Production of liquid hydrocarbon fuels by catalytic conversion of biomass-derived levulinic acid†
Drew J.
Braden
,
Carlos A.
Henao
,
Jacob
Heltzel
,
Christos C.
Maravelias
and
James A.
Dumesic
*
Department of Chemical Engineering, University of Wisconsin-Madison, Madison, Wisconsin 53706, USA
First published on 12th April 2011
Abstract
Levulinic acid derived from ligno-cellulosic biomass has the potential to be utilized as a platform intermediate molecule in the production of renewable liquid fuels for the transportation sector. Herein we report a catalytic process for the conversion of levulinic acid to γ-valerolactone (GVL) using a RuRe/C catalyst that is significantly more active than a traditional Ru/C catalyst. The bimetallic catalyst is active for the reduction of levulinic acid and simultaneous decomposition of formic acid with good stability in the presence of sulfuric acid, the homogeneous catalyst commonly used in the production of levulinic acid from carbohydrates. Results from techno-economic analyses show that the integration of this new process with catalytic decarboxylation of GVL to butene followed by alkene oligomerization could provide a cost-effective route for the conversion of ligno-cellulosic biomass to liquid hydrocarbon fuels.
Introduction
The production of liquid hydrocarbon fuels from renewably grown ligno-cellulosic biomass is a challenge that has been the central focus of recent studies.1 In this respect, strategies for using chemical catalysts to produce liquid hydrocarbon biofuels have focused on identifying chemical transformations of carbohydrate-based molecules that accomplish two general goals: (i) selective deoxygenation of carbohydrates for the production of functional intermediates, and (ii) C–C coupling between these intermediates for the production of higher molecular weight compounds for use in transportation fuels. In the present paper, we address the role of levulinic acid as a functional intermediate for the conversion of cellulose to liquid hydrocarbon fuels. More generally, levulinic acid has been identified by Bozell and Petersen2 as one of the top 10 platform molecules for the production in biorefineries of value-added chemicals and liquid transportation fuels. We describe herein a catalytic process for the conversion of levulinic acid to γ-valerolactone (GVL) in the presence of sulfuric acid, thereby providing a new route for the recovery and recycling of the sulfuric acid used in the cellulose deconstruction step; and, we present results from techno-economic analyses to show that the integration of this new process with catalytic decarboxylation of GVL to butene followed by alkene oligomerization could provide a cost-effective route for the conversion of ligno-cellulosic biomass to liquid hydrocarbon fuels.Previous work has shown that levulinic acid has the potential to be produced at cost effective margins3 by the hydrolysis and subsequent dehydration of hexose sugars, the most prevalent carbohydrate component of ligno-cellulosic biomass, at molar yields higher than 60%,4–6 accompanied by an equal molar amount of formic acid. Thus, catalyst systems have been developed for the conversion of levulinic acid into fuel additives such as GVL, ethyl levulinate (EL), and methyltetrahydrofuran (MTHF).3,7,8 However, because of the inherent blending limits of these fuel additives in hydrocarbon fuels, studies have also addressed catalytic reactions for the conversion of GVL to higher molecular weight hydrocarbons that are fully compatible with gasoline, diesel and jet fuels used today.6,9,10 Serrano–Ruiz and co-workers reported the production of 5-nonanone-based diesel fuel components by the conversion of GVL to valeric acid (VA) combined with ketonization.6 Lange and co-workers reported a strategy for the production of VA-based fuel components, such as alkyl (mono/di) valeric esters, by combination of an intermediate VA production step followed by esterification with small poly-alcohols.9 More recently, Bond, et al. reported a dual reactor approach for the production of C8–C24alkenes from an aqueous solution of GVL, involving decarboxylation of GVL to butene over an acidic silica-alumina catalyst followed by oligomerization of butene isomers over an acidic ion-exchange catalyst (Amberlyst-70).10
While the above catalytic processes are promising approaches for the conversion of GVL to liquid transportation fuels, they all rely on the cost effective production of GVL from biomass-derived levulinic acid. Although several catalyst systems have been reported for the reduction of levulinic acid to GVL,11–14 most studies have used idealistic feeds of levulinic acid and processing conditions that are not amenable for use in a cascade process operating with streams of levulinic acid derived from ligno-cellulosic biomass. Thus, we have studied heterogeneous catalysts for the production of GVL from levulinic acid feedstocks produced by the deconstruction of ligno-cellulosic biomass in aqueous solutions of sulfuric acid, and we have developed an integrated strategy for the conversion of biomass to alkene oligomers. Importantly, we have carried out techno-economic studies and sensitivity analyses that provide insights into the parameters that govern this chemical-catalytic strategy.
