Breast on-a-chip: mimicry of the channeling system of the breast for development of theranostics†‡§
Meggie M.G.
Grafton¶
c,
Lei
Wang¶
a,
Pierre-Alexandre
Vidi¶
a,
James
Leary
*abcd and
Sophie A.
Lelièvre
*ab
aDepartment of Basic Medical Sciences, Purdue University, West Lafayette, IN 47907-2026, USA
bPurdue Center for Cancer Research, Purdue University, West Lafayette, IN 47907-2026, USA
cWeldon School of Biomedical Engineering, Purdue University, West Lafayette, IN 47907-2026, USA
dBirck Nanotechnology Center, Purdue University, Discovery Park, West Lafayette, IN 47907-2026, USA
First published on 14th January 2011
Abstract
Improved detection and therapy of breast neoplasia might benefit from nanodevices traveling inside mammary ducts. However, the decreasing size of branched mammary ducts prevents access to remote areas of the ductal system using a pressure-driven fluid-based approach. Magnetic field guidance of superparamagnetic submicron particles (SMPs) in a stationary fluid might provide a possible alternative but it is critical to first reproduce the breast ductal system to assess the use of such devices for future therapeutic & diagnostic (“theranostic”) purposes. Here we describe the engineering of a portion of a breast ductal system using polydimethylsiloxane (PDMS) microfluidic channels with a total volume of 0.09 μl. A magnet was used to move superparamagnetic/fluorescent SMPs through a static fluid inside the microchannels. Non-neoplastic mammary epithelial S1 cells developed basoapical polarity as a flat monolayer on the PDMS surface when cultured in the presence of laminin 111, and incubation with SMPs did not result in detectable toxicity. Cells could not withstand the fluid pressure if microinjected directly in completed channels. Whereas, they readily covered laminin 111-coated PDMS surfaces when cultured in U-shaped “hemichannels” before completing the channels. This breast-on-chip model represents a critical step towards the mimicry of the tree-like ductal system of the breast for further testing and targeting of SMPs.
Insight boxWe report the development of a new cell culture model that mimics a portion of the breast ductal system. This system will bring the possibility of studying tumor nodules inside ducts of given sizes and testing submicron- and nanoparticles for the detection and treatment of tumor cells using an intraductal approach. Innovation lies in the use of molding technology to produce branched channels of decreasing lumen size and identifying conditions that permit non-neoplastic mammary cell monolayer expansion and differentiation into a basoapically polarized epithelium on polydimethylsiloxane. This model will enable researchers to assess and direct the movement of particles to specific target cells. The production of this breast ductal system on-chip integrates expertise from biologists, engineers and biophysicists. |
Introduction
The human mammary gland is an assembly of several branched ductal systems with channels of decreasing size.1–3 All luminal epithelial cells are thought to be in contact with the central lumen of the ducts, thus early alterations occurring at the apical side of cells (i.e., against the lumen) and small tumors might already be detectable if there is intraductal access. An approach referred to as ductal lavage has been used to collect loose cells from mammary channels with the goal of detecting cancer cells early.4 Although ductal lavage and even nipple aspirates can be successfully used to identify abnormal cells,5 these techniques are likely limited to restricted areas of the ductal system. Indeed, the decreasing size of mammary channels provides too strong a resistance to the fluid movement necessary to collect the content of small distant channels. In addition ductal lavage is associated with discomfort and pain, and risk of perforation.6 Mammary ductoscopy has been implemented as a breast cancer detection method and for assistance in surgery but improvement is still necessary to make it practical.5,7 Fluid pressure would also prevent local therapies with intraductal antineoplastic drugs if original tumor sites were beyond reach.One possible way to access all preneoplastic and tumor sites in the breast might be to use guided superparamagnetic submicron particles (SMP). The fluid can be left static if SMPs are moved through the breast ducts under magnetic fields of appropriate shapes and strengths. Iron-oxide SMPs would provide biocompatible and nontoxic paramagnetic material (i.e., SMPs only acquire magnetic properties in the presence of a magnet);8 these SMPs could also serve as both X-ray and MRI contrast agents. Moreover, fluorescently-labeled magnetic particles could serve to highlight ductal breast cancer cells for subsequent fluorescence-guided surgery. The SMPs can be engineered to target tumor cells and either, carry chemotherapeutic drugs, or trigger heat-induced cell death upon exposure to alternating magnetic field energy that can be readily transduced into the iron-oxide.
