Sequencing transcriptomes in toto
Karin S.
Kassahn†
,
Nic
Waddell†
and
Sean M.
Grimmond
*
Queensland Centre for Medical Genomics, Institute for Molecular Bioscience, Brisbane, Australia. E-mail: s.grimmond@uq.edu.au
First published on 4th February 2011
Abstract
The development of next-generation sequencing technologies has enabled the transcriptome to be measured and characterized at a level which was previously unattainable. Shot gun sequencing of RNAs, or RNA-Seq as it is known, is providing the means to simultaneously survey locus activity, transcript-specific expression, sequence content of transcripts and transcriptome discovery. This article discusses the current state of RNA-Seq, its potential for redefining transcriptomics and some of the challenges associated with this revolutionary technology.
 Karin S. Kassahn | Karin Kassahn's research interests span a broad range of questions in genetics and evolutionary biology. She has pioneered the use of microarray technology to study coral reef ecosystems and has recently joined the Australian cancer genome sequencing initiative as part of the International Cancer Genome Consortium. Her current work focuses on mutation detection in DNA and RNA next-generation sequencing data and the integration of high-throughput sequencing datasets. |
 Nic Waddell | Nic Waddell is a cancer researcher at the Queensland Centre for Medical Genomics. Her research has focused on the identification of genomic aberrations in cancer cells and the classification of tumours based on gene expression and genomic sequence. Her current research is focused on the analysis of next generation sequencing data to identify biomarkers and alternative treatment strategies in cancer. |
 Sean M. Grimmond | Sean Grimmond is Director of the Queensland Centre for Medical Genomics, at the Institute for Molecular Bioscience, The University of Queensland. His research focuses on defining the underlying genetic networks controlling tumorigenesis and mammalian development through whole genome, transcriptome and epigenome sequencing. |
Insight, innovation, integrationThe transcriptome describes the actively expressed genes in a given sample. A survey of the transcriptome is thus crucial for understanding the functional output of the genome in biological systems. Next-generation sequencing of the transcriptome, or RNA-Seq, enables the detection of transcription at an unprecedented scale allowing single nucleotide resolution. It enables identification of novel transcripts and splice variants and can be used to detect allele specific expression, the expression of simple mutations and fusion genes. RNA-Seq approaches are being applied in the study of human diseases including cancer and complement many recent large-scale genome sequencing initiatives. This review describes the current state-of-the-art in transcriptome sequencing and discusses opportunities and challenges when applying this technology. |
Introduction
Over the past decade, there has been a dramatic change in our understanding of transcription from mammalian genomes. Once thought to produce a single transcript per locus, it is now clear that higher eukaryotic genomes are encoded such that a single gene can produce multiple transcripts, dramatically increasing the scale and scope of the genome's transcriptomic and proteomic output. The first detailed report on the potential scale of this transcriptome complexity in mouse and human was compiled by the international Functional ANnoTation Of the Mammals (FANTOM) consortia,1–3 and has been recently verified by the ENCODE project.4 These studies conclusively showed that a combination of alternative splicing, alternative terminator usage, and the employment of 2–3 different promoters per gene drives this expanded repertoire of transcripts (Fig. 1). | ||
| Fig. 1 A diagram showing a USCS genome browser view with gene model, splicing events and wiggle plot. RNA-Seq can be used to quantify gene expression and to identify known and novel splicing events. For example, PSMA4, a gene encoding a subunit of the proteasome complex, is represented by three isoforms in the current RefSeq gene models and by one in the Ensembl gene models. All junctions describing the Ensembl isoform were identified by individual reads mapping across these known junctions (track: observed junctions). If paired-end data is available, the mapping positions of the forward and reverse reads in conjunction with the expected library insert size can be used to predict junctions (track: predicted junctions). The same strategy can also predict novel exon combinations (track: novel junctions). In this example, many of the novel exon combinations are supported by AceView gene models; e.g. the AceView gene models predict an isoform PSMA4.eApr07 which skips exon 5 of the Ensembl transcript ENST00000044462 and the RNA-Seq paired-end data provides evidence that this isoform is expressed. The wiggle plot (positive strand hits) shows the read density across the gene and, in combination with the junction information discussed above, informs about exon usage. In this particular example, expression was largely restricted to known exons, but in other instances RNA-Seq has become a powerful tool to identify novel transcription. For simplicity, only single reads (or read pairs) are shown to represent each junction. | ||
More recently still, our understanding of transcriptional complexity has been further expanded by the discovery of many transcripts which lack an open reading frame.5,6 The function of non-coding RNAs and their role in cis- and trans-regulation of gene activity is now well established; with examples of sense-antisense expression, the expression of long-noncoding RNAs, and the generation of microRNAs (or miRNAs). The latter are now widely reported in the literature and have been shown to interfere with translation of specific targets and potentially play roles in defining locus boundaries and modifying chromatin status.
