 Open Access Article
Open Access ArticleDetermination of uranium isotopic ratio (235U/238U) using extractive electrospray ionization tandem mass spectrometry†
Chunxiao
Liu‡
a,
Bin
Hu‡
b,
Jianbo
Shi
a,
Jianqiang
Li
b,
Xinglei
Zhang
b and
Huanwen
Chen
*b
aState Key Laboratory of Environmental Chemistry and Ecotoxicology, Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences, P.O. Box 2871, Beijing, 100085, P. R. China
bJiangxi Key Laboratory for Mass Spectrometry and Instrumentation, College of Chemistry, Biology and Material Science, East China Institute of Technology, Nanchang, Jiangxi Province 330013, P. R. China. E-mail: chw8868@gmail.com; Fax: (+86)-791-3896-370; Tel: (+86)-791-3896-370
First published on 14th July 2011
Abstract
Extractive electrospray ionization (EESI) mass spectrometry (MS), a typical MS platform for detection of organic compounds, has been used for quantitative detection of uranium isotopic ratio (235U/238U) in uranyl nitrate solution samples, which were prepared from various samples including natural water samples, uranium ore samples and soil samples using nitric acid. This method requires minimal sample pretreatment and no high resolution instruments. The typical time for analysis of a single liquid sample was about 5 min since the prepared sample solutions were directly infused into the EESI source without separation or preconcentration. The relative errors for uranium isotope measurements were in the range of 0.21%–0.25% and the corresponding relative standard deviation (RSD) values were 1.54%–1.81% for the ore samples. The results obtained by EESI-MS for direct analysis of 5 soil samples were validated by using ICP-MS. These findings confirmed that EESI-MS can be employed for quantitative measurement inorganic compounds present in a complex matrix. The fast detection of uranium isotopes shows potential applications of EESI-MS in nuclear research laboratories and the nuclear energy industry.
1. Introduction
Uranium mainly exists as 238U and 235U in nature, and is an important radioactive material with wide military and civilian applications.1–4 The most common isotopes of uranium are 238U and 235U, although 234U is also of low abundance. The natural abundance ratio of the isotopes (235U/238U) is 0.007257. This ratio is a natural rule marking the artificial nuclear activities which change the isotope ratio of uranium. Driven by the ambitious motivation to solve the energy problem, the globally rapid development of nuclear power plants has demanded large amounts of uranium, which sparked the advancement of uranium mining and scientific research on uranium.2 The concentration of uranium isotopes in uranium ores is a principal indicator measuring the quality of ores. The commercial nuclear power plants use uranium fuel that is typically enriched to about 3% 235U.5,6 Therefore, the analytical and separation technologies of uranium are of significance in the uranium industry.6 In addition, the detection of uranium in soil and water is of great interest in biological and environmental science due to its radioactivity.7–9 Solution chemistry of uranium is dominated by uranyl species, which is involved in the processes of nuclear fuel production and waste handling.10–12 Studies of uranyl species helps to monitor and predict their radioactive behaviors in the environment and result in the ultimate control of nuclear waste. Uranium can be dissolved in either an acid or alkali solution; however nitric acid is probably the most commonly used reagent for dissolving uranium ore samples.2,13–15 Thus, development of technologies for the determination of uranium isotopic ratio (235U/238U) in uranyl nitrate is highly desirable.Currently, methods including potentiometric titration,16–18 photometry,19 and polarography20–22etc. are available for detection of uranyl species. Generally these methods are not highly sensitive and are time-consuming. Mass spectrometry (MS) is a relatively universal method with high sensitivity and good selectivity; however, uranyl compounds are inorganic analytes, which are commonly in solutions with complex matrices and are incompatible with most commercially available ionization techniques. Inductively coupled plasma mass spectrometry (ICPMS)23–25 and isotope ratio mass spectrometry (IR-MS) are two widely used mass spectrometric methods for the analysis of uranyl compounds.26 In ICP both organic and inorganic analytes are broken down to elemental species for further MS analysis. This makes ICP-MS difficult to obtain structure information of the analytes. For most liquid samples containing complex matrices, multi-step sample pretreatments such as solvent extraction, ion exchange, and coprecipitation27,28 is usually required prior to ICP-MS experiments. The sample pretreatment steps may decrease detection sensitivity, introduce chemical impurities, and even alter oxidation states of the analytes of interest. Therefore, it would be interesting to develop novel methodologies for the rapid detection of uranium species with high sensitivity and high specificity.