In order to set up meaningful experiments in ductal channels, to test targeting and toxicity of SMPs, it is critical to develop a cell culture system that mimics branched mammary channels. Ways to reproduce the mammary glandular system have been increasingly explored to provide an environment amenable to the better understanding of differentiation and tumor development. Murine cells have been successfully used in the past to recapitulate branching morphogenesis in the presence of epimorphine and growth factors.9 However, this system does not permit the control of the number and size of ducts. Non-neoplastic human mammary epithelial cells have been used repeatedly to produce differentiated glandular units or acini, with cells basoapically polarized, thus mimicking the organization of the smallest mammary glandular structures but they usually fail to develop branched channels using classical 3D culture systems in the presence of basement membrane components.10,11 Recent development in 3D culture design led to the formation of branched ducts from non-neoplastic breast epithelial MCF10A cells on a collagen I basis and/or silk protein scaffold and co-cultures with other cell types present in the breast.12,13 However, like for branching obtained with murine cells, the size, number, and conformation of the branched ductal structures cannot be controlled and this system would be difficult to use for injection of SMPs. Moreover, MCF10A cells seldom make apically polarized epithelia due to the lack of tight junction formation at the apical side of cells.11,14,15
A mammary ductal system starts with a smaller diameter at the orifice, at the nipple, compared to the diameter measured a few millimeters inside the breast where it reaches 0.7 mm on average; then the diameter of the lumena decreases in size as branches form toward the terminal ductal lobular units (TDLUs).3 In order to engineer a controlled ductal system with decreasing diameters, the optimal way might be to culture cells as a monolayer on preformed channels. Elastomeric Polydimethylsiloxane (PDMS) has been utilized to make patterning structures by soft lithography and can be coated with extracellular matrix (ECM) components to allow the culture of different cell types.16 This substratum is most commonly used for microfluidic cell culture and its optical transparency and low autofluorescence properties permit high-resolution imaging through the material. Therefore PDMS appears like an optimal substratum to design branched ductal structures.
Here we have engineered a simple ductal system with branched channels of decreasing size using PDMS as the molded material. SMPs can be directed to move towards the smaller ducts and pulled out of the channels. With proper ECM coating, non-neoplastic HMT-3522 S1 mammary epithelial cells display basoapical polarity on PDMS and can be used to cover the channels.
Results
ECM-coated hydrophobic PDMS material is amenable to the phenotypically normal differentiation of breast epithelial cells
One of the challenges of mimicking the phenotypically normal organization of the breast epithelium is to reproduce the backbone of differentiation, the basoapical polarity axis.11 Another challenge is to apply microscale cell culture necessary to mimic the size of the breast ducts to effectively produce mammary epithelial differentiation in a serum-free medium. In this case the goal is not to produce the well-known spherical three-dimensional (3D) structures referred to as acini, but rather a basoapically polarized monolayer of non-neoplastic cells organized into a tube. PDMS can be used to engineer differently shaped structures on which cells can be cultured.16 However, it has not been applied yet to the culture of non-neoplastic mammary epithelial cells necessary to cover the walls of tiny channels.The first step of our approach was to assess which ECM-based substratum permitted the expansion of the non-neoplastic human mammary epithelial HMT-3522 S1 cells cultured as a monolayer on PDMS. Engelbreth-Holm-Swarm (EHS) sarcoma-extracted ECM material has been commonly used to culture mammary epithelial cells; therefore we coated PDMS with EHS-based Matrigel™ and let it dry overnight before plating the cells. Using dried Matrigel™ should avoid the cell round-up normally obtained with the gel form. Over a 10-day culture period, PDMS surface modification using dried 5% Matrigel™ did not consistently foster cell monolayer expansion while dried 10 and 20% Matrigel™ led to the development of multicellular structures growing in 3D (Fig. 1A). However, PDMS surface modification with dried laminin 111, collagen IV, or a combination of laminin 111 and collagen IV, permitted expansion of the monolayer of S1 cells to 90% confluence within the same culture period. Interestingly, a drip of laminin 111 mixed with cells at the time of plating also led to monolayer expansion, providing a less cumbersome preparation of the cell culture environment (Fig. 1A). Growing monolayers of non-neoplastic mammary S1 cells on laminin 111-coated PDMS was not accompanied with any significant toxicity as shown by Trypan blue exclusion (Fig. 1B).