Results
Levulinic acid reduction over a Ru/C catalyst
The production of levulinic acid from cellulose is most commonly accomplished using aqueous sulfuric acid as the hydrolysis catalyst,5,15 and thus, we aimed at achieving the catalytic reduction of levulinic acid to GVL, combined with decomposition of the formic acid co-product to H2 and CO2, both in the presence of sulfuric acid, as shown in Fig. 1. While reports in literature suggest that ruthenium should be an active metal for the heterogeneously catalyzed reduction of levulinic acid to GVL,12,13 previous studies have reported that ruthenium may not be suitable for use in the presence of sulfuric acid. Osada, et al.16 reported that a Ru/TiO2 catalyst, used for the gasification of lignin to methane, deactivated significantly after soaking the catalyst in a sulfuric acid solution. Similarly, Miyazawa, et al.17 reported that Ru/C, used in combination with Amberlyst-70 for the conversion of glycerol to 1,2-propanediol, was deactivated by sulfur and SO2 evolved from Amberlyst-70 during reaction. Conversely, ruthenium has been identified as a catalytic component that possesses high activity for hydrodesulfurization and hydrogenation reactions used in hydrotreating schemes.18,19 Accordingly, we started our studies using a 5 wt% Ru/C catalyst to carry out the decomposition of formic acid to H2 and CO2 and the simultaneous reduction of levulinic acid to produce GVL.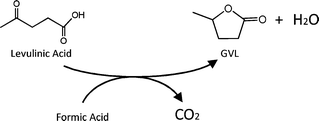 | ||
| Fig. 1 Reaction scheme for formic acid decomposition and levulinic acid reduction for the production of GVL. | ||
Fig. 2 presents the catalytic turnover frequencies (moles species converted per mole of surface metal sites per time) versus time-on-stream for the conversion of levulinic acid and formic acid over a 5 wt% Ru/C catalyst in a flow reactor. Initially, a feed of levulinic and formic acid (0.3 mol L−1 of each acid) was passed over the 5% Ru/C catalyst at 423 K and 35 bar without sulfuric acid in the feed. A minimal co-feed of H2 (5 cm3 min−1 (STP)) was used to ensure that the catalyst remained reduced. Then, after 50 h time-on-stream, sulfuric acid was added to the feed at a level of 0.5 mol L−1. It can be seen that the catalytic activity decreased slowly from the beginning of the experiment until the addition of sulfuric acid (dashed line), followed by a rapid decrease in catalytic activity to a significantly lower level (approximately 3% of the original activity) upon addition of sulfuric acid. Although operating at a low turnover frequency, the results in Fig. 2 demonstrate for the first time that a Ru/C catalyst can operate with stable activity for the production of GVL from levulinic acid (and decomposition of formic acid) in the presence of sulfuric acid. The weight hourly space velocity (WHSV) was decreased by a factor of 3.3 after sulfuric acid addition (at 100 h time-on-stream) to compensate for low levulinic and formic acid conversions (<5% conversion). During these experiments, we observed the formation of an anhydride species comprised of levulinic acid and sulfuric acid, formed through a reversible and catalyst-independent reaction at low WHSV. Similar sulfuric acid anhydride species have been reported in literature20 and the levulinic acid-sulfuric acid anhydride species formed in the current study were confirmed using liquid chromatography coupled with mass spectrometry. High selectivities for production of GVL (>98%) were observed before addition of sulfuric acid, which decreased (to 60–70%) after addition of sulfuric acid due to formation of the aforementioned anhydride species. Measurable quantities were not detected in the reactor effluent of 4-hydroxyvaleric acid, resulting from the partial hydrogenation of levulinic acid, or angelica lactones, resulting from the cyclization and dehydration of levulinic acid.21 After taking into consideration the formation of the anhydride species, the material balance for sulfuric acid closed to within 99%; no measurable amounts of H2S were observed. Analysis of the gas phase showed that the formic acid was almost completely converted to CO2 (>98%), and only trace amounts of methane were observed. Following the addition of sulfuric acid, the material balance for levulinic acid closed to within 95%. The levulinic and formic acid conversions for all points measured were approximately between 5–35% and 15–50%, respectively. No measurable quantities (>1 ppm) of catalyst metals were observed in the liquid reactor effluent, as measured by inductively coupled plasma (ICP) analysis.