Given these recent advances in our understanding of transcriptional complexity in mammals, it is commonly acknowledged that the reference transcriptomes in public databases such as Ensembl or UCSC represent only a fraction of the actual transcript diversity of an organism or even that of a given cell. Based on the amount of transcription from unannotated regions that is commonly observed in RNA-Seq experiments,7–9 the full transcript repertoire may be 25% greater than what is represented by current gene models. In this context, RNA-Seq offers exciting opportunities to quantify gene expression as well as to discover novel, previously undescribed splice variants. As such, RNA sequencing of mRNA was first used to map transcribed sequences in the yeast genome10 and since has been applied to a variety of systems.
The RNA-Seq revolution
Traditionally, microarray expression profiling has been recognized as the premier tool for correlating gene activity and phenotype and allowing rapid discovery of gene pathways involved in biological processes and pathological states. Despite these successes, the capacity of array-based technologies to put recently found transcriptional complexity into a biological context has been constrained by the limitations in array technology. These include: (i) the requirement of extensive prior knowledge of the transcriptome for successful chip design, (ii) the requirement for suitable sequence content in target sequences to ensure clear hybridization results, (iii) the challenge that homologous target sequences cross-hybridize to give non-specific signals, (iv) the difficulty in identifying changes in exon usage as seen with alternative splicing and (v) the limits of sensitivity for rarely expressed transcripts.With the advent of massively parallel next-generation sequencing, it is now possible to assay transcription at a level not previously practicable. For example, RNA quantification based on RNA-Seq is thought to have a greater dynamic range compared to array-based approaches because read counts do not suffer from the same saturation and sensitivity limitations as array fluorescence signals.11,12 Furthermore, compared to array-based approaches, RNA-Seq has the advantage that novel mRNAs, alternative start and polyadenylation sites and splicing events such as exon skipping, alternative 5′ and 3′ splice sites and novel exon usage can be detected.8,10,13 In this review we will describe the options available for RNA-Seq, show how RNA-Seq is being utilized and discuss the advantages and challenges of the technology.
Options for performing RNA-Seq
There are three major sequencing platforms currently available, the SOLiD™ System from Applied Biosystems (Life Technologies), the Genome Analyzer IIx and HiSeq from Illumina and the 454 Genome Sequencer FLX System from Roche. The latter sequencing platform differs from the former two in that it yields significantly fewer sequencing reads per run, however the read lengths are significantly longer. All of the platforms can be applied to genome, epigenome or transcriptome sequencing, although 454 sequencing is much less commonly used for large-scale genome or transcriptome sequencing due to its limited throughput and the associated higher sequencing costs. The basic RNA-Seq workflow for each platform is similar and is comprised of RNA extraction, generally followed by depletion of ribosomal RNA or enrichment of target RNA (e.g. polyA capture or size selection of short RNAs), library preparation, sequencing, mapping of sequence reads to a reference genome and bioinformatic analysis (Fig. 2). Ribosomal RNA represents the majority of RNA within a cell (∼90%), therefore to avoid sequencing predominantly the rRNA it is necessary to first deplete the rRNA or to enrich for the desired RNA population by methods such as polyA enrichment. The library preparation differs slightly depending on the platform. In the case of the SOLiD™ System, RNA is fragmented to a desired size prior to ligation of adapters. A reverse transcription reaction is performed and the resulting cDNA is purified, size selected, then amplified. The amplified DNA is purified and the yield and size distribution of the product is assessed prior to sequencing. There have been some recent advances made to improve RNA-Seq. For example, barcode sequences may be incorporated to distinguish individual samples and thus allow multiple samples to be sequenced in combination. Another advancement is paired-end sequencing, whereby sequencing of either end of an insert of a selected length allows for confirmation and refinement of splicing events (Fig. 1). Libraries may be prepared so that information regarding the original strand of the transcript is preserved (stranded protocols) or where such information is lost during cDNA synthesis (unstranded protocols). In order to identify sense-antisense expression or to characterize novel exons, the use of stranded protocols is preferred. Alternative library preparation methods specifically target small RNAs, such as miRNA. Finally, it is important to consider the desired target sequences when choosing a library preparation protocol as some methods will not detect non-polyadenylated mRNAs.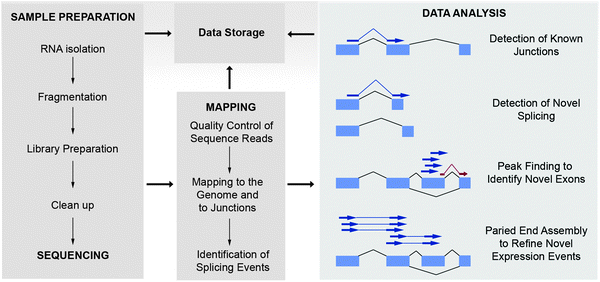 | ||