 | ||
| Fig. 1 Recapitulation of basoapical polarity on ECM-coated PDMS. HMT-3522 S1 cells were cultured on glass or on PDMS coated with dried Matrigel™ (MG at 19.2 and 38.4 μg total proteins/cm2, corresponding to 5 and 10% of the stock solution, respectively), dried laminin 111 (L(dry coat), 5.2 μg proteins/cm2), or dripped with laminin 111 (L(drip) at a final concentration of 133 μg ml−1) for 10 days. (A) Bright field image of cultures of S1 cells. (B) Percentage of cell death measured with Trypan blue exclusion test. L = laminin, (Dunnett, p > 0.05, n = 3). (C)–(E) The distribution of apical polarity marker ZO-1 (red) and basal polarity marker α6-integrin (green) was analyzed by confocal microscopy in S1 cells cultured on PDMS coated with dried laminin-111 (C, D) or with collagen I as negative control for polarity (E). Nuclei were counterstained with DAPI (blue). Serial images from a z stack are shown in C; the direction of optical sectioning is indicated in the cartoon on top. (D) & (E), orthogonal (‘side’) views (top panels) and maximal intensity projections after reslicing in xz (bottom pannels); the orientation of the optical sections is indicated in the cartoon to the left. (F) Schematic representation of a polarized epithelial cell monolayer. Size bars, 50 μm (A) and 5 μm (C)–(E). | ||
Immunostaining for the hemidesmosome component, α6-integrin, and the tight-junction protein, ZO-1, was used to assess proper localization of these basal and apical polarity markers, respectively. Cells cultured on dried or dripped laminin 111, and a combination of dried laminin 111 and dried collagen IV formed a basoapically polarized layer; whereas no polarization was obtained with collagen I, used as negative control (Fig. 1C–E and Table 1). The gold standard for basoapical polarity of epithelial monolayers is to culture cells on filters.17 The cells' display of basoapical polarity when cultured on PDMS with appropriate ECM was similar to cultures on filters dripped with laminin 111 or with a combination of laminin 111 and collagen IV (Table 1; ESI Fig. 1S§). Therefore PDMS surface modification with dried or dripped laminin 111 appears to be the simplest effective option to obtain a monolayer of basoapically polarized mammary epithelial cells.
| PDMS | Filter | |||
|---|---|---|---|---|
| Drip | Dry | Drip | Dry | |
| n.d., not determined; 3D, formation of three-dimensional multicellular structures; B, presence of basal polarity (based on α6-integrin marker); A, presence of apical polarity (based on ZO-1 marker); b, a, absence of basal polarity or apical polarity, respectively. (* nonconsistent results depending on replicate). | ||||
| 5% Matrigel | n.d. | B/A* | n.d. | n.d. |
| 10% Matrigel | n.d. | 3D | n.d. | n.d. |
| 20% Matrigel | n.d. | 3D | n.d. | n.d. |
| Laminin 111 | B/A | B/A | B/A | n.d. |
| Laminin 111 + collagen IV | n.d. | B/A | B/A | n.d. |
| Collagen IV | n.d. | n.d. | B/a | n.d. |
| Collagen I | n.d. | b/a | n.d. | n.d. |
Non-neoplastic mammary epithelial cells cover channels made of dried laminin 111-coated PDMS
A breast ductal system encompasses branches of decreasing lumen size, starting with the largest channels of 700 μm in average diameter3 and ending at TDLUs with channels of 30 μm in average diameter. Soft lithography was used to design rectangle-shaped, PDMS branched channels of decreasing sizes from 50 × 120 μm, to 50 × 60 μm and 50 × 30 μm for height and width, respectively (see methods section) (Fig. 2A). Each channel of a particular width had a length of 5 mm and the whole ductal system could fit on a slide thus, enabling on-chip experiments. The tiny total surface of 7.1 mm2 and total volume of 0.09 mm3 of the on-chip ductal system may hamper the attachment and proliferation of non-neoplastic mammary epithelial cells. Therefore, two methods were compared for the culture of cells inside the channels. In the first method, cells were injected in closed channels using a metered injection system at 0.01 and 0.5 ml h−1, and the medium was exchanged by diffusion by immersing the channel system inside a cell culture medium (Fig. 2B). In the second method, branched hemichannels (U-shaped) were engineered as described in the methods section and cells were cultured on this channeled surface in the presence of laminin 111 (Fig. 2B). Then the channels could be completed at room temperature by adding a PDMS membrane. To correct for the decrease in cell concentration inside the hemichannels or inside the complete channels and hence, the inhibition of cell division that would subsequently occur, the concentration of cells was increased 10 fold compared to usual, to reach 230![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells/cm2.
000 cells/cm2.