In 2004, Cooks et al. raised the concept of ambient mass spectrometry and made a breakthrough towards fast analysis of complex samples with minimal sample pre-treatment.29 Important ambient ionization techniques include desorption electrospray ionization (DESI),29–31 extractive electrospray ionization (EESI),32–34 surface desorption atmospheric pressure chemical ionization (SDAPCI),35–37direct analysis in real time (DART),38,39 low temperature plasma (LTP),40,41etc. Unlike the 2-D techniques (e.g., DESI, DAPCI, DART, etc.) for direct analysis of 2-demensional surfaces, EESI is an ambient ionization technique targeting to direct ionize samples dispensed in a 3-D space.42 In EESI, neutral analytes present in raw samples are ionized by the highly charged primary droplets generated by electrospraying pure solvent (e.g., acetic acid/methanol-water solution) through extraction interactions and collisions, which are mainly completed in the 3-D spatial section formed between the neutral sample introduction channel, the primary reagent ion generation channel, and the ion entrance of the MS instrument.42 The unique design of EESI allows the matrices of samples to be dispersed in a relatively large spatial section. Therefore, EESI tolerates extremely complex matrices and it has been used to analyze complicated mixtures such as milk, urine, explosives, and aerosol drugs.32,33,43 Another advantage of EESI is that samples are isolated from the direct bombardment by charged particles or energetic metastable atoms, which makes EESI attractive for monitoring biological samples in their native forms.44 Due to the specificity achieved by tandem mass spectrometry, EESI-MSn has been successfully applied for the online and real-time analysis in various disciplines including food safety,45drug control,43 environmental science,46etc. However, to date most efforts have been made to use ambient ionization techniques for the rapid detection of organic species; studies employing ambient mass spectrometry for the rapid detection of inorganic compounds are rarely seen in previous literatures.
Previously, we reported the study in which EESI-MSn was implemented to directly analyze uranyl species in natural water samples without any sample pretreatment.46 In this follow-up study, EESI-MSn was used to examine the uranium isotopic ratio (235U/238U) in uranyl nitrate solutions prepared from natural water samples, uranium ore samples and soil samples using nitric acid, requiring no further sample preparation. This approach can not only be used for the rapid detection of radioactive compounds with high tolerance to matrix disturbance, but also is very promising for the accurate uranium isotopic ratio (235U/238U) determination in uranyl nitrate.
2. Experiments and methods
2.1 Reagents and materials
Nitric acid (A.R. grade) was bought from the Chinese Chemical Reagent Co. Ltd. (Shanghai, China). Methanol (HPLC grade) was purchased from Fisher Scientific (Pittsburgh, US). Uranium standard (10 mg L−1, 235U/238U = 0.00725) was purchased from Agilent US. All chemicals were directly used without any pretreatment except dissolution and dilution using deionized water when it was necessary. Three types of natural water samples (lake water, river water 1 and river water 2) were fetched from different geological regions in China. Prior to EESI-MS analysis, nitric acid (6 mol L−1) was added into the natural water samples (10 mL, each) to prepare the analytical solutions, which were of pH 1.0 at the final stage. Approximately 0.1 g soil sample was weighed into a PFA beaker and 3 mL of HF and 1 mL of HNO3 were added carefully. The sample was evaporated to almost dryness. The dissolution step was repeated once again. After evaporation, traces of HF were removed by the successively addition of 2 mL HNO3 followed by evaporation to almost dryness. The residue was dissolved in 15 mL 5% HNO3 and after 3-fold dilution the uranium isotopic ratio was measured by either EESI-MS or ICP -MS. Uranium ore samples were gifts from the China Institute of Atomic Energy (Beijing, China), and the preparation steps were documented with details in the previous literature.47 Briefly, the raw ore samples were ground to particles with sizes less than 75 μm. The fine ore particles (0.1 g) were then decomposed by a mixed acid (nitric acid, hydrofluoric acid and perchloric acid) under heating (80 °C), and then 3 mL of nitric acid (6 mol L−1) were used for extraction. The supernatant was diluted 1000 times using deionized water. The resulting sample was directly infused at a flow rate of 5 μL min−1 for EESI-MS analysis without any further treatment. A methanol/water solution (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) was delivered at a flow rate of 5 μL min−1 to the ESI emitter, by using a syringe pump, for the generation of the charged primary droplets.
:1, v/v) was delivered at a flow rate of 5 μL min−1 to the ESI emitter, by using a syringe pump, for the generation of the charged primary droplets.