![Turnover frequency versus time-on-stream for flow reactor studies of the conversion of formic acid (triangles) and levulinic acid (squares) at 423 K and 35 bar over a Ru/C catalyst (aqueous feed composition: [formic acid] = [levulinic acid] = 0.3 mol L−1. Sulfuric acid was added to the aqueous feed at 50 h time-on-stream (dashed line) [H2SO4] = 0.5 mol L−1).](/image/article/2011/GC/c1gc15047b/c1gc15047b-f2.gif) | ||
| Fig. 2 Turnover frequency versus time-on-stream for flow reactor studies of the conversion of formic acid (triangles) and levulinic acid (squares) at 423 K and 35 bar over a Ru/C catalyst (aqueous feed composition: [formic acid] = [levulinic acid] = 0.3 mol L−1. Sulfuric acid was added to the aqueous feed at 50 h time-on-stream (dashed line) [H2SO4] = 0.5 mol L−1). | ||
Levulinic acid reduction over a RuRe/C catalyst
Based on the stable catalytic activity measured for the conversion of levulinic acid and formic acid in the presence of sulfuric acid over the Ru/C catalyst, we worked towards the identification of other Ru-based materials that might show higher catalytic activity. Bimetallic catalysts that couple typical hydrogenation metals (e.g., Pt, Pd, Ru) with more oxophilic metals (e.g., Re, Mo) have proven to be advantageous for various conversion processes.22 For example, researchers at DuPont demonstrated that Pd–Re and Ru–Re based catalysts are active for the conversion of maleic acid to tetrahydrofuran,23–25 and Pd-Re particles have also been demonstrated to be active for the hydrogenolysis of other carboxylic acids.26 According to the phase diagram for a Ru–Re mixture, the two metals are completely miscible at all temperatures of interest in the current study. Accordingly, we prepared a bimetallic 15 wt% RuRe(3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4)/C catalyst for use in the levulinic reduction reaction.
:4)/C catalyst for use in the levulinic reduction reaction.
The turnover frequencies versus time-on-stream for conversion of levulinic and formic acids (2.2 mol L−1 of each acid) over the RuRe/C catalyst are presented in Fig. 3, in the absence as well as in the presence of sulfuric acid. It can be seen that stable catalytic activity was achieved in the presence of sulfuric acid. In fact, the catalytic activity in the presence of sulfuric acid was higher by a factor of two than in the absence of sulfuric acid. Fig. 3 also shows the rate of GVL formation, from which it can be seen that the RuRe/C catalyst achieves high selectivity for the formation of GVL (>95%) in the presence and in the absence of sulfuric acid. The levulinic acid conversions for all points measured were between 15–40%. Because of the increased activity of the RuRe/C catalyst, reaction kinetics studies of this catalyst were carried out at higher WHSV, and measurable levels of anhydride species comprised of levulinic acid and sulfuric acid were not observed. Formic acid was completely converted for all samples with a high selectivity to CO2 (>99%). The material balance for levulinic acid conversion over the RuRe/C catalyst closed to within 95%, and no measurable quantities of byproducts were observed in the reactor effluent. In addition, no measurable quantities (>1 ppm) of catalyst metals were observed in the liquid effluent streams from the flow reactor. Comparison of the results in Fig. 2 and 3 shows that Re is an effective promoter of Ru for conversion of levulinic and formic acids in the presence of sulfuric acid.
 | ||
| Fig. 3 Turnover frequency versus time-on-stream for the conversion of an equal molar (2.2 mol L−1) solution of levulinic acid (squares) and formic acid (not shown) and the production of GVL (circles) over RuRe/C at 423 K and 35 bar in the presence (solid symbols) and absence (open symbols) of sulfuric acid (0.5 mol L−1). | ||
High conversion catalyst comparison
For the GVL production step to be cost effective on an industrial scale, a high selectivity to GVL must be achieved at a high levulinic acid conversion. Thus, we carried out flow reaction experiments at high levulinic acid conversions (>80%) for both the 5 wt% Ru/C and 15 wt% RuRe(3:4)/C catalysts using a concentrated levulinic and formic acid feed (2.2 mol L−1 of each acid) in the presence of sulfuric acid (0.5 mol L−1) at 423 K. (As discussed below, this feed composition is typical of that expected from the deconstruction of cellulose.) For the high conversion experiments, the steady state GVL production rate for the Ru/C catalyst (0.0028 mmol min−1 g-cat−1) is more than 10 times lower than the steady state GVL production rate for the RuRe/C catalyst (0.034 mmol min−1 g-cat−1). We also note that the steady-state rate for the Ru/C catalyst is significantly lower than the rate measured using the lower concentration feed (0.3 mol L−1) at a lower conversion (approximately 0.02 mmol min−1 g-cat−1).