| Fig. 2 A schematic overview of the steps involved in RNA-Seq. RNA is extracted and incorporated into a library prior to purification and sequencing. The sequence reads undergo quality control and are then mapped to a reference genome and to a library of exon junctions. The expression level of transcripts is determined using tag counts and analyses are performed to determine known and novel splicing events. Paired-end sequencing can be used to validate and refine novel expression events detected with RNA fragment libraries. | ||
The volume of data which is generated from a single RNA-Seq run is large and significant computing infrastructure is necessary to store, map and analyze these data, although future advances in compute technology are likely to meet the demands of RNA-Seq experiments very soon. Nevertheless, at the present time, many laboratories do not have access to the computing infrastructure and bioinformatic resources necessary to perform next-generation sequencing. As an alternative to local compute clusters, analysis pipelines that use cloud computing are being developed. Crossbow (http://bowtie-bio.sourceforge.net/crossbow/index.shtml) is a pipeline that runs the read aligner Bowtie14 and the SNP calling software, SoapSNP15 on the Amazon EC2 compute cluster. As next-generation sequencing technologies become more readily available and more widely utilized, we expect that there will be an increase in the use and development of such virtual compute environments. Data security for computing in cloud environments will become of paramount importance, especially for studies handling sensitive patient data.
Estimating the abundance of known transcripts and detection of splice variants
In RNA-Seq experiments, transcript expression levels are typically inferred by the number of tags that describe a certain transcript sequence. How to accurately quantify transcript expression levels from RNA-Seq data is an active area of research16 and different tools for RNA quantification are currently being developed. To describe the average transcript activity within a sample, RPKM, or the number of mapped reads per kilobase of exon per million mapped reads,7 has become a common approach. More recently, modified RPKM values or read counts that take the mappability of different transcript regions into account have been used.17 This approach removes biases in RNA quantification that are purely a result of the different level of ‘uniqueness’ of different transcripts.17 Normalized gene locus expression levels may then be determined using software such as ERANGE.7 For determining differential expression between two RNA-Seq samples, the application DEGseq may be used,18 although many groups also develop their own in-house analysis pipelines.19Several factors have been shown to affect quantification, such as the depth of sequencing and the length of the transcript to be quantified. The depth of sequencing will determine the ability to detect and quantify rare transcripts,7 while the length of the transcript affects the probability with which tags are detected and hence the statistical power to detect differential expression.20 Specifically, longer transcripts are over-represented among differentially expressed transcripts compared to shorter transcripts.20 Given that some gene classes tend to be composed of genes of longer length, this transcript length bias may affect downstream interpretation of the types of pathways dysregulated in comparisons of different experimental treatments. Development of more-sophisticated statistical analysis approaches for RNA-Seq data will thus be of paramount importance.
These challenges left aside, RNA expression quantification from RNA-Seq is typically performed in the following analysis steps. Initially, RNA-Seq reads are mapped to the genome as well as to a library of exon junctions. To this end, several software applications are available with RNA-Mate having become a popular choice for mapping SOLiD sequencing reads,21 while BWA22 and Bowtie23 are popular choices for mapping Illumina sequencing reads. Novel exons may be identified by the presence of a cluster of tags that map outside known exons.
