Multi-scale modelling and simulation in systems biology†
Joseph O.
Dada
ab and
Pedro
Mendes
abc
aManchester Centre for Integrative Systems Biology, MIB, UK
bSchool of Computer Science, Oxford Road, The University of Manchester, UK
cVirginia Bioinformatics Institute, Blacksburg, VA
First published on 6th January 2011
Abstract
The aim of systems biology is to describe and understand biology at a global scale where biological functions are recognised as a result of complex mechanisms that happen at several scales, from the molecular to the ecosystem. Modelling and simulation are computational tools that are invaluable for description, prediction and understanding these mechanisms in a quantitative and integrative way. Therefore the study of biological functions is greatly aided by multi-scale methods that enable the coupling and simulation of models spanning several spatial and temporal scales. Various methods have been developed for solving multi-scale problems in many scientific disciplines, and are applicable to continuum based modelling techniques, in which the relationship between system properties is expressed with continuous mathematical equations or discrete modelling techniques that are based on individual units to model the heterogeneous microscopic elements such as individuals or cells. In this review, we survey these multi-scale methods and explore their application in systems biology.
 Joseph O. Dada | Joseph Olufemi Dada is a member of IEEE. He received the BEng (First Class Honours) and MEng degrees in electrical engineering from University of Ilorin, Nigeria, and his PhD in computer and electrical engineering from University of Duisburg-Essen, Germany. He has taught at the University of Ilorin and Akure Federal University of Technology, Nigeria and was a Research Associate at the University of Duisburg-Essen, Germany before he joined the service of University of Manchester in 2005 as a Research Associate. His current research interests include application of web services in systems biology, multi-agent systems, multi-scale modelling and simulation of systems biology. |
 Pedro Mendes | Pedro Mendes is Professor of Computational Systems Biology at the University of Manchester and the Virginia Bioinformatics Institute, Virginia Tech. He is the Deputy Director of the Manchester Centre for Integrative Systems Biology and one of the PIs of the COPASI biochemical simulation software project. He has as a Biochemistry Major from the University of Lisbon, and obtained a PhD in 1994 from the University of Wales Aberystwyth. His broad research interests cover the entire range of systems biology including bottom-up and top-down strategies, computational methods, and specific biochemical pathways, such as iron metabolism, and oxidative stress. |
Insight, innovation, integrationThe understanding of biological system functions requires integration of processes interacting across a range of spatial and temporal scales. This requires modelling at different scales, model reduction and model integration. The task of simulating large scale integrated models that cut across these diverse scales is computationally very expensive unless the multi-scale nature of the biological problems is exploited in a systematic way. We highlight the importance of computational multi-scale methods in systems biology and the need for the development of modelling and simulation frameworks that would enable automated multi-scale biological models assembly for systems biology problems. It is only by combining multi-scale modelling strategies with advances in computational technology that the aims of systems biology can fully be achieved. |
1. Introduction
Biological systems are made up of many spatial and temporal scales, each rich and complex. Biology at each scale integrates information from strata above and below.1 There are different types of scale in biological systems. Southern and colleagues used the organisation of biological systems to classify the biological processes in them into a hierarchy of spatial scales and called this “levels of biological organisation”, which range from gene, to proteins, individual biological cells, tissues, organs, and up to the individual organism that interacts with environment.2 Associated with this spatially based organisation are the temporal scales of biological processes that range from microsecond (∼10−6 s) for molecular interactions to 80 years (∼109 s) for the average human life expectancy.3 These diverse scales coupled with intra- and inter-scale interactions make a biological system extremely complex (e.g.cell made up of millions of molecules, tissue made up of billions of cells, etc.).To understand the behaviour of a biological system, whether it is natural or engineered, requires models that integrate the various interactions that occur on these diverse spatial and temporal scales4 (Fig. 1). Successful physiological analysis requires an understanding of the functional interactions between the key components of cells, organs, and systems, as well as how these interactions change in disease states.5 The complexity of biological systems has made the use of mathematical and computational models to describe and analyse their behaviours and functions an active area of research in recent years. The use of models and experimental data to study how the intra- and inter-scale interactions give rise to their collective behaviours and how they form relationships with their environments is a central theme of systems biology research. While synthetic biology has been mostly focused on microorganisms, it is expected that this activity will expand to higher organisms with more complex hierarchical levels of organisation and then it will also require multi-scale modelling. To some extent it is perhaps already a current need, since even microbial cultures can display this type of multi-scale organisation, such as in biofilms.
 | ||
| Fig. 1 Spatial and temporal scales in biology. | ||
In order to interpret the complexity of biological systems, two major strategies can be employed: ‘top-down’ or ‘bottom-up’. The ‘top-down’ approach starts with the observation of biological characteristics in the intact biological system and then construct theories that would explain the observed behaviours, eventually uncovering the underlying mechanisms. On the other hand, the ‘bottom-up’ approach begins with the study of the system components in isolation and then integrates the behaviour of each component in order to predict the behaviour of the entire system. Another emerging approach is the ‘middle out’ approach, which starts with an intermediate scale (usually the biological cell, the basic unit of life) that is gradually expanded to include both smaller and larger spatial scales.3
To fully achieve the objectives of systems biology, models at different scales must be coupled together to produce integrated models across multiple scales. This needs to be carried out with appropriate methods that integrate models representing the different scales involved. The resulting complex integrated models, however, are often computationally expensive and difficult to solve numerically even with the incredible advances in computational power. This is a typical characteristic of multi-scale problems that require the use of appropriate multi-scale algorithms.
There is a range of multi-scale modelling methods that could potentially be employed in systems biology. These methods can be classified as either continuum or discrete methods, based on the strategy they use to integrate the various scales. Historically, continuum methods have been used to describe the precise behaviour of macroscopic systems subject to physical laws such as fluids, solids, and celestial objects.6 The continuum strategy usually expresses the relationship between system properties in form of continuous mathematical equations, which are then solved numerically under a range of conditions. These equations are most often a mean-field approximation that summarizes the result of the interactions at the lower level into an equation at the higher level. In contrast to the continuum strategy, the discrete approach does not attempt to summarize the low level properties in a continuous function, but rather constructs a model of a complex system by explicitly representing discrete units at the lower level and their interactions. The discrete strategy is appropriate for complex systems such as social, financial, economic, and also biological systems, which are all composed of heterogeneous unitary elements such as individuals or cells whose behaviours cannot be easily related to macroscopic outcomes.6 In these situations, discrete methods such as Agent-Based Modelling (ABM) may be more appropriate. Both approaches have their own strengths and shortcomings and a choice between them should depend on the biological questions that are being addressed.
This review focuses on the multi-scale modelling and simulation in systems biology. We begin by describing the multi-scale methodologies that are playing or will play important roles in multi-scale problems in systems biology. We then review various multi-scale models in biology that have been developed or under development. This will follow by the description of the emerging multi-scale simulation environments, before we conclude with discussion and future challenges.