2.2 Instrumental setup
Besides of the reference experiments which were performed using ICP-MS (Agilent 7500ce, USA), all the other experiments were carried out using a LTQ-XL mass spectrometer (Finnigan, San Jose, CA, USA) equipped with a homemade EESI source. The schematic illustration of EESI-MS for the detection of radioactive species is shown in Fig. 1, which has been constructed in a way similar to that described in our previous study.46 Because the radioactive samples were involved in the experiments, there is a safety issue concerned with this study. Therefore, the EESI source was sealed to the LTQ mass spectrometer so that no material could be released into ambient air during the whole analysis. In the negative ion detection mode, the primary charged particles were generated in the ESI channel using a high voltage (−3.5 kV). After ionization in EESI, analyte ions were introduced into the LTQ mass analyzer through the ion guide system for mass analysis. The distance (a) between the spray tips of the EESI source and the MS inlet was 5 mm, and the distance (b) between the end-tips of the two sprays was 2 mm. The angle (α) between the electrospray beam and the MS inlet of the LTQ-MS instrument was 150°, and the angle (β) between the two spray beams was 60°. The temperature of the heated capillary of the LTQ-MS was maintained at 200 °C. No further optimization was performed to the heated capillary, the tube lenses, the conversion dynodes, the detectors, etc. All the full scan mass spectra were recorded using Xcalibur software with an average time of 30 s. Collision induce dissociation (CID) experiments were performed by applying 30% collision energy for 30 ms to the precursor ions. The precursor ions of interest were isolated using a mass window of 1.2 mass/charge (m/z) units. CID mass spectra were recorded using an average time of 5 min at the maximum. Compounds of interest were identified with MS and CID data matching against the authentic standards.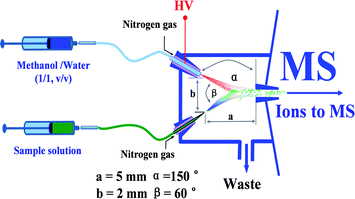 | ||
| Fig. 1 Schematic diagram of the EESI source for detection of radioactive species. Note that the diagram is not proportionally scaled. | ||
2.3 Special safety remarks
The handling of radioactive substances requires special permission. The whole EESI source has to be built as a ventilation system coupled to the LTQ-MS instrument. It is very important to make sure that no radionuclides are emitted into laboratory atmosphere. The waste from the EESI source and the exhaust of the LTQ-MS should be carefully collected for the special waste handling.3. Results and discussion
3.1 Detecting 238U and 235U in uranium ore extracts by EESI-MSn
Fig. 2 shows the EESI-MS mass spectrum for the detection of uranyl nitrate. Uranyl nitrate forms [UO2(NO3)3]− complex ions in nitric acid aqueous solution. In the negative ion detection mode, EESI-MS generated a base peak at m/z 456 (Fig. 2) due to the [238UO2(NO3)3]− ions. The relative abundance of 238U is so high that it is difficult to directly observe other uranium isotope ions in the MS spectrum. However, when the mass spectrum was zoomed enough, the peaks of [235UO2(NO3)3]− ions and [234UO2(NO3)3]− ions were clearly detected, with appropriate abundance ratios. The inset of Fig. 2 shows both peaks corresponding to the ions of m/z 453 ([235UO2(NO3)3]−) and m/z 456 ([238UO2(NO3)3]−), showing sound peak shapes for both peaks, regardless of the significant difference in the abundances.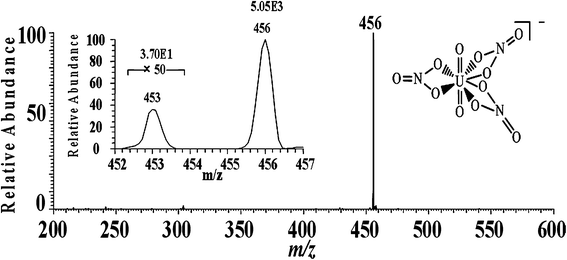 | ||