Given our incomplete knowledge of eukaryotic transcriptome diversity, the library of exon junctions against which sequence reads are mapped in a first instance commonly includes all theoretically possible exon combinations for a given gene locus, so that novel combinations of known exons can be identified in the sample. Nevertheless, mapping will still fail if the read spans a junction that is not represented in the junction library or one that involves one or two novel exons. To overcome these challenges, the software QPALMA was developed for de novo junction mapping.24 This software uses information from known splice sites, including intron length models, to train a support vector machine that can then be applied to identify novel junctions in a test sample. However, the software requires the availability of a set of known exon junctions for training and has long computational run times. More recently, an ab initio method for the detection of splice sites was developed, TopHat, that does not rely on a training set of known splice sites and which has favorable computational run times.25 TopHat first maps reads to the genome and assembles sequence reads into putative exons based on read coverage in a stretch of contiguous sequence. Exon junctions are then identified by first comparing against known splice junctions and then considering all pairings of known splice donor and acceptor sites within a region. Non-canonical donor and acceptor sites are not considered at present. In this context, the availability of paired-end and mate pair data can help reduce the number of possible splice combinations that need to be considered and thus significantly speed up analysis time as well as potentially provide more accurate exon junction predictions. Finally, a comprehensive RNA-Seq analysis package, Alexa-Seq, has been recently published that allows identification and quantification of alternative transcripts.26 Alexa-Seq uses an internal database to store ‘expression features’ that can be defined by any gene model. RNA-Seq reads are then mapped against these features to identify known and predicted transcript isoforms. Differentially expressed features can also be determined using this software; this feature-based approach can be used to determine the relative use of different transcript isoforms in a given system.
An alternative approach to transcriptome discovery is based on the de novoassembly of transcriptomes and would appear particularly useful for the identification of exon skipping, intron retention and novel, alternative splicing events. Recent advances in this area use de Bruijn graphs and overlapping k-mers to assemble short reads into contigs.27,28 However, sequencing errors significantly complicate the analysis and also result in very long computational analysis times. A recent software application, ABySS, tries to reduce the computational load by using a parallel implementation of the algorithm.27 The approach was able to assemble approximately 30 million bases of human transcriptome sequence into some 800![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 contigs, representing some 1 percent of the genome.27 Despite these advances, the de novoassembly of mammalian transcriptomes remains challenging and significant advances in this area may have to await a new generation of next-gen sequencing technologies capable of producing longer sequence reads.
000 contigs, representing some 1 percent of the genome.27 Despite these advances, the de novoassembly of mammalian transcriptomes remains challenging and significant advances in this area may have to await a new generation of next-gen sequencing technologies capable of producing longer sequence reads.
The adaptation and application of RNA-Seq approaches
RNA-Seq approaches have been adapted in a broad range of transcriptomic studies. For example, RNA-Seq data has been used to detect single nucleotide variations in expressed exons.29Allele-specific expression (ASE) which is an important transcriptional regulation mechanism involved in processes such as X-inactivation30 and gene imprinting,31 may be associated with these SNP variants,32 causing unequal expression of the two alleles. Current PCR and array methods to detect ASE are difficult to implement due to PCR bias and cross hybridization, respectively. RNA-Seq of padlock captured single nucleotide polymorphisms targeting 27000 SNPs found that 11–22% of the heterozygous mRNA-associated SNPs showed ASE in 4 different cell lines.33 An alternate strategy using a paired-end sequencing approach was able to attain enough sequencing depth to assay 1371 transcripts for ASE and the authors were able to identify ASE of a variant which in a GWAS study had previously been associated with multiple sclerosis.34 Nevertheless, some biases in ASE quantification potentially remain in RNA-Seq experiments, as read mapping can be affected by sequence variation such as SNPs.35 The authors show that masking the reference sequence for known SNPs in combination with simulation studies to identify regions prone to mismapping can be used effectively to filter out false positive ASE candidates and this approach was used to identify ASE across a cohort of HapMap individuals.19 In contrast, the extent to which such approaches underestimate ASE due to mapping artifacts is not known to date.
A large proportion of the published RNA-Seq studies are from sequencing small RNA cDNA libraries derived from the small size selected fraction of total RNA. MicroRNA and other small non-coding RNAs are involved in transcriptional regulation.36 RNA-Seq has been used to characterize novel and known short RNAs in a variety of systems37–39 and tools for discovering new miRNAs in deep sequencing data have been developed.40,41 RNA-Seq of the short RNAs has also led to the identification of a new class of small RNAs called tiRNAs or transcription initiation RNAs.42 These tiRNAs are approximately 18nt in length and associated with highly expressed RNA transcripts and RNA polymerase II binding sites.