2. Multi-scale methods
The aim of multi-scale modelling is to represent the behaviour of complex systems across a wide range of spatial and temporal scales. This requires the use of cost-effective and efficient computational techniques. Over the past decades, several multi-scale methods been developed in other fields of science to meet this requirement. In this section, we introduce these methodologies with the aim to later discuss how they could be adapted to systems biology.2.1 Quasi-continuum method
The Quasi-Continuum (QC) method is a multi-scale method for solving problems of interest at the atomistic scale by dramatically reducing the cost of computation by a coarse-graining procedure. The ideal behind this method is that only a certain fraction, relatively small, of a problem requires full atomic detail while the rest can be modelled using the assumption of continuum mechanics that reduce the degrees of freedom and computational demand.7The QC method consists of coarse-graining an atomistic domain by selecting a small subset of the total number of atoms, called representative atoms. In certain critical areas of the problem domain where interesting physical processes are occurring, such as dislocation nucleation, dislocation intersections, and crack propagation, etc., all the atoms are selected as representative atoms (referred to as non-local atoms), whereas far away from the critical regions where the deformation is more uniform at the fine scale, a group of atoms are enslaved to move under the influence of the representative atoms at the nodes of a tetrahedral element (referred to as local atoms) enclosing the group of atoms.8
2.2 Hybrid quantum mechanics–molecular mechanics method
The hybrid Quantum Mechanics–Molecular Mechanics (QM-MM) method9 is a popular molecular simulation method that combines the power of both QM and MM calculations. It uses quantum mechanical model in regions of chemical reaction, and classical molecular mechanics models elsewhere. This approach has been used for calculating protein function and studying the chemical processes in proteins.102.3 Equation-free multi-scale method
A key feature of complex systems is the emergence of macroscopic, coherent behaviour from the interactions of microscopic agents, e.g. molecules, cells, individuals in a population, between themselves and with their environment. This means that macroscopic rules (description of behaviour at a high level) can somehow be deduced from microscopic ones (description of behaviour at a much finer level).11 Equation Free Multi-scale (EFM) method is a framework for computer-aided multi-scale analysis, which enables models at a fine (microscopic/stochastic) level of description to perform modelling tasks at a coarse (macroscopic, systems) level. These macroscopic modelling tasks, yielding information over long time and large space scales, are accomplished through appropriately initialized calls to the microscopic simulator for only short times and small spatial domains.12This approach contrasts with other traditional multi-scale modelling approaches, which first involve the derivation of macroscopic evolution equations (balances closed through constitutive relations). The evolution equations are then solve using analytical and numerical techniques. The EFM approach can bypass the derivation of the macroscopic evolution equations when these equations conceptually exist but are not available in closed form. It consists of a set of techniques; coarse projective integration, gap-tooth scheme and patch dynamic that are briefly described below.
2.4 Heterogeneous multi-scale method
The heterogeneous multi-scale method (HMM), proposed in ref. 15 is a general framework for the efficient numerical computation of multi-scale problems. HMM relies on an efficient coupling between the macroscopic and microscopic models. It uses the microscopic solver to supply the necessary data for the macroscopic solver in cases when the macroscopic model is not explicitly available or invalid. HMM starts with a macro-scale solver, taking into account the available information about the macro-scale process, and use the micro-scale model to provide the missing macro-scale data. The micro-scale model is usually constrained with the (local) macro state of the system.HMM also provides a methodology for designing new methods for a large variety of multi-scale problems.15 These problems are classified into four types by Weinan and colleagues: (Type A) these are problems that contain isolated defects or singularities such as cracks, dislocations, shocks and contact lines. For these problems, the microscopic model is only necessary near defects or singularities. Further away it is adequate to use some macroscopic model. Such a combined macro-micro strategy should satisfy the minimum requirement if the micro-scale model is limited to a small part of the computational domain; (Type B) these are problems for which a closed macroscopic model should exist for a properly selected set of macroscopic variables, but the macro-scale model is not explicit enough to be used directly as an efficient computational tool; (Type C) these are problems that have features of both type A and type B; (Type D) these are problems that exhibit self-similarity in scales. Problems in each category share some common features that can be used when designing multi-scale methods. For type A and type B problems, the special feature is scale separation. Statistical self-similarity is the special feature of type D problem.15
2.5 Multi-grid method
Multi-Grid (MG) method developed by Brandt is a fast and efficient way of solving a wide class of integral and partial differential equations. It requires a series of problems to be solved on a hierarchy of grids with different mesh sizes in order to reduce the problem execution time.16 The original MG was later extended by Brandt17 taking into consideration different nature of the models at different scales.2.6 Multi-scale agent-based modelling method
Agent-Based Modelling (ABM), also known as Individual Based Modelling (IBM), is a computational modelling technique that is object-oriented, rule-based, discrete event and discrete-time. Agent-based modelling has its origins in the fields of ecology, social science, and anthropology.18 In this review, we refer to ABM to mean modelling paradigms that include cellular automata and software agents. ABM falls into the ‘middle-out’ modelling approach and the most obvious level of biologically-oriented agent-based modelling is that which uses cells as the primary agent level partly because of the obvious analogy of cells being autonomous agents since they are the units of biological organization. ABM is based on the rules and interactions between the components (“agents”) of a system, simulating them in a “virtual world” to create an in silico experimental model.19 They have an intrinsically modular structure via the grouping of components into classes based on similar rules. This type of modelling methodology is quiet useful to capture the emergent behaviours of a system from the behaviour of individual components of the system based on simple rules and interactions between the system's components. The basic approach in ABM is to model and simulate the behaviour of the system's constituent units (the agents) and their interactions, capturing emergence from the bottom up when the simulation is run.20 This makes it well suitable for use at any level of biological organisation from molecular to population.There are many ABM multi-scale approaches for modelling biological systems. Walker and Southern3 classified these approaches into three categories: (i) cellular-continuum approaches in which biological sub-scales are represented in the model as a field of values representing concentrations that are considered to be in steady-state. In this case, single time scale is incorporated in order to represent cellular behaviours; (ii) spatially hierarchical approaches which represent sub-cellular components explicitly as a lower hierarchy of agent in the model, but without separation of biological time scales; (iii) temporally separated approaches that integrate a second model which represent processes that occur on the a faster time scale. Processes are usually represented by mathematical differential equations that are solved using numerical simulators such as COPASI.21 Full details of these approaches can be found in ref. 3 The choice of an approach for multi-scale modelling depends on the type of biological problems to be addressed.
The major drawback of ABM method is its high computational requirements. The determination of emerging macroscopic system behaviours by simulating the behaviour of all the interacting components can be extremely computational intensive, particularly for higher eukaryotes, where even single organs are composed of large numbers of cells. Efficient techniques for reduction in the computational cost are therefore necessary to help overcome this limitation. Despite this limitation, the important of ABM as modelling paradigm in different areas of biological and biomedical problems has been recognised in recent years. We explore the application areas in the next section.