| Fig. 2 EESI-MS mass spectrum for the detection of uranyl nitrate. The inset shows the mass spectrum of isotopic peaks. | ||
To confirm the identification, CID experiments were performed by applying 30% collision energy to the precursor ions of m/z 456. As shown in Fig. 3a, a small amount of precursor ions lost NO and generated a peak at m/z 426 with low abundance (1%). Most of the precursor ions (m/z 456) lost NO2 to form fragment ions [238UO3(NO3)2]− of m/z 410 (Fig. 3a). [238UO3(NO3)2]− has a proposed symmetric structure with double tetra-atomic rings (Fig. 3a) which are beneficial to host negative charges. Thus, the fragment ions at m/z 410 are relatively stable and the relative abundance of this peak is the highest in Fig. 3a. EESI-MS3 experiments were further performed targeting the precursor ions of m/z 410. As a result, three different fragment ions were produced at m/z 348, 366, and 380 (Fig. 3b). The fragment peaks at m/z 348 and 380 are due to the loss of NO3 and NO from [238UO3(NO3)2]− (m/z 410), respectively. The fragment ions of m/z 366 were generated because of the ion-molecule interactions between the intermediate ionic species at m/z 348 and H2O. Water, with a small amount in the ion trap, has lone pair electrons; meanwhile U in [OUO2NO3]− (m/z 348) has empty orbits. Therefore, it is not surprising to form the [H2O + OUO2NO3]− complex species during the CID experiments The resultant complex ions of m/z 366 are unstable, and it readily loses H2O to generate a peak at m/z 348 as shown in the inset of Fig. 3b. In the EESI-MS4 experiments, 30% of collision energy was applied to the ions of m/z 348 and we obtained a fragment peak at m/z 302 with low abundance as (Fig. 3c). This is because the precursor ions (m/z 348) lost NO2 to produce [UO4]− which is the final product of tandem mass spectrum. Again, the peak at m/z 366 is detected due to the adduct ions formed between the ions of m/z 348 and H2O in the ion trap. The fragmentation patterns and ionic structures in this study are in good agreement with previous studies,48–50 which confirms the successful detection of uranyl nitrate in the extract samples of uranium ore.
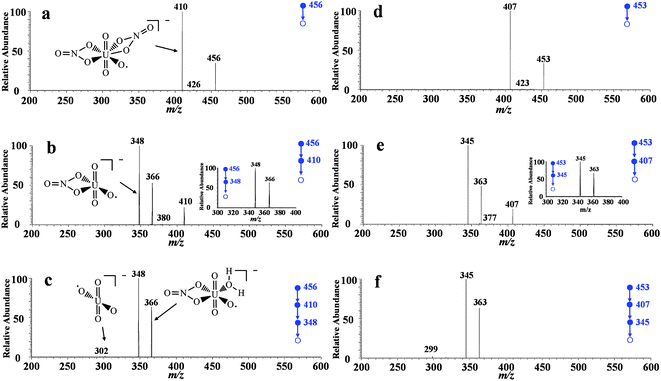 | ||
| Fig. 3 Tandem mass spectra of uranyl nitrate prepared from uranium ore samples. a) EESI-MS2 experiments targeting the peak at m/z 456; b) EESI-MS3 experiments targeting the peak at m/z 410, and the inset shows the CID experiments on m/z 366; c) EESI-MS4 experiments targeting the peak at m/z 348; d) EESI-MS/MS experiments targeting the peak at m/z 453; e) EESI-MS3 experiments targeting the peak at m/z 407, and the inset shows the CID experiments on m/z 363; f) EESI-MS4 experiments targeting the peak at m/z 345. | ||
To exclude false positive signals and further confirm the fragmentation patterns, CID experiments with the same conditions as above (for 238U) were performed on the uranyl species but targeting those containing 235U. The precursor ions with 235U were m/z 3 less than the corresponding ions having 238U. The EESI-MSn (n = 2, 3, 4) spectra were shown in Fig. 3d, 3e, and 3f, respectively. The experimental data show that the fragmentation patterns are in good consistence for uranyl species containing 235U or 238U. For example, the precursor ions of m/z 407 produced the characteristic fragment ions such as ions of m/z 345, 363, and 377 in the EESI-MSn (n = 2, 3, 4) experiments, which are all 3 units less than the corresponding fragments derived from the 238U species. From another point of view the results herein confirmed the existence of uranium isotope 235U.