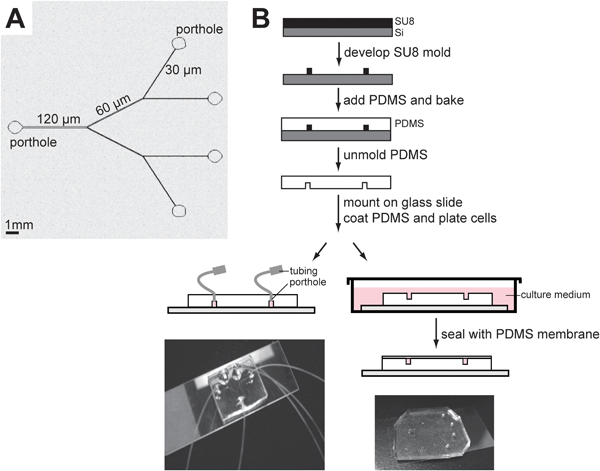 | ||
| Fig. 2 Engineering of PDMS channels on a chip. A microchannel system was molded in PDMS, coated with laminin 111, and used as substrate for the culture of HMT-3522 S1 cells. (A) Schematic of the branched channel system. (B) Two independent approaches were developed: In the first approach (left, drawing and picture of the system), PDMS microchannels were sealed onto a glass coverslip, coated with dried or dripped laminin 111, and used for the culture of S1 cells in a closed environment. Cells were injected through tubing connected to the portholes using a syringe pump and the medium was changed by immersion. In the second approach (right), cells were cultured in an open ‘hemichannel’ system (top side of microchannel left open). The channels can be completed using a PDMS membrane on the day of the experiment. | ||
In the first culture method, a laminin drip had to be used in the preformed channels since dried laminin was clogging some of the channels. Unfortunately, cells that were injected into the channels seemed unable to survive. In the second cell culture method, precoating the hemichannels with dried laminin 111 allowed cells to expand on the PDMS walls more effectively compared to laminin 111 dripped at the time of cell plating, without cells clogging the channel (Fig. 3A–D). Cells displayed basal and apical polarity, including those in the flat monolayer covering the side walls of the channels (Fig. 3E–F). Therefore, using such a small channel environment required the culture of cells on the hemichannels, before completing the ductal system with the PDMS membrane.
 | ||
| Fig. 3 Culture of mammary epithelial cells in branched PDMS hemichannels in the presence of laminin 111. HMT-3522 S1 cells were cultured in presence of laminin 111 (L) in complete microchannels or in hemichannels according to Fig. 2B. Laminin 111 was either coated and dried on PDMS (dry) or diluted in the H14 culture medium and dripped on the cell population at the time of plating (drip). (A) Bright field images. Individual round cells are indicated by arrowheads in a complete channel. Cell clumping in hemichannels with the drip method is indicated by the arrow. (B)–(D) Confocal analysis of DAPI-stained S1 cells in the terminal branch from a hemichannel coated with dried laminin 111. Maximal intensity projection of a z-stack taken at low magnification is shown together with tiled bright field micrographs of the hemichannel (B). Maximal intensity projection of a z-stack taken at high magnification (C) with orthogonal view at the level of the dotted line (D). (E) & (F) Confocal analysis of basal polarity marker α6 integrin (green) and apical polarity marker ZO-1 (red) in S1 monolayer located on a side wall of the hemichannel coated with dried laminin 111. Nuclei are counterstained in blue. Size bars, 20 μm (B) and 5 μm (C, E, F). | ||
Fluorescent superparamagnetic submicron particles can be used with the PDMS-based channeled system and are nontoxic to the mammary epithelium
One of the goals of the ductal system on a chip is to provide a model to study the use of SMPs for future detection and treatment applications. SMPs of 0.86 μm in average diameter were loaded into complete cell-free microfluidic channels to assess their movement in this constrained environment. These relatively large particles were chosen so that individual particles can be visualized amidst live cells using phase and fluorescence microscopy. Also, SMPs will not require huge magnetic fields to move them since, all else being equal, the force driving the particles within the channels will be proportional to the cube of the diameter of the magnetic material portion of the particle. Furthermore, it is important that these SMPs be “superparamagnetic” (and not simply “paramagnetic”) which means that they become magnetic only in the presence of a magnetic field. When the magnetic field is taken away the SMPs are free to disperse locally and, if targeted to surface markers on specific cells, bind to their targets. After injection, the SMPs showed Brownian movement, as expected. Upon application of the magnet, SMPs could be collected on one side of the channel and directed to enter specific channels of decreasing size (Fig. 4; ESI Movies 1–3§). | ||
| Fig. 4 Movement of fluorescent magnetic submicron particles within channels. Time lapse micrographs showing the movement of SMPs in a 50 × 30 μm (height × width) channel. A pulling force was exerted using a wedge shaped magnet. The arrowhead points to one particle followed through the images taken at 700 ms intervals. | ||
For toxicity assays, cells were cultured on PDMS coated with dried laminin 111 and incubated with different concentrations of SMPs. A preliminary assay suggested that the solution containing SMPs was nontoxic, as shown by Trypan blue exclusion, when using a volume that corresponded to that added if SMPs were 10 fold the concentration of cells. This result was confirmed in several rounds of experiments showing no significant toxicity when incubating SMPs for two days in their solvent at 1, 5 and 10 fold the cell concentration at time of incubation. Confocal microscopy revealed that SMPs did not seem to go inside the cells, and there was no detectable effect of SMPs on basoapical polarity when used as a 10 fold concentration compared to cells, as shown by immunostaining for α6-integrin and ZO-1 (Fig. 5A–D). SMPs were used as raw materials in these experiments and therefore stuck nonspecifically to the cells and parts of PDMS not covered by cells. This close encounter between SMPs and cells was necessary to ascertain that the SMPs per se (i.e., without addition of any specific targeting molecule) would not be affecting cells. Appropriate coating of SMPs with targeting molecules will be necessary to effectively control their travel to target cells upon application of a magnetic field in future experiments.