2.7 Complex automata
Complex Automata (CxA) is a general modelling framework developed in ref. 22 for multi-scale systems using Cellular Automata (CA), Lattice Boltzmann Models (LBM) and Agent Based Models (ABM) as building blocks. CxA is based on the ideal that a multi-scale system can be decomposed into N single-scale CA that mutually interact across the scales. A system is first decomposed into its subsystems and a Scale Separation Map (SSM) is built on which each subsystem can be represented as area according to its spatial and temporal scales as shown in Fig. 2. The SSM is defined as two dimensional map with the horizontal axis coding for the temporal scales and vertical axis coding for the spatial scales.23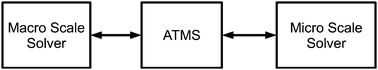 | ||
| Fig. 2 Operation of Adaptive Tabulation Multi-Scale Approach (ATMS). ATMS stores calculated ODEs solution from micro scale solver in memory. On request from macro scale solver, ATMS returns ODEs solution that meets the specified tolerance, otherwise a direct integration of the ODEs is carried out and the solution is returned to the macro scale solver, which is also added into memory. | ||
CxA allows the decomposition of the original CA into a number of single-scale CA and let the CAs exchange information in such a way that the overall behaviour of the multi-scale process is reproduced as accurate as possible. Processes having well separated scales are easily identified as components of the multi-scale model.22 The interaction across the scales (i.e. exchange of information) is through appropriate coupling mechanisms, which can either be Sub-Domain Coupling (SDC) or Hierarchical-Model Coupling (HMC) method. In the case of SDC, adjacent spatial domains are described by different models on space-time grids of possible different resolution, while some parameters or variables of the main model are first computed locally on the fly by a finer scale model when HMC is used.23
2.8 Multi-scale numerical scheme
Multi-Scale Numerical Scheme (MSNS) is an efficient technique developed by Whiteley24 for solving bidomain equations by exploiting the multi-scale nature of the problems modelled by these equations. Bidomain model consist of elliptic differential equation and a parabolic partial differential equation, coupled at each point in space with a large systems of stiff, non-linear ordinary differential equations. These type of equations are usually used to model cardiac electrophysiology. Solution of these equations is computationally expensive for three-dimensional heart model. The large computation time is due to the stiffness of the ordinary differential equations and parabolic partial differential equation.Processes modelled by the electrophysiological model are characterized by a wide variety of time scales and wide range of length scales. The numerical algorithm uses the observation that only a very small number of quantities vary on a short time scale and a short length scale, and computes only these quantities at a high resolution. The key to implementing this algorithm is the use of two meshes. A fine mesh is used to approximate the rapidly varying variables. Other variables are computed on a coarser mesh. Linear interpolation is then used to transfer the slower variables onto the fine mesh when required. MSNS algorithm was reported to give an increase in computational efficiency of more that two orders of magnitude.24
2.9 Adaptive tabulation multi-scale approach
The dynamic simulation of many types of models involve direct integration of a set of ordinary differential equations (ODEs) for a give time step. In combustion chemistry this leads to a situation in which there are a very large number of ODEs with similar initial conditions that require being evaluated millions of times, leading to enormous computational costs. Adaptive Tabulation Multi-Scale (ATMS) approach named in situ adaptive tabulation by Pope was then developed25 to tackle this type of multi-scale problem: instead of solving all of the ODEs for every initial condition, ATMS stores previously-calculated solutions, and when a new solution of the ODEs is required for similar initial conditions, the set of the previously calculated values is searched and an approximate solution that satisfies a specified error tolerance is returned. Fig. 2 shows the inter-relationship between the solvers and the ATMS.3. Multi-scale models in biology
A multi-scale model can be defined as a composite model that incorporates more elementary models from several levels of biological organisation or temporal scales.2 There has been an increasing interest in multi-scale modelling in biological systems, leading to the development of several multi-scale models covering various biological processes that use some of the multi-scale methods described above. In this section, we describe briefly some of these biological systems and processes, and the associated multi-scale models3.1 Multi-scale models of tumour growth
Tumour refers to complex phenomena that cut across different biological scales. It develops when genetic and epigenetic changes disrupt the processes that maintain cellular homeostasis by regulating cell division, growth, movement and apoptosis. The formation of new blood vessels is necessary for tumours to progress to the rapid phase of vascular growth. The inter-related processes act over a spectrum of spatial and temporal scales. The spatial scales range from the sub-cellular to the cellular and macroscopic levels, while the time scales vary from seconds (or less) for signal transduction to months (and years) for tumour-doubling times.26 The desire to understand the complexity of tumour has given rise to various multi-scale models to describe its growth. These models are either continuum-based or ABM-based.An example of the continuum-based models is the multi-scale model for avascular tumour growth developed by Jiang and colleagues, which spans three distinct biological scales. At the cellular level, a discrete lattice Monte Carlo model describes the cell growth, proliferation, death, and intercellular adhesion. A simplified Boolean protein expression regulatory network controls the cell cycle arrest at the sub-cellular level, while at the extracellular level, a system of differential equations describes the diffusion, consumption, and production of nutrient, metabolites, growth promoters and inhibitors. The three levels were integrated together and data from experiments with multicellular spheroids using the mouse mammary tumour cells were used to determine the parameters of the model simulations.27 The simulation results show a good agreement with experimental data from mouse mammary tumour spheroids.
Another continuum-based multi-scale tumour growth model is the mathematical model of avascular tumour growth developed by Ribba and colleagues to investigate the effect of inhibitors of metalloproteinases (MMPi) treatment on cancer growth.28 This consists of an age-structured model that distinguishes between proliferation and quiescent cells. The cell cycle model is embedded in a macroscopic model of the spatial tumour dynamics. The model simulation results predict the lack of efficacy of MMPi in advance cancer patients. However, unlike the earlier model, this model is theoretical and has not been validated with experimental data.
There are also ABM-based multi-scale models of tumour growth. The work of Wang and colleagues that developed multi-scale agent-based non-small cell lung cancer is a good example. The model consists of a 3D environment with which cancer cells interact while processing through phenotypic changes. They integrated a transforming growth factor (TGF) into in silico model as a second extrinsic input in addition to the epidermal growth factor (EGF).29 Their simulation and analysis results demonstrate that changes in TGFβ can modulate the EGF downstream cascade, thereby affecting the motility of tumour cells through signalling cross-talk.