3.2 Determining the isotopic ratio (235U/238U)
The isotopic ratio between 235U and 238U is fixed in natural uranium samples. For example, the abundance of 238U and 235U is 99.2742% and 0.7204% in the ore, respectively; thus, the theoretical isotopic ratio of 235U/238U is 0.007257 in this study. This ratio also serves as a natural rule characterizing the technologies for mineral dressing and processing by monitoring the abundance ratio of 235U/238U.51 The determination of isotopic ratio by MS is gaining more and more interest, because the characteristic signals of isotopes obtained from tandem mass spectrometry can ensure high accuracy for the detection. In MS1 spectrum, false positive signals can be produced by matrices, which reduce the reliability of measurements. Multi-stage tandem mass spectrometry generates characteristic fragment ions, which can be employed to exclude false positives. As discussed above, the fragmentation patterns of uranyl species contained 235U and 238U are almost the same (Fig. 3), thus the characteristic signals produced under the same conditions from the two isotope species can be used to measure the abundance ratio of the corresponding species. Generally, signals have better specificity in higher stage tandem mass spectrometry, but the intensities of signals will be reduced as well. In this study, the highest stage CID experiments were EESI-MS4, because no sample preconcentration was performed prior to the sample analysis.At the corresponding tandem MS stage, the EESI-MS spectra recorded by directly infusing the acidified water samples into the EESI source were featured by the same mass spectral patterns shown in Fig. 3, although the total uranium contents varied from 0.69 μg L−1 to 1.2 μg/L. The isotope ratios of 235U and 238U of the uranium ore samples were calculated using the abundances of the characteristic fragment ions obtained in the MS4 spectra. The analytical results are summarized in Table 1. Note that the river water samples 1 and 2 were obtained from a uranium mining area, where the total uranium content was slightly higher than that in natural ground water samples. The isotope ratios of 235U and 238U might change as that indicated by the values found in the EESI-MS measurements, however, this remains valid using other advanced techniques. The RSD values were relatively large, because the total uranium amounts were low in all the water samples. These results show that the isotope ratios of 235U and 238U in the natural water samples can be easily measured by the introduction of the natural water samples containing sufficient nitric acid.
| Sample | Concentration (μg L−1)a | Measured values (m/z 363/366) | Theoretical values (235U/238U) | Relative error (%)b | RSD (n = 7) |
|---|---|---|---|---|---|
| a Values detected by a photometer, ref: ZHANG Xing-lei, HUA Rong, HUAN Yanfu, LUO Mingbiao, ZHANG Xie, CHEN Huanwen, Uranium Geology, 2009, 25(9):312–315. b Relative error (%) = |C − CT| × 100%/CT, C is the (235U/238U) ratio measured by EESI-MS and CT is the theoretical value of (235U/238U) ratio. | |||||
| Lake water | 0.69 | 0.007224 | 0.007257 | −0.45 | 4.35 |
| River water 1 | 0.89 | 0.006353 | 0.007257 | 1.3 | 6.12 |
| River water 2 | 1.2 | 0.006779 | 0.007257 | 1.5 | 5.36 |
Similarly, the ore sample solutions were also directly analyzed by EESI-MSn without further sample treatment. As calculated by using the abundances of the characteristic fragment ions obtained in multi-stage tandem mass spectrometry, the isotopic ratio of 235U/238U for the #1 sample obtained in the EESI-MSn (n = 1–4) experiments was 0.7401, 0.7369, 0.7357, and 0.7272, respectively. Accordingly; the 235U/238U ratio obtained in EESI-MSn (n = 1–4) experiments for #2 sample was 0.7409, 0.7375, 0.7362 and 0.7275, respectively. These data shows that as n increases the measured isotopic ratio values approach the theoretical value (0.7257). Table 2 shows the results of measuring uranium isotopic ratios (235U/238U) using the characteristic fragment ions detected in EESI-MS4. With seven measurements, the RSD is about 1.54–1.81%, and the relative error is 0.21–0.25%. The slight positive bias observed was probably a result of the impurity of the signals obtained in the MS4 experiments, because the analyte was not purified before it was analyzed by the EESI-MS experiments. This bias can be theoretically reduced by increasing the stage of tandem MS experiment, however, at the current stage, the values obtained are in agreement with the data previously obtained from China land samples.52
| Samples | Measured values (m/z 363/366) | Theoretical values (235U/238U) | RSD (%) (n = 7) | Relative errors (%)a |
|---|---|---|---|---|
| a Relative error (%) = |C − CT| × 100%/CT, C is the (235U/238U) ratio measured by EESI-MS and CT is the corresponding theoretical value of (235U/238U) ratio. | ||||
| 1 | 0.007272 | 0.007257 | 1.54 | 0.21 |
| 2 | 0.007275 | 0.007257 | 1.81 | 0.25 |
In order to validate the EESI-MS method for 235U and 238U isotope ratio measurement, extra experiments were performed using both ICP-MS and EESI-MS for another set of 5 soil samples containing a wide variety of uranium. As shown in Table 3, for all the samples tested, EESI-MS provided analytical results comparable to those obtained by ICP-MS for measuring the 235U and 238U isotope ratios, no matter if the uranium content in the soil sample was at high levels (∼3.1 ppm) or trace concentrations (∼2.6 ppb). For example, the EESI-MS method provided RSD values ranged between 1.25–3.26%, which were considerably comparable to those (0.71–1.46%) obtained by ICP-MS. The RSD obtained by EESI-MS was notably larger than those of ICP-MS, probably because the EESI-MS was not a commercialized instrument, which produced more serious uncertainty than the commercial ICP-MS instrument. In comparison with ICP-MS, the relative error values obtained for all the 5 samples were ranged between −4.5–2.8%, which were also acceptable for most applications. These findings confirm that the EESI-MS method established here provides useful analytical results, which are comparable to those obtained by ICP-MS, for direct 235U and 238U isotope ratio measurement.