 | ||
| Fig. 5 Lack of toxicity of fluorescent magnetic submicron particles. SMPs were incubated for 48 h with S1 cells cultured on PDMS coated with dried laminin 111 at a concentration of 1, 5, or 10 particles per cell. (A) Fluorescent SMPs (green, top panel) and corresponding bright field images of cells (bottom panel). (B) Orthogonal view from a confocal z-stack showing cell nuclei (DAPI, blue), cell membranes (DiI, red) and fluorescent SMPs (green). The maximal intensity projection after reslicing in xz is shown at the bottom. (C) Percentage of cell death measured by Trypan blue staining after incubation of S1 cells with the SMPs. (Dunnett, p > 0.05, n = 3). (D) Orthogonal view of immunostaining for apical polarity marker ZO-1 (red) in S1 cell monolayer incubated with SMPs (10 particles per cell, green, arrow). Size bars, 50 μm (A), 5 μm (B and D). n.s., nonspecific. | ||
Discussion
We have shown that non-neoplastic mammary epithelial cells can cover laminin 111-coated hydrophobic PDMS and that the resulting epithelium displays basoapical polarity. This was a critical step in the design of the channel system as we have demonstrated previously that apical polarity is extremely labile and sensitive to culture conditions.11 Indeed, the polarity status of the epithelium might impact future studies with antibody-coated SMPs necessary for targeting to specific cell types using cell surface markers. Laminin 111 was found to be sufficient to foster the development of basoapical polarity, confirming long-time knowledge that this ECM molecule acts as a powerful inducer of breast epithelial differentiation under different cell culture conditions.18 Surprisingly, the EHS extract that contains more basement membrane ECM components than the sole laminin 111 could not provide consistent results. It is possible that the concentration of materials contained in this extract, some of which are not well determined, influences the spreading of cells and that it is more amenable for the formation of three-dimensional breast epithelial structures than flat monolayers of polarized cells.A critical aspect of the design of a breast ductal system is to develop a model that contains branched channels of decreasing size. Based on our review of sections of normal looking breast tissue, the lumen size at the level of TDLUs should be around 30 μm in average diameter.11 This is in the range of the channel size that was mimicked by the smallest branches of the ductal system built with PDMS if we take into account the thickness of mammary epithelial cells (∼10 μm) cultured on the channels' surface. Producing such small channels was accompanied with technical challenges. Indeed, it is not surprising that our cells did not survive when injected in complete channels, likely as a consequence of shear stress. In other reports using cell injection in microfluidic systems, the entrance channel was much wider and the feeding system used parallel conduits.19,20 Culturing cells in hemichannels (U-shaped) as successfully reported here, followed by the completion of the channel with a PDMS coverslip (the latter could be covered with cells), is an acceptable solution to deal with channels of very small diameter. Our results show that even in the smallest channels, the cells could effectively cover the hemichannel on all of its three surfaces without inducing intrachannel cell clogs (see Fig. 3). The absence of piling up of cells responsible for clogs in the hemichannels was observed with dried laminin 111; however, clogs were present when laminin 111 was dripped, although on PDMS coverslips both dried and dripped laminin 111 led to the development of a flat monolayer of cells. This might be due to the narrow indent used to create the hemichannel, which could entice cells to pile up more easily when surrounded by dripped laminin 111. Importantly, with hemichannels, it will be possible to seed tumor nodules within the ductal system at specific locations to study SMPs targeting.
In this model of a breast ductal system we did not include myoepithelial cells at the basal side of the luminal cells nor terminal ductal lobular units at the ends of the narrowest channels although these represent normal features of the mammary epithelium. Instead, we focused on the production of a basoapically polarized monolayer of luminal cells because this device is intended to mimic the luminal portion of the ductal breast system. One of our goals is to introduce tumor cells and nodules in the microchannels that mimic the ductal breast system, the development of which is reported here, to test SMPs for neoplasia detection and treatment purposes. SMPs were found to travel effectively under the power of a custom wedge shaped magnet. Critically, no cytotoxity or effect of the SMPs on the differentiation of the breast epithelium, as measured by the distribution of basoapical polarity markers, could be detected even after two days of incubation.