Catalyst characterization
The Ru/C and RuRe/C catalysts used for the production of GVL were analyzed using various characterization techniques. The number of surface metal sites was determined for reduced Ru/C and RuRe/C catalyst samples by CO chemisorption. The total number of surface metal sites increased from 150 to 247 μmol sites g−1 following the impregnation of Re on the Ru/C catalyst (4:3 molar ratio of Re to Ru).
Temperature programmed reduction (TPR) experiments were carried out on fresh Re/C, Ru/C and RuRe/C catalyst samples. Results for the H2 uptake as a function of temperature are shown in Fig. 4. The H2 uptake for the Ru/C catalyst is centered at approximately 400 K, whereas the Re/C catalyst showed a broader H2 uptake that was centered at a higher temperature (approximately 650–700 K). Interestingly, the RuRe/C catalyst showed a H2 uptake that was centered at approximately 450 K, a temperature that is slightly higher than for Ru/C but much lower than Re/C. This finding suggests that Ru catalyzes the reduction of rhenium oxide. Thus, Ru and Re are suggested to be in intimate contact on the carbon support for the RuRe/C catalyst under reducing conditions.
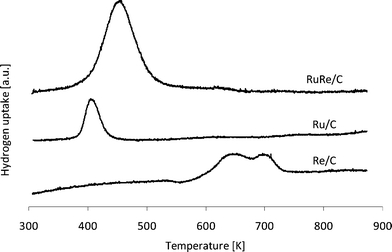 | ||
| Fig. 4 TPR profiles for fresh Re/C, Ru/C and RuRe/C catalyst samples. | ||
X-Ray diffraction (XRD) experiments were carried out on Ru/C and RuRe/C catalyst samples before and after use in flow reaction kinetics experiments, both in the presence and absence of sulfuric acid. Diffraction peaks corresponding to Ru and Re particles were not observed for all catalyst samples measured. Thus, the Ru and Re particles are well-dispersed on the carbon support before and after the reaction.
Integrated biofuel production strategy
Using experimental results for the production of GVL over a RuRe/C catalyst in the presence of sulfuric acid at high conversions of levulinic acid, we developed an integrated biofuel production strategy that involves the complete utilization of ligno-cellulosic biomass. The framework of our chemical catalytic approach is based on the biochemical process model originally developed by the National Renewable Energy Laboratory (NREL) for the conversion of corn stover to ethanol.27–30 A schematic representation for our integrated conversion strategy is presented in Fig. 5, and includes processing steps required to handle each component of biomass (e.g., hemi-cellulose, cellulose and lignin). The hemi-cellulose fraction of the biomass is first removed through dilute acid pretreatment that produces an aqueous stream of hemi-cellulose monomers (mainly xylose when starting with corn stover).31,32 We then pass the solid biomass containing the cellulose and lignin fractions to a reactor for cellulose deconstruction to levulinic and formic acids in aqueous sulfuric acid, followed by our cascade catalytic process for the production of liquid hydrocarbon transportation fuels. The recovered xylose fraction, the insoluble lignin fraction and the solid humins (i.e., insoluble polymerization products formed during cellulose deconstruction) are sent to a boiler/turbogenerator for the production of process heat and electricity. | ||
| Fig. 5 Schematic diagram for the integrated production of liquid alkene oligomers from ligno-cellulosic biomass. | ||
Fig. 6 shows a schematic flowsheet of our cascade process to convert the cellulose portion of ligno-cellulosic biomass to liquid hydrocarbon fuels. The conversion of cellulose to an equal molar mixture of levulinic and formic acids can be carried out (in a batch reactor) by the progressive addition of cellulose at 423 K to an aqueous solution of sulfuric acid (0.5 mol L−1), as recently reported,6 achieving high levels of levulinic and formic acids (>2 mol L−1). The yield of levulinic and formic acids from the cellulose deconstruction is approximately 55%, with the remainder of the partially deoxygenated carbohydrate monomers forming humins.6 Following the cellulose deconstruction step, the insoluble lignin and humins formed during the reaction are separated from the aqueous stream using a settling tank. The aqueous stream of levulinic and formic acids, containing sulfuric acid, can be converted to GVL at essentially quantitative yields over the 15% RuRe(3:4)/C catalyst (catalytic reactor 1) at 423 K. The increased hydrophobic nature of GVL enables its separation from the aqueous layer by liquid–liquid extraction with an acetate solvent, as reported previously.6 We selected butyl acetate over other acetate solvents (e.g., propyl, isobutyl and isoamyl acetates), because it is relatively inexpensive and has adequate selectivity for this extraction process. Results for GVL extraction using butyl acetate as the solvent are reported in the supporting information in Table S4.