RNA-Seq has also been applied to gain a better insight into the processes of transcription and translation. Ribosome profiling, whereby sequencing is performed on ribosomal protected transcripts which are actively being translated has the potential to quantify the proteins that a cell produces and provides a unique opportunity to assay protein translation.43 Global run-on sequencing (GRO-seq) is able to map and quantify nascent RNA which is associated with transcriptionally engaged polymerases.44 This assay highlighted the complexity of transcription by revealing the presence of transcriptionally active polymerases situated upstream and in opposite orientation to the gene. RNAs produced by these divergent polymerases were identified in low concentrations in the small RNA fraction and likely have a role in transcriptional regulation.
Current challenges in RNA-Seq
While RNA-Seq has already found a diverse range of applications, many challenges remain. To address some of these challenges the major biotechnology companies are continuously seeking to improve their sequencing chemistry to increase accuracy and read length and decrease run time. Longer read lengths and paired-end or long-mate pair approaches are expected to help with read mapping. It is common that only 40% of reads of an individual RNA-Seq experiment map to the reference genome. The remaining reads likely represent chimeric PCR products, empty adapters, and other library artifacts. However, some of these unmapped reads may represent: (i) novel transcripts and will thus only be discovered using alternative approaches such as de novo transcriptome assembly; (ii) sequence variants which can significantly affect the accuracy of read mapping; (iii) fusion transcripts as the result of chromosome structural rearrangements.45,46 Longer read lengths and paired-end or long-mate pair approaches are expected to help with read mapping, however it is presently unknown to what extent approaches that rely on mapping against a reference genome will be able to identify sequence variants in highly rearranged genomes, such as cancer genomes. The quality of the reference genome sequence and the junction library against which the reads are mapped are also of paramount importance in this context.With regards to quantifying the expression levels of individual transcripts, diagnostic junctions are only indicative of the presence of a specific isoform, but on their own can not provide accurate isoform quantification. For example, when diagnostic events for more than one variant transcript are identified it is impossible to be certain they haven't both arisen from a novel transcript that has not been previously observed. Furthermore, because some alternative exons are found in multiple transcripts, reads mapping to such exons can not be unambiguously assigned to a single RNA.
Technical challenges associated with the library generation method also need to be resolved. It is well recognized that biases and sequence errors can be introduced during RNA fragmentation and cDNA synthesis. Another area requiring improvement is the amount of starting material required for RNA-Seq experiments. Recent studies involving direct sequencing of RNA47 and single blastocyst transcriptome sequencing48 have shown the potential of single molecule sequencing and new library approaches to improve input requirements.
The future of RNA-Seq
RNA-Seq has already had a significant impact on the detection of post-transcriptional modifications and the characterization of novel transcripts and splicing events. It has enabled the discovery and analysis of different aspects of the transcriptome and has shed light on the processes of transcription and translation. While many challenges remain, researchers are continuously developing novel ways in which to apply RNA-Seq and new tools and pipelines to analyze the data. RNA-Seq requires a large computing infrastructure and bioinformatics support, but as these facilities and tools become more readily available the impact of RNA-Seq will most certainly increase. Several recent large scale genome sequencing initiatives already routinely include RNA-Seq in their analysis pipelines, including the International HapMap project, the ENCODE consortium and the International Cancer Genome Consortium (ICGC).49 In the not too distant future we will hence have a catalogue of transcriptome variation across populations, cell-types, and disease-states at an unprecedented resolution. In many of these studies RNA-Seq experiments are complemented by genome re-sequencing and methylation studies so that an integrated view of the genomic variants and their expression and pathogenicity can be obtained. In the near future, we are also likely to see RNA-Seq experiments being applied more-broadly to non-model organisms. Furthermore, the impending arrival of long read (1–10 kb) reads from single molecule sequencing stands to revolutionize transcriptomics, as whole transcripts will be sequenced in a single pass. Based on these observations, we predict that during the next few years RNA-Seq will continue to grow rapidly and become the key tool in discovery, classification and differential expression analysis of the transcriptome in all aspects of the biosciences.References
- P. Carninci, T. Kasukawa, S. Katayama, J. Gough, M. C. Frith and N. Maeda, et al. , Science, 2005, 309, 1559–1563 CrossRef CAS.