The proposed model of the mammary ductal tree will be an asset to the initial steps of the design of nanomedicine techniques. For instance, magnetic fields could be used to provide a force to move antibody-targeted fluorescent and large SMPs through static fluid channels covered by mammary epithelial cells. In the long-run the SMPs will be designed to be “theranostic” meaning that they can be used for both diagnostic and therapeutic purposes. The SMPs could serve as X-ray contrast agents that will enhance mammograms, providing greater contrast between tumors and fibrous breast tissue. In principle, if SMPs are targeted against specific surface markers of neoplastic cells in the ducts, free SMPs can be “washed” away, by reversing the direction of the magnetic field, to leave only SMPs specifically bound. The SMPs bound to neoplastic cells can also serve as Magnetic Resonance (MR) imaging contrast agents, possibly enabling higher resolution MRI to determine the exact 3D location, size, and volume of the tumor for subsequent surgery. Since the SMPs are also fluorescent, they could be used to guide surgeons for real-time fluorescence-guided surgery to more completely remove tumors that may not be visible under normal surgery. Additionally, the SMPs could also contain anticancer agents that could be delivered directly to the tumor, hence decreasing the patient's exposure to total-body chemotherapy. All of these are examples of applications of SMPs for the fight against breast cancer, and the breast ductal system that we have developed will be a critical stepping stone to the development of effective SMPs.
Methods
Cell culture
Non-neoplastic human mammary epithelial cells (HMT-3522 S121) were cultured at 37 °C in 5% CO2 in H14 medium consisting of DMEM/F12 (Sigma, St Louis, MO) with 250 ng ml−1insulin (Boehringer Mannheim, Indianapolis, IN), 10 μg ml−1 transferrin (Sigma), 2.6 ng ml−1sodium selenite (BD Biosciences, Bedford, MA), 10−10 M estradiol (Sigma), 1.4 μM hydrocortisone (BD Biosciences), 5 μg ml−1 luteotropic hormone (Sigma), and 10 ng ml−1epidermal growth factor (EGF; BD Biosciences) for 10 days with medium changed every two to three days.Laminin 111 and collagen IV (BD Biosciences, Discovery Labware) were used at a final concentration of 133 μg ml−1 and 20 μg ml−1, respectively, shown to induce polarity in S1 cells.11 When let to dry overnight, the laminin 111 coat corresponded to 5.2 μg proteins/cm2 of PDMS. Matrigel™ (BD Biosciences Discovery Labware) was used at a final concentration of 5, 10 or 20% (corresponding to 19.2, 38.4, or 76.8 μg total proteins/cm2, respectively). When dried, these ECM substrata were placed at 37 °C overnight before plating cells. When laminin 111 and collagen IV were combined, they were dried sequentially (starting with laminin 111) over two consecutive nights. For the drip method, the substrata were mixed at the desired final concentration in 50% of the final volume of the medium and dripped over the cells plated a few minutes before in 50% of the final volume of the medium.
Toxicity test
S1 cells were cultured directly on PDMS coverslips covered with different ECM substrata for 10 days to assess toxicity. For experiments involving SMPs, S1 cells were cultured for eight days with dried or dripped laminin 111 before incubating for 48 h with 1, 5 and 10 times as many SMPs (Catalog # ME03F, Bangs Laboratories, Fishers, IN) as S1 cells present on day 8, or with the SMP-free preserving solution used at a volume corresponding to 10, 5 or 1 times as many SMPs. The number of cells at day 8 was estimated by detaching and counting cells from one well of the 12-well plate used for the experiment. For the toxicity tests, after medium removal, cells were incubated with Trypan blue (Sigma) for five minutes and washed in PBS. Blue-stained dead cells were scored out of a total of 500 cells.Fluorescence immunostaining
S1 cells were incubated for 10 min in a permeabilization buffer [0.5% Triton X-100 in cytoskeleton buffer (100 mM NaCl, 300 mM sucrose, 10 mM pipes, pH 6.8, 5 mM MgCl2)] with protease and phosphatase inhibitors [1 mM Pefabloc (Roche Diagnostics, Indianapolis, IN), 10 μg ml−1aprotinin (Sigma), 250 μM NaF], before fixation in 4% paraformaldehyde (Sigma) and immunostaining as previously described.22 Primary antibodies were polyclonal rabbit anti-human ZO-1 (Invitrogen, Carlsbad, CA) and monoclonal rat anti-human α6-integrin CD49f (Millipore, Billerica, MA). Secondary antibodies were donkey anti-Rabbit IgG (H+L) and donkey anti-Rat IgG (H+L) (Jackson ImmunoResearch, West Grove, PA). Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI; Sigma) and specimens were mounted in ProLong® Gold antifade reagent (Invitrogen).PDMS handling and making of channels
Photoresist masks were made on 4′′ silicon wafers following standard photolithographic procedures. Briefly, SU-8 2025 (Microchem, Newton, MA) was spun to a 50 μm depth onto a cleaned 4′′ silicon wafer. The photoresist was exposed to the designed mask and then developed. Further procedural details were as described previously.23 The completed wafer was baked and treated with 97% trichlorosilane vapors for 30 min. PDMS elastomer and curing agent (Dow Corning Sylgard 184, Ellsworth Adhesives, Germantown, WI) were mixed in a 10:1 ratio and debubbled for 20 min. The PDMS was then poured over the silicon–photoresist mold and cured overnight at room temperature. Individual chips were cut from the mask and a conical 20 gauge needle was used to bore holes for tubing. A 10:1 ratio of PDMS to curing agent was also spread thinly on a 5′′ Petri dish and cured overnight at room temperature. The thin PDMS was cut to cover a large rectangular coverslip as the base of the chip. Both the PDMS coated coverslips and the PDMS chips were oxidized using a Corona treater. The PDMS surfaces were exposed to the localized oxygen plasma for approximately 30 s each. To make complete channels, each chip was pressed together with a coverslip and any bubbles were rubbed out. The bound chips were then baked at 70 °C for 30 min to stabilize the bond. Tygon tubing was inserted into the inlet and outlet ports and sealed with 10:1 ratio uncured PDMS.