† Following the extraction of GVL, the sulfuric acid aqueous solution is recycled back to the deconstruction reactor. Essentially all of the butyl acetate (>99%) can be recovered by simple distillation. The GVL product can then be upgraded to alkene oligomers using the dual reactor catalytic conversion process developed by Bond, et al.,10 in which an aqueous solution of GVL (60 wt%) is first decarboxylated to form a mixture of butene isomers over a silica-alumina catalyst (catalytic reactor 2) at 648 K and 36 bar. Essentially all of the water is condensed in a separator at 373–383 K and 36 bar and recycled to be mixed with more GVL. The butene, in the presence of CO2, can then be oligomerized to larger alkene oligomers over an Amberlyst-70 catalyst (catalytic reactor 3) at 443 K and 36 bar. The oligomers can be condensed at ambient conditions leaving a relatively pure steam of CO2 at 36 bar. Based on the study by Bond, et al., we assume that 99% of the GVL produced from levulinic acid is converted to alkene oligomers (of carbon length C8–C24). The distribution of oligomers produced, as reported by Bond et al.,10 is included in the supporting information (Table S3†).
 | ||
| Fig. 6 Schematic flowsheet for the cascade conversion of cellulose to liquid alkene oligomers. | ||
Techno-economic analysis
We performed techno-economic analyses based on ASPEN simulation models. The process model specifications were based in part on values reported by NREL for the conversion of corn stover to ethanol.27–30 Our goal was to establish a common basis for the comparison of alternative biofuel production strategies. To that end, we initially used the same corn stover feedstock and processing rate (2000 MT dry corn stover per day), similar process equipment whenever possible, the same costs for feedstock, chemicals and utilities, and the same economic parameters (e.g., reference index year, income tax rate, return on investment, depreciation). Due to the large plant capacity, the plant design consists of three identical processing trains operating in parallel and each train has multiple parallel unit operations (e.g., reactors, distillation columns). The details of our process model are included in the supporting information.†In addition to examining the overall feasibility of our proposed chemical catalytic strategy, we also studied the significance of using an alternative raw material. To that end, we developed two processing scenarios. In scenario 1, corn stover is used as raw material, while in scenario 2, loblolly pine, an important and prevalent softwood that grows readily in the southern United States, is used. It is important to note that the hemi-cellulose fraction of loblolly pine is rich in C6sugars,33 instead of C5sugars which are abundant in the hemi-cellulose of corn stover (see Table 1). Thus, for the case of loblolly pine, a higher fraction of the original biomass can be transformed into valuable liquid fuels using the proposed route. Furthermore, the use of loblolly pine as a feedstock leads to a simplified flowsheet in which the dilute acid pretreatment step used to extract C5sugars from the hemi-cellulose fraction of the corn stover is no longer needed. Additionally, the small fraction of C5sugars in the raw material leaves the process as humins along with the lignin. In both cases the unused materials (humins, lignin, and recovered C5sugars in the case of corn stover) are sent to the boiler/turbo generation unit to produce heat and electricity. In scenario 1, we assume that 85% of the C5 fraction of the hemi-cellulose is recovered, concentrated (using excess heat), and sent to the reboiler/turbogenerator. Using catalytic activities reported in literature, we determined the catalyst amounts required in the batch reactor (cellulose deconstruction reactor),6 reactor 2 (GVL decarboxylation reactor)10 and reactor 3 (butene oligomerization reactor).10 Experimental results for the high conversion study discussed above were used to determine the required amount of RuRe/C catalyst for reactor 1 (levulinic acid reduction reactor) which used inlet feed concentrations that are similar to those used in the experimental investigation above. Finally, to facilitate the comparison between the two scenarios, the processing capacity in scenario 2 was adjusted so that the two scenarios have the same C6sugar throughput and therefore the same oligomer production capacity.