Other multi-scale models in the areas of tumour growth include the multi-scale mathematical model of solid tumour growth which couples an improved model of tumour invasion with a model of tumour-induced angiogenesis by Macklin and colleagues,30 and multi-scale ABM model of brain cancer by Zhang and colleagues.31
3.2 Multi-scale models of organs
A biological organ is a group of tissues joined in structure unit to perform a specific function or functions (e.g. Heart, brain, lungs, etc.). Here we consider heart as an illustrative example. The mechanisms underlying heart function are characterised by a range of spatial scales organization as shown in Fig. 3. Various processes at the tissue, cellular, sub-cellular and lower levels occur at multiple time scales influencing the behaviour of the heart. Heart multi-scale models are usually based on the continuum modelling approaches using various mathematical models at different scales and for physical processes. Some of these models include Noble model32 or Fenton–Karma model33 for the membrane potential and calcium transient, and Fitzhugh–Nagumo34 and Hodgkin–Huxley35 models of ion channels at the cellular scale (see ref. 2 for details of multi-scale modelling of the ion channel), monodomain model for the action potential propagation at the tissue level, Lin–Yin constitutive law36 for modelling the heart mechanics and bidomain model developed over 40 years ago for the cardiac electrophysiology is still extensively being used. See ref. 37 for detailed review of the techniques that are currently employed to model cardiac electrical activity. At the organ level, finite element (FE) meshes are usually constructed from experimental measurements of the heart of dogs,38,39 pigs40 and rabbits.41 Imaging technology also allows the generation of anatomically accurate representation of the human heart to be constructed from MRI42 and CT scans.43The model of the heart therefore consists of an integrated system of models from different scales. Various simulators have been developed to simulate the heart model (see section 4 for description of multi-scale simulators). A major problem in simulating the heart model is the high computational requirements. Some of the above described multi-scale methods will play an important role in the reduction of computational cost associated with such complex integrated model. For example, MSNS method developed by Whiteley24 allows efficient simulation of the bidomain model at the tissue level.
The important role of multi-scale modelling in understanding biological systems and processes has given rise to the collaborative IUPS Physiome44 project (Renal, Heart, Giome, Epitheliome and Living Human Physiome projects). The Physiome project intends to create a public domain framework for computational physiology, including the development of modelling standards, computational tools and web-accessible databases of models of structure and function at all spatial scales. Its main goal is to establish a publicly accessible framework for handling the hierarchy of computational models, associated experimental data and publications, that will help integrate knowledge, from the genomic and proteomic levels to whole organ and body scale, into an understanding of physiological function for intact organisms.45
Another similar project to the Physiome project is the Virtual Physiological Human (VPH), which is an initiative, supported by the European Commission (EC), that seeks to develop an integrative model of human physiology at multiple scales from the whole body through the organ, tissue, cell and molecular levels. This is expected to be a methodological and technological framework that, once established, will enable the investigation of the human body as a single complex system.46 The distributed architecture approach using wokflow management systems to link various models into an integrated digital human model proposed by Kell47 supports the ideals of VPH.
There many other organ system models. An example is the multi-scale model of the human musculoskeletal systems for accurate prediction of the risk of bone fracture developed by Viceconti and colleagues.48 This model consists of the interconnection of five interdependent sub-models that consist of the continuum, the boundary condition, the constitutive equation, the remodelling history and the failure criterion sub-models. The model already found some clinically relevant applications, especially in the analysis of joint prostheses.
Another challenging multi-scale modelling of an organ system is that of the lung. Chakraborty and colleagues developed a multi-scale model for pulmonary oxygen uptake, which consists of coupling together of three-dimensional convection–diffusion–reaction model at the micro-scale (red blood cell), the convective–diffusion models at the meso- (capillary) and macro- (lung) scales.49
A general multi-scale model for in vivo tissue exchanges and intra-organ metabolism (GENTEX)50 is a whole organ model of the vascular network providing intra-organ flow heterogeneity and accounts for substrate transmembrane transport, binding and metabolism in erythrocytes, plasma, endothelial cells, interstitial space and cardiomyocytes. The model is a convection–diffusion–transport–reaction model allowing exchanges and metabolism among three cell types, red blood cells (RBC), endothelium and parenchymal cells, the interstitial space and the plasma space.51
 | ||
| Fig. 3 Hierarchy of scales involved in modelling the heart. | ||
3.3 Multi-scale models of epithelium tissues
Epithelium tissues serve as protective covers to the whole surface of the body. It is made up of cells closely packed and ranged in one or more layers. Many multi-scale models of epithelium tissues have been developed. The three dimensional multi-scale computational model of human epidermis using ABM, which was developed by Adra and colleagues52 is good example of multi-scale model of epithelium tissue. This model consists of three interacting and integrated layers made up of agent-based model that captures the biological rules governing the cells in the human epidermis, a COPASI21 model that simulates the expression and signalling of TGF-1 at the sub-cellular level and a mechanical layer embodied by a numerical physical solver responsible for resolving the forces exerted between cells at the multi-cellular level. The agent-based modelling framework Flame53 was used to set up the 3D multi-scale model. The validity of the basic biological rules at the cellular level and the sub-cellular mechanisms used in the COPASI were tested by qualitatively comparing the simulation results of an intact epidermis with published research. The model successfully simulated many of the described behaviours of keratinocytes and TGF-β1 sub-cellular mechanisms.The Epitheliome Project54 is part of the Physiome Project that intends to developed a computational model of cell behaviour within the context of tissue architecture, differentiation, wound repairs and malignancy. The details of the methodology for integrating ABM and continuum models of cellular behaviour in the project are discussed in ref. 54
3.4 Other multi-scale models in biology
The work of Yan and colleagues55 is another good example of how to link various models at different scales. They developed a 3D multi-scale numerical model for analysing stresses and deformations of cell when the tissue construct is subjected to macro-scale mechanical loads. Using the FE at the macro-level, they incorporated the multi-cellular model, which contains the micro-level model of the single cell modelled as two-phase inclusion. The displacements mapped from the multi-cellular volume to the single cell model enabled the analysis of the deformations of substructures within the cell to be carried out. At the macro-scale, the 3D numerical model was validated for mechanical characterization of alginate-cell discs. The predicted results capture the trend of the damage cell observed from the experimental study. This work clearly demonstrated the integration of experimental data and modelling.Cristofolini and colleagues adopted the “middle out” approach and a continuum modelling method to model the human fermur. They focused on the organ-level model of the fracture of the femur, while integrating body-, tissue- and cell-level models. The higher level (the body level) provides bone loading conditions, lower level (tissue level) provides constitutive equations and failure criteria and the cellular level accounts for the bone remodelling. Like the model of Yan and colleagues, they used FE to model the organ-level bone structures. The validation of the model with the experimental data was carried out to quantify the model's sensitivity and accuracy. As highlighted in this work, a large amount of information is available at all scales but only the organ-level models are really mature in this perspective.56
Another multi-scale modelling is from Villa and colleagues who use quasi-continuum method to model and simulate the interaction between DNA and the lac repressor. The simulation treats the lac repressor and the surrounding complement of water molecules in full atomistic detail while the looped DNA region is treated using elasticity theory.57
Others notable multi-scale models in biology include the model of the physical and biological processes implicated in the coronary artery in-stent restenosis using CxA multi-scale method by Evan and colleagues,58 model of cell-cell contact-mediated signalling in cancer cell invasion,59 model of in vitro wounded epithelial cell monolayers60 and the work of An18 that used a series of linked ABMs to model inflammatory disease.