SMP handling and coating
Fluorescent superparamagnetic microspheres (0.86 μm average diameter) were chosen to be large enough to be moveable with a magnetic field (the amount of ponderomotive force provided by the external magnetic field is directly proportional to the volume of the magnetic material in the SMP), reduce random Brownian movement, and permit observation with conventional fluorescence microscopy (i.e., diameter above the optical imaging resolution limit). The SMPs were injected into the PDMS channels using a 1 ml syringe and 30 gauge blunt tip needle. Once the SMPs were in the channels, flow was allowed to stop as no further pressure was applied to the syringe. A permanent magnet (Quadrant Magnetics, Louisville, KY) with NdFeB (“Neo 42”) material with a target poleface field of approximately 300 mT was used to drag the SMPs through the channels.Imaging
Bright field images of cell monolayers were taken with an Olympus IX70 epifluorescence microscope (Olympus, Center Valley, PA). Fluorescently stained samples were analyzed using a Zeiss (Oberkochen, Germany) LSM 710 confocal microscope system equipped with a 100×/1.4NA oil immersion objective. Images were captured using the Zen software (Zeiss) and processed with ImageJ (ttp://rsbweb.nih.gov/ij/index.html). Video recording of SMP movements was performed on an inverted fluorescence microscope (Nikon Diaphot) at 20× magnification using a Retiga EXi camera (QImaging, Surrey, BC, Canada). The Image-Pro Plus software was used to capture images every 50 ms.Statistical analyses
Data are presented as means ± SEM and statistical comparisons were performed using GraphPad Prism 3.0 software (GraphPad Software Inc, San Diego, CA). Nonpaired t-test was used for comparison of two groups whereas one-way ANOVA with Dunnett post-hoc test was employed for comparisons between three or more groups of samples. A p < 0.05 was considered significant.Conclusion
This first attempt to reproduce the mammary ductal system in vitro demonstrates the importance of combining engineering and biology expertise in order to achieve appropriate settings for epithelial differentiation in a defined environment, i.e., here the tiny branched channels. Our ultimate goal is to use this breast-on-chip system to coculture phenotypically normal and diseased cells and tumor nodules, and assess the targeting of SMPs to specific cells while these particles migrate within ducts that mimic the luminal breast environment.Acknowledgements
This work is supported by a Congressionally-Directed Medical Research Program grant # W81XWH-09-1-0354 to SAL and JL, and a Novartis Foundation Fellowship to PAV.References
- B. A. P. Cooper, in On the Anatomy of the Breast, ed. Longman, Orme, Green and Brown, Longmans, London, 1840 Search PubMed.
- S. M. Love and S. H. Barsky, Anatomy of the nipple and breast ducts revisited, Cancer, 2004, 101, 1947–1957 CrossRef.
- J. E. Rusby, E. F. Brachtel, J. S. Michaelson, F. C. Koerner and B. L. Smith, Breast duct anatomy in the human nipple: three-dimensional patterns and clinical implications, Breast Cancer Res. Treat., 2007, 106, 171–179 CrossRef.
- E. Evron, W. C. Dooley, C. B. Umbricht, D. Rosenthal, N. Sacchi, E. Gabrielson, A. B. Soito, D. T. Hung, B. Ljung, N. E. Davidson and S. Sukumar, Detection of breast cancer cells in ductal lavage fluid by methylation-specific PCR, Lancet, 2001, 357, 1335–1336 CrossRef CAS.
- I. Locke, G. Mitchell and R. Eeles, Ductal approaches to assessment and management of women at high risk for developing breast cancer, Breast Cancer Res., 2004, 6, 75–81 CrossRef.
- J. Tondre, M. Nejad, A. Casano, D. Mills and S. Love, Technical enhancements to breast ductal lavage, Ann. Surg. Oncol., 2008, 15, 2734–2738 Search PubMed.
- K. Uchida, H. Fukushima, Y. Toriumi, K. Kawase, I. Tabei, A. Yamashita and H. Nogi, Mammary ductoscopy: current issues and perspectives, Breast Cancer, 2009, 16(2), 93–6 Search PubMed.