| Component | Corn Stover | Loblolly Pine |
|---|---|---|
| C6carbohydrates | 41% | 55% |
| C5carbohydrates | 24% | 9% |
| Lignin | 18% | 32% |
| Other | 17% | 4% |
| Cost (US$ dry-MT−1) | 83 | 57 |
Capital cost comparison
The capital costs for the two aforementioned oligomer production scenarios as well as the ethanol production process analyzed by NREL are shown in Table 2. The same capital costs were used for processing steps that are similar to both the production of alkene oligomers and the production of ethanol (e.g., feedstock handling, pretreatment, storage, utilities). The cost of the boiler/turbogenerator for the oligomer production process is estimated using the standard cost scaling equation (eqn (1)),34 which relates the unit capacity (e.g. kmol h−1carbon sent to boiler/turbogenerator) to its cost, using reference values. | (1) |
| Process Sections | Ethanol process | Oligomer process scenario 1 | Oligomer process scenario 2 | |||
|---|---|---|---|---|---|---|
| (MM$) | (%) | (MM$) | (%) | (MM$) | (%) | |
| a Taken from study by Kazi, et al.30 b Estimated from value reported by Kazi, et al.30 c Determined using Aspen Icarus. d Includes other direct (e.g., instrumentation, insulation) and indirect (e.g., engineering, contingency) costs. | ||||||
| Feedstock handlinga | 10.9 | 6.6 | 10.9 | 5.4 | 10.9 | 7.3 |
| Pretreatmenta | 36.2 | 22.1 | 36.2 | 17.9 | ||
| Saccharification/fermentationa | 21.8 | 13.3 | ||||
| Distillation/solids recoverya | 26.1 | 15.9 | ||||
| Wastewater treatmentb | 3.5 | 2.1 | 2.8 | 1.4 | 2.9 | 1.9 |
| Storage a | 3.2 | 2.0 | 3.2 | 1.6 | 3.2 | 2.2 |
| Boiler/turbogeneratorb | 56.1 | 34.2 | 89.9 | 44.4 | 72.1 | 48.5 |
| Utilitiesa | 6.3 | 3.8 | 6.3 | 3.1 | 6.3 | 4.2 |
| Cellulose deconstructionc | 12.3 | 6.1 | 12.7 | 8.5 | ||
| GVL Productionc | 7.8 | 3.9 | 7.7 | 5.1 | ||
| Cooling/CO2 separationc | 1.5 | 0.8 | 1.5 | 1.0 | ||
| GVL extraction/H2SO4 recyclec | 8.4 | 4.1 | 8.2 | 5.5 | ||
| Butene productionc | 12.8 | 6.3 | 12.8 | 8.6 | ||
| Butene & CO2 separationc | 0.9 | 0.4 | 0.9 | 0.6 | ||
| Butene oligomerizationc | 8.8 | 4.4 | 8.8 | 5.9 | ||
| Oligomer condensationc | 0.7 | 0.4 | 0.7 | 0.5 | ||
| Total installed equip. cost | 164.1 | 202.4 | 148.6 | |||
| Total project investmentd | 377.4 | 465.5 | 341.7 | |||
The capital costs for all unit operations are given in Table 2. First, we note that the cost of the boiler/turbogenerator unit in the three processes ranges between 34 and 49% of the total capital cost, making it the most expensive unit. Thus, an accurate estimation of its capital cost as a function of its capacity, using eqn (1), is important. Second, the results in Table 2 show that the total installed equipment cost for scenario 1 (corn stover) is 23% ($38.3 million) higher than the ethanol process. This difference is due mainly to the larger boiler/turbogenerator ($33.8 million of difference). Third, compared to the ligno-cellulosic ethanol strategy, the cost of scenario 2 (loblolly pine) is approximately 9% lower ($15.5 million). This difference is because the process in scenario 2 has no preprocessing section and a smaller turbo generation unit due to the higher loblolly pine mass fraction used to generate fuels (i.e., a lower biomass fraction sent to the boiler).
Finally, we note that the cost of the equipment required to catalytically convert cellulose into alkene oligomers in both scenarios ($53 million) is similar to the cost of the equipment used for the saccharification/fermentation and distillation/solids recovery sections in the ethanol process ($48 million).