It must be noted that there are large body of literature on multi-scale models in biology using both the ABM and continuum based approaches. Space restriction will not permit us to discuss all these models. We therefore summarise some other recent multi-scale models developed with these approaches in Table 1. Interested readers are referred to ref. 3, 61 and 62 for detailed review of multi-scale modelling using ABM.
| Model | Objective | Key achievements | Type | Ref. |
|---|---|---|---|---|
| Virtual tissues | To predict histopathological outcomes from alterations of cellular phenotypes that are controlled by chemical-induced perturbations in molecular pathways. | The behaviours of thousands of heterogeneous cells in tissues were simulated discretely. | ABM | 6 |
| Xenopus laevis explant model | To integrate intracellular signalling information with multi-cell behaviours in the context of a spatially heterogeneous tissue environment. | Permit the testing and identification of key systems-level hypotheses regarding how signalling proteins affect overall tissue-level behaviour during morphogenesis in an experimentally verifiable system. | ABM (TS) | 61 |
| A multi-scale model of juxtacrine EGFR-MAPK signalling | To develop an ODE model of juxtacrine EGFR-ligand activation of the MAPK intracellular pathway and to couple this to an agent-based representation of individual cells. | Results show that mean experimental data obtained from analysing entire cell populations is an oversimplification, and should not be extrapolated to deduce the signal:response paradigm of individual cells. | ABM (TS) | 63 |
| Agent-oriented In Silico Liver (ILS) | To study the hepatic disposition and metabolism of antipyrine, atenolol, labetalol, diltiazem, and sucrose administered alone or in combination. | ISL Simulations were validated separately and together against in situ data and prior physiologically based pharmacokinetic (PBPK) predictions. | ABM | 64 |
| Model of thrombus development | To study the formation of a thrombus (clot) in a blood vessel | Able to study in detail the growth of the thrombus, where initially activated plattelets arrive at the from of thrombus. | ABM (TS) | 65 |
| Three-dimensional multi-scale tumour model | Simulating gene-protein interaction profiles, cell phenotypes and multicellular patterns in brain cancer. | The simulation results show that over time proliferative and migratory cell populations not only oscillate but also directly impact the spatio-temporal expansion patterns of the entire cancer system. | ABM | 66 |
| A multi-scale model of dendritic cell education and trafficking in the lung | To improve understanding of the dynamic role of dendritic cells in the lung. | Provides an essential aid in understanding the impact of a dynamically changing lung microenvironment on the ability of dendritic cells to orchestrate adaptive immunity. | CM | 67 |
| Multi-scale model of follicular development | To accounts for the changes in the total cell number, growth fraction and global maturity of both ovulatory and degenerating follicles for various intensities of the selection rate. | The model predicted selection process outputs (mono- or poly-ovulation, anovulation) are consistent with physiological knowledge. | CM | 68 |
| Multi-scale in silicoleukocytes model | To represent the dynamics of rolling, activation, and adhesion of individual leukocytesin vitro | The emergent behaviour in silico closely match the observed behaviour in vitro | ABM | 69 |
| Multi-scale model of amelogenesis | To simulate organogenesis that uses a single cell response function to define the behaviour of individual cells in an organ-scale simulation of a large cell population. | The calibrated model using human data predicts wavelengths in the mouse incisor and an ordering transition at the chimpanzee cingulum. | CM | 70 |
4. Multi-scale simulators
There is presently a shortage of modelling and simulation frameworks for both continuum and ABM approaches of modelling multi-scale problems in biological systems. Most of the available continuum based multi-scale simulation environments were developed for specific problems. Only a few of them are general frameworks that can be applied to any multi-scale problems in systems biology. The most advanced multi-scale simulators come from the heart modelling community. A model of the heart is an integrated system of models that include cellular, tissue and organ models. Several packages have been developed for modelling and simulation of the heart by various groups around the world. Some of these packages include a simulator for coupling the electrophysiology and mechanics of the heart developed at the University of Auckland71 a cardiac simulation package developed by Usyk and McCulloch72 at the University of California San Diego, and a ventricle simulator developed by Watanabe and colleagues73 at the University of Tokyo.In order to overcome some of the problems usually associated with individual group based simulators, Pitt-Frances and colleagues74 started the development of Cancer, Heart and Soft Tissue Environment (CHASTE). CHASTE is targeted at the cardiac electrophsiology and heart modelling and is being developed with and by researchers from both areas with as much code modularity and re-use as possible. It is a library of computational biology software that began as an experiment in the use of agile programming methods.74CHASTE is designed to run in parallel to help reduce the computational time taken to simulate very large computational problems. Recently, the MSNS numerical algorithm developed by Whiteley24 for solving the bidomain equations has been incorporated into CHASTE.75
The problem of lack of multi-scale simulation frameworks is not limited to the continuum based modelling approaches. Many of the ABM biological models described above were developed using custom code or a combination of custom code and general purpose ABM development toolkits. Among the most popular ABM toolkits are Repast (http://repast.sourceforge.net/), Swarm (http://www.swarm.org), NetLogo (http://ccl.northwestern.edu/netlogo/), Mason76 and Flame.53 Railsback and colleagues77 provide a comprehensive review and comparison of these ABM modelling toolkits.
The Multi-scale Systems Immunology (MSI) is an open source multi-scale modelling framework written in C++ and Python. It is based on the computational framework for modelling immunological interactions.78 The main purpose of MSI is to run large scale cell-based immune simulations. The software implements a modular design that allows for flexible configuration of components and initialization of parameters, thus allowing simulations to be run that model processes occurring over different temporal and spatial scales. It uses the relational database to store the parameters that characterise the biological entities (e.g.cell types such as macrophages, neutrophils, fibroblasts, etc.). The database is closely integrated with model classes in the code to enable biological entities to be instantiated by the database name lookup.
The Basic Immune Simulator (BIS) developed by Folcik and colleagues is an ABM based simulator for studying the interaction between innate and adaptive immunity. The innate immune response is essential for immunity to bacterial, fungal and parasitic infections, which is followed by the adaptive immune response that is responsible for fighting disease.79 The BIS uses three “zones” to represent tissue spaces where the immunological processes occur. Zone 1 is the site of initial tissue challenge with pathogen. Zone 2 is an abstract representation of a lymph node or the spleen, where lymphocytes reside and proliferate. Zone 3 is an abstract representation of the lymphatic and blood circulation, the conduits for travel for the cells of the immune system.19BIS was developed using an open-source Repast software library.
Another notable emerging multi-scale computational framework is the Multi-Scale Coupling Library and Environment (MUSLE). MUSLE is a multi-scale computational framework currently being develop under the Complex Autonoma Simulation Technique (COAST) project.58 It is based on the scale separation map conceptual framework developed in the CxA multi-scale modelling technique.22 The purpose of MUSLE is to reduce the common problems associated with multi-scale models coupling and to facilitate the development of simulations that consist of several sub-models. The core of MUSLE implementation is the Distributed Space Time Coupling Library that implements the CxA graph and scale separation map using an agent based approach. MUSLE is being build upon JADE library, a multi-agents based simulation environment. It aims to provide a flexible and powerful way for model developers to integrate new kernels as well as legacy code.
