huang, W. Schnelle, Z. Hu and R. Kniep, Z. Anorg. Allg. Chem., 2003, 629, 1778–1786 CrossRef CAS.
- Z. Stoeva, R. I. Smith and D. H. Gregory, Chem. Mater., 2006, 18, 313–320 CrossRef CAS.
- Y. Liu, K. Horikawa, M. Fujiyosi, N. Imanishi, A. Hirano and Y. Takeda, Electrochim. Acta, 2004, 49, 3487–3496 CrossRef CAS.
- T. Shodai, Y. Sakurai and T. Suzuki, Solid State Ionics, 1999, 122, 85–93 CrossRef CAS.
- S. Suzuki, T. Shodai and J. Yamaki, J. Phys. Chem. Solids, 1998, 59, 331–336 CrossRef CAS.
- F. Bernhardt and R. Hoppe, Z. Anorg. Allg. Chem., 1993, 619, 969–975 CAS.
- W. Burow, J. Birx, F. Bernhardt and R. Hoppe, Z. Anorg. Allg. Chem., 1993, 619, 923–933 CAS.
- A. Möller, Z. Anorg. Allg. Chem., 2001, 627, 2625–2629 CrossRef.
- A. Möller, M. A. Hitchman, E. Krausz and R. Hoppe, Inorg. Chem., 1995, 34, 2684–2691 CrossRef.
- M. Sofin and M. Jansen, Z. Anorg. Allg. Chem., 2001, 627, 2115–2117 CrossRef CAS.
- M. G. Barker, A. J. Blake, P. P. Edwards, D. H. Gregory, T. A. Hamor, D. J. Siddons and S. E. Smith, Chem. Commun., 1999, 1187–1188 RSC.
- J. B. Ducros, S. Bach, J. P. Pereira-Ramos and P. Willmann, Electrochim. Acta, 2007, 52, 7035–7041 CrossRef CAS.
- M. T. Weller, S. E. Dann, P. F. Henry and D. B. Currie, J. Mater. Chem., 1999, 9, 283–287 RSC.
- G. Kresse and J. Furthmuller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter, 1996, 54, 11169–11186 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396–1396 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- A. Rabenau and H. Schulz, J. Less Common Met., 1976, 50, 155–159 CrossRef CAS.
- Z. Stoeva, B. Jäger, R. Gomez, S. Messaoudi, M. B. Yahia, X. Rocquefelte, G. B. Hix, W. Wolf, J. J. Titman, R. Gautier and P. Herzig, J. Am. Chem. Soc., 2007, 129, 1912–1920 CrossRef CAS.
- K. Miwa, N. Ohba, S. Towata, Y. Nakamori and S. Orimo, J. Alloys Compd., 2005, 404–406, 140–143 CrossRef CAS.
- M. Gupta and R. P. Gupta, J. Alloys Compd., 2007, 446–447, 319–322 CrossRef CAS.
- D. H. Gregory, P. M. O'Meara, A. G. Gordon, D. J. Siddons, A. J. Blake, M. G. Barker, T. A. Hamor and P. P. Edwards, J. Alloys Compd., 2001, 317–318, 237–244 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- J. R. Walker and C. R. A. Catlow, J. Phys. C: Solid State Phys., 1980, 6, 32–34.
| This journal is © The Royal Society of Chemistry 2011 |