- T. K. Jain, M. K. Reddy, M. A. Morales, D. L. Leslie-Pelecky and V. Labhasetwar, Biodistribution, Clearance, and Biocompatibility of Iron Oxide Magnetic Nanoparticles in Rats, Mol. Pharmaceutics, 2008, 5, 316–327 CrossRef CAS.
- Y. Hirai, A. Lochter, S. Galosy, S. Koshida, S. Niwa and M. J. Bissell, Epimorphin functions as a key morphoregulator for mammary epithelial cells, J. Cell Biol., 1998, 140, 159–169 CrossRef CAS.
- S. A. Lelièvre and M. J. Bissell, Three dimensional cell culture: The importance of context in regulation of function, in Encyclopedia of Molecular Cell Biology and Molecular Medicine (EMCBMM), ed. R. A. Meyers, Weinheim: Wiley-VCH, 2nd edn, 2005, vol. 14, pp. 383–420 Search PubMed.
- C. Plachot, L. S. Chaboub, H. A. Adissu, L. Wang, A. Urazaev, J. Sturgis, E. K. Asem and S. A. Lelièvre, Factors necessary to produce basoapical polarity in human glandular epithelium formed in conventional and high throughput three-dimensional cell culture: Example of the breast epithelium, BMC Biol., 2009, 7, 77 CrossRef.
- E. Dhimolea, M. V. Maffini, A. M. Soto and C. Sonnenschein, The role of collagen reorganization on mammary epithelial morphogenesis in a 3D culture model, Biomaterials, 2010, 31, 3622–3630 CrossRef CAS.
- X. Wang, L. Sun, M. V. Maffini, A. Soto, C. Sonnenschein and D. L. Kaplan, A complex 3D human tissue culture system based on mammary stromal cells and silk scaffolds for modeling breast morphogenesis and function, Biomaterials, 2010, 31, 3920–3929 CrossRef CAS.
- V. C. Fogg, C. J. Liu and B. Margolis, Multiple regions of Crumbs3 are required for tight junction formation in MCF10A cells, J. Cell Sci., 2005, 118, 2859–2869 CrossRef CAS.
- J. M. Underwood, K. M. Imbalzano, V. M. Weaver, A. H. Fischer, A. N. Imbalzano and J. A. Nickerson, The ultrastructure of MCF-10A acini, J. Cell. Physiol., 2006, 208, 141–148 CrossRef CAS.
- L. Kim, Y. C. Toh, J. Voldman and H. Yu, A practical guide to microfluidic perfusion culture of adherent mammalian cells, Lab Chip, 2007, 7, 681–694 RSC.
- S. Fuller, C. H. von Bonsdorff and K. Simons, Vesicular stomatitis virus infects and matures only through the basolateral surface of the polarized epithelial cell line, MDCK, Cell, 1984, 38, 65–77 CrossRef CAS.
- T. Gudjonsson, L. Ronnov-Jessen, R. Villadsen, M. J. Bissell and O. W. Petersen, To create the correct microenvironment: three-dimensional heterotypic collagen assays for human breast epithelial morphogenesis and neoplasia, Methods, 2003, 30, 247–255 CrossRef CAS.
- L. Kim, M. D. Vahey, H. Y. Lee and J. Voldman, Microfluidic arrays for logarithmically perfused embryonic stem cell Culture, Lab Chip, 2006, 6, 394–406 RSC.
- Y. C. Toh, C. Zhang, J. Zhang, Y. M. Khong, S. Chang, V. D. Samper, D. van Noort, D. W. Hutmacher and H. Yu, A novel 3D mammalian cell perfusion-culture system in microfluidic Channels, Lab Chip, 2007, 7, 302–309 RSC.
- P. Briand, O. W. Petersen and B. Van Deurs, A new diploid nontumorigenic human breast epithelial cell line isolated and propagated in chemically defined medium, In Vitro Cell. Dev. Biol., 1987, 23, 181–188 CrossRef CAS.
- S. A. Lelièvre, V. M. Weaver, J. A. Nickerson, C. A. Larabell, A. Bhaumik, O. W. Petersen and M. J. Bissell, Tissue phenotype depends on reciprocal interactions between the extracellular matrix and the structural organization of the nucleus, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 14711–14716 CrossRef CAS.
- D. C. Duffy, J. C. McDonald, O. J. A. Schueller and G. M. Whitesides, Rapid Prototyping of Microfluidic Systems in Poly(dimethylsiloxane), Anal. Chem., 1998, 70, 4974–4984 CrossRef CAS.
Footnotes |
| † Published as part of an Integrative Biology themed issue in honour of Mina J. Bissell: Guest Editor Mary Helen Barcellos-Hoff. |
| ‡ This article is dedicated to Mina J. Bissell for her visionary influence on multidisciplinary research and the necessary development of meaningful cell culture systems. |
| § Electronic supplementary information (ESI) available. See DOI: 10.1039/c0ib00132e |
| ¶ The authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2011 |