- Y. Okazaki, M. Furuno, T. Kasukawa, J. Adachi, H. Bono and S. Kondo, et al. , Nature, 2002, 420, 563–573 CrossRef.
- H. Suzuki, A. R. Forrest, E. van Nimwegen, C. O. Daub, P. J. Balwierz and K. M. Irvine, et al. , Nat. Genet., 2009, 41, 553–562 CrossRef CAS.
- E. Birney, J. A. Stamatoyannopoulos, A. Dutta, R. Guigo, T. R. Gingeras and E. H. Margulies, et al. , Nature, 2007, 447, 799–816 CrossRef.
- J. S. Mattick and I. V. Makunin, Hum. Mol. Genet., 2006, 15(Spec No 1), R17–29 CrossRef CAS.
- P. P. Amaral, M. E. Dinger, T. R. Mercer and J. S. Mattick, Science, 2008, 319, 1787–1789 CrossRef CAS.
- A. Mortazavi, B. A. Williams, K. McCue, L. Schaeffer and B. Wold, Nat. Methods, 2008, 5, 621–628 CrossRef CAS.
- M. Sultan, M. Schulz, H. Richard, A. Magen, A. Klingenhoff and M. Scherf, et al. , Science, 2008, 321, 956–960 CrossRef CAS.
- N. Cloonan, A. Forrest, G. Kolle, B. Gardiner, G. Faulkner and M. Brown, et al. , Nat. Methods, 2008, 5, 613–619 CrossRef CAS.
- U. Nagalakshmi, Z. Wang, K. Waern, C. Shou, D. Raha and M. Gerstein, et al. , Science, 2008, 320, 1344–1349 CrossRef CAS.
- J. C. Marioni, C. E. Mason, S. M. Mane, M. Stephens and Y. Gilad, Genome Res., 2008, 18, 1509–1517 CrossRef CAS.
- Z. Wang, M. Gerstein and M. Snyder, Nat. Rev. Genet., 2009, 10, 57–63 CrossRef CAS.
- B. T. Wilhelm, S. Marguerat, S. Watt, F. Schubert, V. Wood and I. Goodhead, et al. , Nature, 2008, 453, 1239–1243 CrossRef CAS.
- B. Langmead, C. Trapnell, M. Pop and S. L. Salzberg, GenomeBiology, 2009, 10, R25 CrossRef.
- B. Langmead, M. C. Schatz, J. Lin, M. Pop and S. L. Salzberg, GenomeBiology, 2009, 10, R134 CrossRef.
- S. Pepke, B. Wold and A. Mortazavi, Nat. Methods, 2009, 6, S22–32 CrossRef CAS.
- R. Koehler, H. Issac, N. Cloonan and S. M. Grimmond, Bioinformatics, 2010 DOI:10.1093/bioinformatics/btq640.
- L. Wang, Z. Feng, X. Wang and X. Zhang, Bioinformatics, 2009, 26, 136–138.
- J. K. Pickrell, J. C. Marioni, A. A. Pai, J. F. Degner, B. E. Engelhardt and E. Nkadori, et al. , Nature, 2010, 464, 768–772 CrossRef CAS.
- A. Oshlack and M. Wakefield, Biol. Direct, 2009, 4, 14 Search PubMed.
- N. Cloonan, Q. Xu, G. J. Faulkner, D. F. Taylor, D. T. P. Tang and G. Kolle, et al. , Bioinformatics, 2009, 25, 2615–2616 CrossRef CAS.
- H. Li and R. Durbin, Bioinformatics, 2009, 25, 1754–1760 CrossRef CAS.
- B. Langmead, C. Trapnell, M. Pop and S. Salzberg, GenomeBiology, 2009, 10, R25 CrossRef.
- F. De Bona, S. Ossowski, K. Schneeberger and G. Ratsch, Bioinformatics, 2008, 24, i174–180 CrossRef.
- C. Trapnell, L. Pachter and S. L. Salzberg, Bioinformatics, 2009, 25, 1105–1111 CrossRef CAS.
- M. Griffith, O. L. Griffith, J. Mwenifumbo, R. Goya, A. S. Morrissy and R. D. Morin, et al. , Nat. Methods, 2010, 7, 843–847 CrossRef CAS.