| Samples | Concentration of uranium (μg L−1)a | Measured value (235U/238U) | Relative error (%)c | |||
|---|---|---|---|---|---|---|
| ICP-MS | EESI-MS | |||||
| 235U/238U | RSD % (n = 7) | (m/z 363/366) | RSD % (n = 7)b | |||
| a The concentration of uranium measured by ICP-MS. b The RSD measured by EESI-MS. c Relative error (%) = (CEESI − CICP) × 100%/CICP, CEESI is the (235U/238U) ratio measured by EESI-MS and CICP is corresponding (235U/238U) ratio measured by ICP-MS in an international atomic energy agency lab in China. | ||||||
| #1 | 2.634 | 0.006838 | 1.33 | 0.006829 | 3.26 | −0.14 |
| #2 | 2708 | 0.007531 | 1.46 | 0.007745 | 1.84 | 2.8 |
| #3 | 16.44 | 0.003631 | 1.10 | 0.003701 | 1.46 | 1.9 |
| #4 | 3024 | 0.006939 | 1.43 | 0.00663 | 1.25 | −4.5 |
| #5 | 7.366 | 0.004343 | 0.81 | 0.004323 | 1.89 | −0.46 |
| Standard | 10.00 | 0.007155 | 0.71 | 0.007054 | 2.44 | −1.4 |
3.3 Reproducibility and accuracy
A syringe pump was used to continuously infuse the uranium samples for real-time EESI-MS4 analysis. As a result, no large variation was seen in the selected ion current chromatography (Fig. 4). Also, the MS spectra pattern maintained the same during the analysis (5 min). In addition, Table 1–3 show that the higher stage (n = 1–4) tandem mass spectrometry has better reproducibility and improved accuracy, probably because multi-stage tandem mass spectrometry can minimize disturbance from false positive signals. Our results demonstrated that EESI-MS4 has acceptable reproducibility and stability, which are suitable for the real-time monitoring of radioactive species.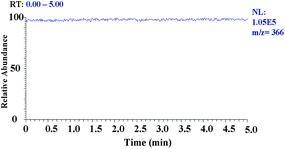 | ||
| Fig. 4 Selected ion chromatogram of fragment ion signal (m/z 366). | ||
4. Conclusions
The detection and analysis of radioactive species plays an important role in different areas including public safety, environmental science, nuclear geo-science, nuclear plant management and maintenance, etc.Radionuclide forms various complexes in the natural environment and chemical industry, and thus sensitive and reliable methods are required to detect them both qualitatively and quantitatively. This study showed that EESI-MS4 can detect uranium isotopes in water, soils and uranium ores with minimal sample pretreatment. The method has advantages of high reproducibility, fast analysis speed, acceptable accuracy and only requires a widely available ion-trap MS instrument. The short measurement time (5 min for each sample) is very attractive to laboratories for analysis of large numbers of samples. In addition, the approach established in this study can easily be adapted into nanoEESI43,45 for in situ analysis using miniature mass spectrometers, which makes this method very promising for the analysis of uranium samples in the field. If the sample can be introduced by means such as neutral desorption sampling,32,53 the technique reported here can be used in extreme conditions (e.g., highly radioactive environment) for remote online analysis.Acknowledgements
This work was jointly supported by the Innovation Method Fund of China (2008IM040400), National Natural Science Foundation of China (No. 21005024) and another grant from HIT (IMJQ10070004).References
- J. K. Gibson and J. Marcalo, Coord. Chem. Rev., 2006, 250, 776–783 CrossRef CAS.
- W. C. Woodmansee, Uranium, U.S. Dept. of the Interior, Bureau of Mines, 1976 Search PubMed.