5. Discussion and future challenges
Systems biology aims to describe and understand the operation of complex biological systems and ultimately to develop predictive models of human disease.80 This objective requires integration of knowledge from diverse biological components and data that span several spatial and temporal scales into models of the system as a whole. We described approaches that can be used to tackle multi-scale problems in systems biology including both continuum and ABM based multi-scale modelling approaches. We discussed the emerging multi-scale models for various biological systems and processes. These models use different strategies for modelling at different scales and for coupling between scales.Most of the continuum based models discussed are usually simulated using the traditional FE and/or finite differences (FD) or finite volume (FV) methods. The use of parallel processing techniques is also employed in some cases to reduce the computational cost. As argued in ref. 81, three dimensional FE and FM models are far too computational intensive for a whole systems simulation at once. Techniques are needed to link the FE and FV models to one- or two-dimensional models. The use of multi-scale methods (e.g. GTS, HMM, PD) described above need to be exploited in addition to the parallelisation to help in the computational cost reduction. Effort in this direction is demonstrated recently in the work of Whiteley24 that explored the multi-scale nature of the heart to develop a technique (MSNS described above) for solving bidomain heart model efficiently.
Most of the ABM based models described above are bi-scale models. Only few models span entire continuum from gene to organism. This may be due to the fact that methods for representing multiple spatial scales (e.g. MG, GTS, HMM) have been developed mainly for continuum type (e.g. FE, FD) and their application to models representing dynamic biological processes, such as tissue growth and regeneration, may not be straightforward.3
One major challenge in multi-scale modelling in systems biology is how to couple together the various models that are available at different spatial and temporal scales. These models have been developed for specific purposes and their behaviours when integrated with others have not yet been fully studied. This is in addition to the fact that models at different scales may be of different types (e.g. deterministic or stochastic models or ABM). Methods for integrating stochastic processes into a deterministic framework (for example the formulation of stochastic master equations) for chemical reactions that occur on multiple time scales are already available but these methods have not been applied to any large-scale systems biology problems.2 Even if all the models of a biological system at different scales can easily be integrated, the task of simulating the integrated models is computational very expensive unless the multi-scale nature of biological systems is exploited in specific ways to take advantage of computational efficiencies. Coupling between scales has been a major issue in multi-scale problems in many scientific domains. Three basic challenges need to be tackled in coupling micro-scale and macro-scale models. These include (i) the choice of a suitable initial and boundary condition for the fine scale domains, that correctly reflects macro-scale information; (ii) using the fine scale calculations to derive the macroscopic information and identification of sub-domains in which macro-scale model are not sufficient or existed in a closed form, and a fine scale model is required. We described various multi-scale methods that are capable of overcoming these challenges and application of these methods to problems in biology is beginning to emerge. In addition to these computational methods, the coupling of models across scales can also benefit from model reduction. Model reduction is a strategy desired for simplifying large complex models, which often arise from multi-scale approaches. As argued in ref. 81, there is a need for automated methods to help in analysing a complex model defined at a particular spatial and temporal scale in order to compute the parameters of a simpler model that captures the model behaviour relevant to the scales above. Although not providing a completely automated approach, the methods discussed in ref. 82 could be part of such a methodology.
Another important difficulty in multi-scale modelling is their high computational costs, which is a problem in both the continuum and ABM approaches. It may be more pronounce in the ABM approaches because of the large numbers of agents involved when using ABM for realistic biological systems. Efficient techniques for reduction of computational costs are therefore necessary to be able to use the full power of these approaches. High performance computing, parallel processing, grid computing and similar technologies can help, but these technologies require specific software to be written in order to take full advantage of the hardware. One way to tackle this challenge is to combine various multi-scale methods described above. For example, we are currently exploring a combination of ATMS with ABM to reduce the computational demand.
Availability of suitable modelling frameworks and simulation environments will likely be the most important factor in the wider adoption of multi-scale modelling in systems biology. Most of the present modelling and simulation tools are developed for single scale modelling and alone are not sufficient for multi-scale modelling. Some will however play major roles in the new emerging frameworks, as simulation of multi-scale models will probably involve assembly of many of these simulators in an integrative manner, each individual being used in its own area of specialisation. We have discussed some of the emerging frameworks, such as BIS, MIS, CHASTE and MUSCLE, however, to our knowledge, the use of these frameworks is still restricted to their inventors. It remains to be seen whether these frameworks will be taken up by the systems biology community. The biggest hurdle is perhaps the construction of an easy to use, yet powerful, software interface to facilitate its use by non-specialists. This was indeed the problem with biological modelling and simulation in general in the past.
The roles of standards in multi-scale modelling and simulation can also not be underestimated. Standards such as SBML,83 CellML84 and FieldML85 are playing major roles in the multi-scale modelling frameworks being developed in the Physiome projects.81 Furthermore, the simulation of large scale multi-scale models will likely have to span several organisations, thus requiring integration and management of models and data. Web and Grid services offer flexible means of integrating often incompatible applications and tools for data acquisition, registration, storage, provenance, organization, analysis and presentation.86 In addition to the use of these technologies, markup languages such as System Biology Results Markup Language (SBRML)87 will be required for associating experimental results with models for passing to analysis tools, and for recording the results of analysis for validation, archiving or comparison.
The need for multi-scale modelling and simulation techniques to help identify and elucidate mechanistic interactions between various components of a biological system is clear and in particular for higher eukaryotes. The development of the necessary techniques will depend to a large extent on meeting the above challenges, which will require collaborative efforts involving various experts from different disciplines such as biology, computer science, mathematics and engineering.
Acknowledgements
This work has been supported by the European Commission within the seventh Framework Programme, theme 1 Health through the UNICELLSYS project (Grant 201142) and BBSRC/EPSRC through the Manchester Centre for Integrative Systems Biology (BB/C008219/1). We thank Juergen Pahle and our colleagues in the MCISB for helpful discussions.References
- S. Schnell, R. Grima and P. K. Maini, Am. Sci., 2007, 95, 134–142 Search PubMed
.