- I. Birol, S. D. Jackman, C. B. Nielsen, J. Q. Qian, R. Varhol and G. Stazyk, et al. , Bioinformatics, 2009, 25, 2872–2877 CrossRef CAS.
- D. R. Zerbino and E. Birney, Genome Res., 2008, 18, 821–829 CrossRef CAS.
- I. Chepelev, G. Wei, Q. S. Tang and K. J. Zhao, Nucleic Acids Res., 2009, 37, e106 CrossRef.
- J. C. Chow, Z. Yen, S. M. Ziesche and C. J. Brown, Annu. Rev. Genomics Hum. Genet., 2005, 6, 69–92 CrossRef CAS.
- P. P. Luedi, F. S. Dietrich, J. R. Weidman, J. M. Bosko, R. L. Jirtle and A. J. Hartemink, Genome Res., 2007, 17, 1723–1730 CrossRef CAS.
- L. A. Hindorff, P. Sethupathy, H. A. Junkins, E. M. Ramos, J. P. Mehta and F. S. Collins, et al. , Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 9362–9367 CrossRef CAS.
- K. Zhang, J. B. Li, Y. Gao, D. Egli, B. Xie and J. Deng, et al. , Nat. Methods, 2009, 6, 613–618 CrossRef CAS.
- G. A. Heap, J. H. M. Yang, K. Downes, B. C. Healy, K. A. Hunt and N. Bockett, et al. , Hum. Mol. Genet., 2010, 19, 122–134 CrossRef CAS.
- J. F. Degner, J. C. Marioni, A. A. Pai, J. K. Pickrell, E. Nkadori and Y. Gilad, et al. , Bioinformatics, 2009, 25, 3207–3212 CrossRef CAS.
- W. Filipowicz, L. Jaskiewicz, F. A. Kolb and R. S. Pillai, Curr. Opin. Struct. Biol., 2005, 15, 331–341 CrossRef CAS.
- M. Bar, S. K. Wyman, B. R. Fritz, J. L. Qi, K. S. Garg and R. K. Parkin, et al. , Stem Cells, 2008, 26, 2496–2505 Search PubMed.
- C. Ender, A. Krek, M. R. Friedlander, M. Beitzinger, L. Weinmann and W. Chen, et al. , Mol. Cell, 2008, 32, 519–528 CrossRef CAS.
- Q. H. Zhu, A. Spriggs, L. Matthew, L. J. Fan, G. Kennedy and F. Gubler, et al. , Genome Res., 2008, 18, 1456–1465 CrossRef CAS.
- M. R. Friedlander, W. Chen, C. Adamidi, J. Maaskola, R. Einspanier and S. Knespel, et al. , Nat. Biotechnol., 2008, 26, 407–415 CrossRef.
- M. Hackenberg, M. Sturm, D. Langenberger, J. M. Falcon-Perez and A. M. Aransay, Nucleic Acids Res., 2009, 37, W68–W76 CrossRef CAS.
- R. J. Taft, E. A. Glazov, N. Cloonan, C. Simons, S. Stephen and G. J. Faulkner, et al. , Nat. Genet., 2009, 41, 572–578 CrossRef CAS.
- N. T. Ingolia, S. Ghaemmaghami, J. R. S. Newman and J. S. Weissman, Science, 2009, 324, 218–223 CrossRef CAS.
- L. J. Core, J. J. Waterfall and J. T. Lis, Science, 2008, 322, 1845–1848 CrossRef CAS.
- M. F. Berger, J. Z. Levin, K. Vijayendran, A. Sivachenko, X. Adiconis and J. Maguire, et al. , Genome Res., 2010, 20, 413–427 CrossRef CAS.
- J. Z. Levin, M. F. Berger, X. Adiconis, P. Rogov, A. Melnikov and T. Fennell, et al. , GenomeBiology, 2009, 10, R115 CrossRef.
- F. Ozsolak, A. R. Platt, D. R. Jones, J. G. Reifenberger, L. E. Sass and P. McInerney, et al. , Nature, 2009, 461, 814–U873 CrossRef CAS.
- F. C. Tang, C. Barbacioru, Y. Z. Wang, E. Nordman, C. Lee and N. L. Xu, et al. , Nat. Methods, 2009, 6, 377–U386 CrossRef CAS.
- The International Cancer Genome Consortium, Nature, 2010, 464, 993–998 CrossRef.
Footnote |
| † These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2011 |