- G. S. Groenewold, A. K. Gianotto, K. C. Cossel, M. J. Van Stipdonk, D. T. Moore, N. Polfer, J. Oomens, W. A. de Jong and L. Visscher, J. Am. Chem. Soc., 2006, 128, 4802–4813 CrossRef.
- S. Handley-Sidhu, M. J. Keith-Roach, J. R. Lloyd and D. J. Vaughan, Sci. Total Environ., 2010, 408, 5690–5700 CrossRef CAS.
- B. K. Sovacool, J. Contemporary Asia, 2010, 40, 369–400 Search PubMed.
- M. A. Temnikov, Sov. At. Energy, 1980, 48, 153–158 Search PubMed.
- E. Ansoborlo, O. Prat, P. Moisy, C. Den Auwer, P. Guilbaud, M. Carriere, B. Gouget, J. Duffield, D. Doizi, T. Vercouter, C. Moulin and V. Moulin, Biochimie, 2006, 88, 1605–1618 CrossRef CAS.
- J. D. Van Horn and H. Huang, Coord. Chem. Rev., 2006, 250, 765–775 CrossRef CAS.
- Z. Karpas, A. Lorber, E. Elish, R. Kol, Y. Roiz, R. Marko, E. Katorza, L. Halicz, J. Riondato, F. Vanhaecke and L. Moens, Health Phys., 1998, 74, 337–345 CAS.
- A. Earnshaw and N. Greenwood, Chemistry of the Elements, Butterworth-Heinemann, 1997 Search PubMed.
- D. G. Brookins, Geochemical Aspects of Radioactive Waste Disposal, Springer, 1984 Search PubMed.
- G. Nocton, P. Horeglad, V. Vetere, J. Pecaut, L. Dubois, P. Maldivi, N. M. Edelstein and M. Mazzanti, J. Am. Chem. Soc., 2010, 132, 495–508 CrossRef.
- J. S. Gamare, K. V. Chetty, S. K. Mukerjee and S. Kannan, Anal. Sci., 2009, 25, 1167–1170 CrossRef CAS.
- M. H. Khan, S. Shahida and A. Ali, Radiochim. Acta, 2008, 96, 35–40 CrossRef CAS.
- M. Nakahara and Y. Sano, Radiochim. Acta, 2009, 97, 727–731 CrossRef CAS.
- C. V. Karekar, K. Chander, G. M. Nair and P. R. Natarajan, J. Radioanal. Nucl. Chem., 1986, 107, 297–305 CAS.
- Y. Kato and M. Takahashi, Bunseki Kagaku, 1976, 25, 841–846 CrossRef CAS.
- J. Slanina, F. Bakker, A. J. P. Groen and W. A. Lingerak, Fresenius' Z. Anal. Chem., 1978, 289, 102–105 CrossRef CAS.
- U. Rohr, L. Meckel and H. M. Ortner, Fresenius' J. Anal. Chem., 1994, 349, 412–423 CAS.
- S. K. Das, A. V. Kulkarni and R. G. Dhaneshwar, Analyst, 1993, 118, 1153–1155 RSC.
- R. L. Deutscher and A. W. Mann, Analyst, 1977, 102, 929–933 RSC.
- D. Nibou and S. Lebaili, Quim. Anal., 1997, 16, 147–152 Search PubMed.
- S. D'Ilio, N. Violante, O. Senofonte, C. Majorani and F. Petrucci, Anal. Methods, 2010, 2, 1184–1190 RSC.
- Z. Varga, R. Katona, Z. Stefanka, M. Wallenius, K. Mayer and A. Nicholl, Talanta, 2010, 80, 1744–1749 CrossRef CAS.
- Z. Varga, M. Wallenius and K. Mayer, J. Anal. At. Spectrom., 2010, 25, 1958–1962 RSC.
- S. Li and S. Hou, J. Nucl. Radiochem., 1992, 14, 126–129 CAS.
- S. McSheehy and M. Sperling, Spectroscopy, 2009, 24, 14 Search PubMed.
- B. Kuczewski, C. M. Marquardt, A. Seibert, H. Geckeis, J. V. Kratz and N. Trautmann, Anal. Chem., 2003, 75, 6769–6774 CrossRef CAS.
- Z. Takats, J. M. Wiseman, B. Gologan and R. G. Cooks, Science, 2004, 306, 471–473 CrossRef CAS.
- A. Venter and R. G. Cooks, Anal. Chem., 2007, 79, 6398–6403 CrossRef CAS.