- J. Southern, J. Pitt-Francis, J. Whiteley, D. Stokeley, H. Kobashi, R. Nobes, Y. Kadooka and D. Gavaghan, Prog. Biophys. Mol. Biol., 2008, 96, 60–89 CrossRef CAS
- D. C. Walker and J. Southgate, Briefings Bioinf., 2009, 10, 450–461 Search PubMed
- J. Twycross, L. R. Band, M. J. Bennett, J. R. King and N. Krasnogor, BMC Syst. Biol., 2010, 4, 34 CrossRef
- D. Noble, Science, 2002, 295, 1678–1682 CrossRef CAS
- I. Shah and J. Wambaugh, J. Toxicol. Environ. Health B Crit. Rev., 2010, 13, 314–328 CrossRef CAS
- M. Gunzburger and Y. Zhang, Multiscale Model. Simul., 2010, 8(2), 571–590 CrossRef
-
E. B. Tadmor and R. E. Miller, in Hand Book of Material Modelling, ed. S. Yip, Springer Netherlands, 2007, vol. 2(13), 663–682 Search PubMed
- A. Warshel and M. Levitt, J. Mol. Biol., 1976, 103, 227–249 CAS
- S. L. Kamerlin, M. Haranczyk and A. Warshel, J. Phys. Chem. B, 2009, 113, 1253–1272 CrossRef CAS
- I. G. Kevrekidis and G. Samaey, Annu. Rev. Phys. Chem., 2009, 60, 321–344 CrossRef CAS
- I. G. Kevrekidis, C. W. Gear, J. M. Hyman, P. G. Kevrekidis, O. Runborg and C. Theodoropoulos, Comm. Math. Sci., 2003, 1(4), 715–762 Search PubMed
- G. Samaey, I. G. Kevrekidis and D. Roose, Sixth International Congress on Industrial Applied Mathematics, 2007, 7(1), 1025803–1025804 Search PubMed
- J. M. Hyman, Comput. Sci. Eng., 2005, 7(3), 47–53 CrossRef
- E. Weinan and B. Engquist, Comm. Math. Sci., 2003, 1, 87–132 Search PubMed
- A. Brandt, Math. Comput., 1977, 31(138), 333–390 CrossRef
-
A. Brandt, in Multiscale and Multiresolution Methods: Theory and Applications, ed. T. Barth, Springer-Verlag, 2001 Search PubMed
- G. An, Theor. Biol. Med. Modell., 2008, 5, 11 Search PubMed
- G. An, Q. Mi, J. Dutta-Moscato and Y. Vodovotz, Wiley Interdiscip. Rev.: Syst. Biol. Med., 2009, 1(2), 159–171 Search PubMed
- E. Bonabeau, Proc. Natl. Acad. Sci. U. S. A., 2002, 99(Suppl 3), 7280–7287 CrossRef CAS
- S. Hoops, S. Sahle, R. Gauges, C. Lee, J. Pahle, N. Simus, M. Singhal, L. Xu, P. Mendes and U. Kummer, Bioinformatics, 2006, 22, 3067–3074 CrossRef CAS
- A. G. Hoekstra, J.-L. Falcone, A. Caiazzo and B. Chopard, Lect. Notes Comput. Sci., 2008, 5191, 192–199 Search PubMed
- A. Hoekstra, E. Lorenz, J.-L. Falcone and B. Chopard, Int. J. Multiscale Comput. Eng., 2007, 5, 491–502 CrossRef
- J. P. Whiteley, Ann. Biomed. Eng., 2008, 36, 1398–1408 CrossRef
- S. Pope, Combust. Theory Modell., 1997, 1, 41–63 Search PubMed
- J. M. Osborne, A. Walter, S. K. Kershaw, G. R. Mirams, A. G. Fletcher, P. Pathmanathan, D. Gavaghan, O. E. Jensen, P. K. Maini and H. M. Byrne, Philos. Trans. R. Soc. London, Ser. A, 2010, 368, 5013–5028 CrossRef CAS
- Y. Jiang, J. Pjesivac-Grbovic, C. Cantrell and J. P. Freyer, Biophys. J., 2005, 89, 3884–3894 CrossRef CAS
- B. Ribba, O. Saut, T. Colin, D. Bresch, E. Grenier and J. P. Boissel, J. Theor. Biol., 2006, 243, 532–541 CrossRef CAS
- Z. Wang, C. M. Birch, J. Sagotsky and T. S. Deisboeck, Bioinformatics, 2009, 25, 2389–2396 CrossRef CAS
- P. Macklin, S. McDougall, A. R. A. Anderson, M. A. J. Chaplain, V. Cristini and J. Lowengrub, J. Math. Biol., 2009, 58, 765–798 CrossRef
- L. Zhang, Z. Wang, J. A. Sagotsky and T. S. Deisboeck, J. Math. Biol., 2009, 58, 545–559 CrossRef
- D. Noble, A. Varghese, P. Kohl and P. Noble, Can. J. Cardiol., 1998, 14, 123–134 CAS
- F. Fenton and A. Karma, Chaos, 1998, 8, 20–47 CrossRef
- R. Fitzhugh, Biophys. J., 1961, 1, 445–466
- A. L. Hodgkin and A. F. Huxley, J. Physiol., 1952, 117, 500–544 CAS
- D. H. Lin and F. C. Yin, J. Biomech. Eng., 1998, 120, 504–517 CrossRef CAS
- G. Plank, L. Zhou, J. L. Greenstein, S. Cortassa, R. L. Winslow, B. O'Rourke and N. A. Trayanova, Philos. Trans. R. Soc. London, Ser. A, 2008, 366, 3381–3409 CrossRef
- P. M. Nielsen, I. J. L. Grice, B. H. Smaill and P. J. Hunter, Am. J. Physiol., 1991, 260, H1365–H1378 CAS
- I. J. LeGrice, B. H. Smaill, L. Z. Chai, S. G. Edgar, J. B. Gavin and P. J. Hunter, Am. J. Physiol., 1995, 269, H571–H582 CAS
- C. Stevens and P. J. Hunter, Prog. Biophys. Mol. Biol., 2003, 82, 229–241 CrossRef
- F. Vetter and A. McCulloch, Prog. Biophys. Mol. Biol., 1998, 69, 157–183 CrossRef CAS
- H. Watanabe, S. Sugiura, H. Kafuku and T. Hisada, Biophys. J., 2004, 87, 2074–2085 CrossRef CAS
- L. Xia, M. Huo, Q. Wei, F. Liu and S. Crozier, Phys. Med. Biol., 2005, 50, 1901–1917 CrossRef
- P. J. Hunter, E. J. Crampin and P. M. F. Nielsen, Briefings Bioinf., 2008, 9, 333–343 Search PubMed
- E. J. Crampin, N. P. Smith and P. J. Hunter, Histochem. J., 2004, 35, 707–714 CrossRef
- M. Viceconti, G. Clapworthy and S. V. S. Jan, J. Physiol. Sci., 2008, 58, 441–446 Search PubMed
- D. B. Kell, IUBMB Life, 2007, 59, 689–695 Search PubMed
- M. Viceconti, F. Taddei, S. V. S. Jan, A. Leardini, L. Cristofolini, S. Stea, F. Baruffaldi and M. Baleani, Clin. Biomech., 2008, 23, 845–852 CrossRef
- S. Chakraborty, V. Balakotaiah and A. Bidani, J. Theor. Biol., 2007, 244, 190–207 CrossRef
- J. B. Bassingthwaighte, G. M. Raymond, J. D. Ploger, L. M. Schwartz and T. R. Bukowski, Philos. Trans. R. Soc. London, Ser. A, 2006, 364, 1423–1442 CrossRef CAS
- J. B. Bassingthwaighte, G. M. Raymond, E. Butterworth, A. Alessio and J. H. Caldwell, Ann. N. Y. Acad. Sci., 2010, 1188, 111–120 CrossRef
- S. Adra, T. Sun, S. MacNeil, M. Holcombe and R. Smallwood, PLoS One, 2010, 5, e8511 CrossRef
- P. Richmond, D. Walker, S. Coakley and D. Romano, Briefings Bioinf., 2010, 11, 334–347 Search PubMed
- R. Smallwood and M. Holcombe, IEEE Xplore, 2006, 816–819 Search PubMed
- K. C. Yan, K. Nair and W. Sun, J. Biomech., 2010, 43, 1031–1038 CrossRef
- L. Cristofolini, F. Taddei, M. Baleani, F. Baruffaldi, S. Stea and M. Viceconti, Philos. Trans. R. Soc. London, Ser. A, 2008, 366, 3319–3341 CrossRef
- E. Villa, A. Balaeff, L. Mahadevan and K. Schulten, Simulation, 2004, 2(4), 527–553 CAS
- D. J. W. Evans, P. V. Lawford, J. Gunn, D. Walker, D. R. Hose, R. H. Smallwood, B. Chopard, M. Krafczyk, J. Bernsdorf and A. Hoekstra, Philos. Trans. R. Soc. London, Ser. A, 2008, 366, 3343–3360 CrossRef CAS
- I. Ramis-Conde, D. Drasdo, A. R. A. Anderson and M. A. J. Chaplain, Biophys. J., 2008, 95, 155–165 CrossRef CAS
- D. C. Walker, G. Hill, S. M. Wood, R. H. Smallwood and J. Southgate, IEEE Trans. NanoBiosci., 2004, 3, 153–163 CrossRef CAS
- B. C. Thorne, A. M. Bailey and S. M. Peirce, Briefings Bioinf., 2007, 8, 245–257 Search PubMed
- F. Azuaje, Briefings Bioinf., 2010, 1–14 Search PubMed
- D. C. Walker, N. T. Georgopoulos and J. Southgate, BMC Syst. Biol., 2008, 2, 102 CrossRef CAS
- L. Yan, G. E. P. Ropella, S. Park, M. S. Roberts and C. A. Hunt, Pharm. Res., 2008, 25, 1023–1036 CAS
- Z. Xu, N. Chen, M. M. Kamocka, E. D. Rosen and M. Alber, J. R. Soc. Interface, 2008, 5, 705–722 CrossRef
- L. Zhang, C. A. Athale and T. S. Deisboeck, J. Theor. Biol., 2007, 244, 96–107 CrossRef CAS
- D. J. Klinke, Ann. Biomed. Eng., 2007, 35, 937–955 CrossRef
- N. Echenim, D. Monniaux, M. Sorine and F. Clément, Math. Biosci., 2005, 198, 57–79 CrossRef CAS
- J. Tang, K. F. Ley and C. A. Hunt, BMC Syst. Biol., 2007, 1, 14 CrossRef
- B. Cox, J. Theor. Biol., 2010, 262, 58–72 CrossRef
- D. Nickerson, N. Smith and P. Hunter, Europace, 2005, 7, S118–127 CrossRef
- T. P. Usyk and A. D. McCulloch, J. Cardiovasc. Electrophysiol., 2003, 14, S196–S202 Search PubMed
- H. Watanabe, S. Sugiura, H. Kafuku and T. Hisada, Biophys. J., 2004, 87, 2074–2085 CrossRef CAS
- J. Pitt-Francis, M. O. Bernabeu, J. Cooper, A. Garny, L. Momtahan, J. Osborne, P. Pathmanathan, B. Rodriguez, J. P. Whiteley and D. J. Gavaghan, Philos. Trans. R. Soc. London, Ser. A, 2008, 366, 3111–3136 CrossRef
- M. O. Bernabeu, R. Bordas, P. Pathmanathan, J. Pitt-Francis, J. Cooper, A. Garny, D. J. Gavaghan, B. Rodriguez, J. A. Southern and J. P. Whiteley, Philos. Trans. R. Soc. London, Ser. A, 2009, 367, 1907–1930 CrossRef
- S. Luke, C. Cioffi-Revilla and L. Panait, Simulation, 2005, 81(7), 517–527 CrossRef
- S. F. Railsback, S. L. Lytinen and S. K. Jackson, Simulation, 2006, 82(9), 609–623 CrossRef
- F. Mitha, T. A. Lucas, F. Feng, T. B. Kepler and C. Chan, Source Code Biol. Med., 2008, 3, 6 Search PubMed
- V. A. Folcik, G. C. An and C. G. Orosz, Theor. Biol. Med. Modell., 2007, 4, 39 Search PubMed
- E. C. Butcher, E. L. Berg and E. J. Kunkel, Nat. Biotechnol., 2004, 22, 1253–1259 CrossRef CAS
- J. Bassingthwaighte, P. Hunter and D. Noble, Exp. Physiol., 2009, 94, 597–605 Search PubMed
- I. Surovtsova, N. Simus, T. Lorenz, A. König, S. Sahle and U. Kummer, Bioinformatics, 2009, 25, 2816–2823 CrossRef CAS
- M. Hucka, A. Finney, H. M. Sauro, H. Bolouri, J. C. Doyle, H. Kitano, A. P. Arkin, B. J. Bornstein, D. Bray, A. Cornish-Bowden, A. A. Cuellar, S. Dronov, E. D. Gilles, M. Ginkel, V. Gor, I. I. Goryanin, W. J. Hedley, T. C. Hodgman, J.-H. Hofmeyr, P. J. Hunter, N. S. Juty, J. L. Kasberger, A. Kremling, U. Kummer, N. L. Novère, L. M. Loew, D. Lucio, P. Mendes, E. Minch, E. D. Mjolsness, Y. Nakayama, M. R. Nelson, P. F. Nielsen, T. Sakurada, J. C. Schaff, B. E. Shapiro, T. S. Shimizu, H. D. Spence, J. Stelling, K. Takahashi, M. Tomita, J. Wagner, J. Wang and S. B. M. L. Forum, Bioinformatics, 2003, 19, 524–531 CrossRef CAS
- P. F. Nielsen and M. D. Halstead, Conf. Proc. IEEE Eng. Med. Biol. Soc., 2004, 7, 5411–5414 Search PubMed
- G. R. Christie, P. M. F. Nielsen, S. A. Blackett, C. P. Bradley and P. J. Hunter, Philos. Trans. R. Soc. London, Ser. A, 2009, 367, 1869–1884 CrossRef
- P. M. A. Sloot and A. G. Hoekstra, Briefings Bioinf., 2010, 11, 142–152 Search PubMed
- J. O. Dada, I. Spasić, N. W. Paton and P. Mendes, Bioinformatics, 2010, 26, 932–938 CrossRef CAS
Footnote |
| † Published as part of an iBiology themed issue on Synthetic Biology: Guest Editor Professor John McCarthy. |
| This journal is © The Royal Society of Chemistry 2011 |