- H. W. Chen, N. N. Talaty, Z. Takats and R. G. Cooks, Anal. Chem., 2005, 77, 6915–6927 CrossRef CAS.
- H. W. Gu, S. P. Yang, J. Q. Li, B. Hu, H. W. Chen, L. L. Zhang and Q. Fei, Analyst, 2010, 135, 779–788 RSC.
- H. W. Chen, A. Venter and R. G. Cooks, Chem. Commun., 2006, 2042–2044 RSC.
- H. W. Chen, A. Wortmann, W. H. Zhang and R. Zenobi, Angew. Chem., Int. Ed., 2007, 46, 580–583 CrossRef CAS.
- H. W. Chen, J. H. Lai, Y. F. Zhou, Y. F. Huan, J. Q. Li, X. Zhang, Z. C. Wang and M. B. Luo, Chin. J. Anal. Chem., 2007, 35, 1233–1240 CAS.
- S. P. Yang, B. Hu, J. Q. Li, J. Han, X. Zhang, H. W. Chen, Q. Liu, Q. J. Liu and J. Zheng, Chin. J. Anal. Chem., 2009, 37, 691–694 CAS.
- S. P. Yang, J. H. Ding, J. Zheng, B. Hu, J. Q. Li, H. W. Chen, Z. Q. Zhou and X. L. Qiao, Anal. Chem., 2009, 81, 2426–2436 CrossRef CAS.
- F. J. R. Varela and O. Savadogo, J. Electrochem. Soc., 2005, 152, A1755–a1762 CrossRef CAS.
- W. Schwack and G. Morlock, Dtsch. Lebensm.-Rundsch., 2010, 106, 77–81 Search PubMed.
- Y. Zhang, X. X. Ma, S. C. Zhang, C. D. Yang, Z. Ouyang and X. R. Zhang, Analyst, 2009, 134, 176–181 RSC.
- J. D. Harper, N. A. Charipar, C. C. Mulligan, X. R. Zhang, R. G. Cooks and Z. Ouyang, Anal. Chem., 2008, 80, 9097–9104 CrossRef CAS.
- H. Chen, B. Hu and X. Zhang, Chin. J. Anal. Chem., 2010, 38, 1069–1088 CAS.
- H. W. Gu, B. Hu, J. Q. Li, S. P. Yang, J. Han and H. W. Chen, Analyst, 2010, 135, 1259–1267 RSC.
- H. W. Chen, S. P. Yang, M. Li, B. Hu, J. Q. Li and J. Wang, Angew. Chem. Int. Ed., 2010, 49, 2358–2361 CAS.
- B. Hu, X. J. Peng, S. P. Yang, H. W. Gu, H. W. Chen, Y. F. Huan, T. T. Zhang and X. L. Qiao, J. Am. Soc. Mass Spectrom., 2010, 21, 290–293 CrossRef CAS.
- M. B. Luo, B. Hu, X. Zhang, D. F. Peng, H. W. Chen, L. L. Zhang and Y. F. Huan, Anal. Chem., 2010, 82, 282–289 CrossRef CAS.
- J. Song, Chemical Analysis of Uranium Ores, Atomic Energy Press, Beijing. 2006 Search PubMed.
- B. Hu, H. W. Chen, X. Zhang, S. P. Yang and S. H. Feng, Acta Chim. Sin., 2009, 67, 1331–1335 Search PubMed.
- M. Sokalska, M. Prussakowska, M. Hoffmann, B. Gierczyk and R. Franski, J. Am. Soc. Mass Spectrom., 2010, 21, 1789–1794 CrossRef CAS.
- G. S. Groenewold, J. Oomens, W. A. de Jong, G. L. Gresham, M. E. McIlwain and M. J. Van Stipdonk, Phys. Chem. Chem. Phys., 2008, 10, 1192–1202 RSC.
- H. G. Wood, Science and Global Security, 2008, 16, 26–36 Search PubMed.
- Y. Liu and S. Fu, Chin. Sci. Bull., 1997, 42, 814–817 CrossRef CAS.
- H. W. Chen and R. Zenobi, Nat. Protoc., 2008, 3, 1467–1475 Search PubMed.
Footnotes |
| † This article was presented at the 2010 Asia Pacific Winter Conference on Plasma Spectrochemistry, Chengdu, China, November 26–30, 2010. |
| ‡ These authors contributed equally to this manuscript. |
| This journal is © The Royal Society of Chemistry 2011 |