Operating cost comparison
A comparison of the operating costs for the biochemical production of ethanol and the catalytic production of alkene oligomers is shown in Fig. 7. We assumed a price of $83 per dry MT for corn stover28 and $57 per dry ton for loblolly pine ($26 per green short ton−1 assuming 50% moisture) based on a 50 mile collection radius specific to south eastern Georgia.35 The “other raw materials” includes the sulfuric acid and lime used in the pretreatment of corn stover (to release the hemi-cellulose), wastewater treatment chemicals and polymers, and the boiler and cooling tower chemicals. Fixed costs include employee salaries, insurance, overhead and maintenance. Waste disposal costs include gypsum disposal following the sulfuric acid neutralization during biomass pretreatment and other solid waste disposal (e.g., ash). Using current market prices, the purchase cost of precious catalyst metals (i.e.Ru and Re) required for scenarios 1 and 2 is $33 million. We assume that the catalyst is leased and has a salvage value at the end of the life of the project. Thus, the catalyst cost is included as an operating cost for the refurbishing and continued maintenance of the GVL production catalyst (see supporting information for more details†). It should be noted that the costs for the SiO2/Al2O3 catalyst (used in GVL decarboxylation) and Amberlyst-70 catalyst (used in butene oligomerization) are negligible compared to the RuRe/C catalyst, and hence they are not considered in this analysis. | ||
| Fig. 7 Annual operating costs for the biochemical production of ethanol (reported by Kazi, et al.,30 blue), alkene oligomer production scenario 1 (corn stover, red), and scenario 2 (loblolly pine, green). Total annual costs: ethanol process = 140 MM$, oligomer scenario 1 = 90 MM$, oligomer scenario 2 = 46 MM$. | ||
As shown in Fig. 7, the feedstock cost is the most significant fraction of operating costs in all strategies. Nevertheless, the feedstock cost as well as the cost of “other raw materials” in scenario 2 are significantly lower than the corresponding costs in scenario 1 and the bioethanol strategy. This difference is due to a lower raw material price (in US$ dry-MT−1) and the higher C6sugar content, which means that less feedstock is required to produce the same amount of fuel. The cost of “other raw materials” is lower because scenario 2 has no preprocessing stage. Also, we note that the cost of the enzyme for the biochemical production of ethanol is considerable and surpasses any variable cost for the production of alkene oligomers.
Another aspect in the production of alkene oligomers is the co-production of electricity and process heat. To estimate the output of the larger boiler/turbogenerator, reference values previously reported for ethanol production29 were scaled using simple mass and energy balances. Our analysis indicates that oligomer production scenarios 1 and 2 consume approximately 30% of the boiler/turbogenerator heating, and provide an electricity surplus which translates into an additional annual revenue of approximately 24 and 20 MM$ yr−1, respectively.
Revenue-cost balance
Using the calculated capital and operating costs, a minimum selling price (MSP) for the produced fuels can be determined. To make the two oligomer routes comparable to the ethanol process, fuel production (i.e., ethanol and oligomers) was expressed in terms of gallons of gasoline equivalent (GGE). The calculated MSP value for the NREL ethanol process30 is $5.13 GGE−1, while the values for oligomer production scenarios 1 and 2 are equal to $7.78 GGE−1 and $4.31 GGE−1, respectively. Fig. 8 shows how the total production cost (based on the aforementioned capital and operational cost calculations) is balanced by the sales of electricity and fuels. First, we note that the total production cost of the ligno-cellulosic ethanol route is higher than in both oligomer scenarios. This difference is due mainly to the larger operating costs (e.g., cost of cellulases – see Fig. 7). However, the NREL strategy leads to higher fuel production and sales. In particular, it leads to higher fuel sales than scenario 1 (although the two routes process the same amount of feedstock) since its biomass-to-fuels yield is higher. The NREL strategy also leads to higher production than in scenario 2, despite the high biomass-to-fuels yield of the latter, because it processes 2000 MT of corn stover per day versus 1365 MT of loblolly pine per day. Second, we note that the production cost in scenario 2 is substantially lower than in scenario 1, due to lower capital (Table 2) and operational (Fig. 7) costs. It is important to stress here that the amounts of fuel produced in the two alkene oligomer scenarios are identical (recall that they convert the same amount of C6sugars), but the MSP for scenario 2 is lower ($4.31 GGE−1vs. $7.78 GGE−1), which leads to a lower revenue from sales at the break-even point. | ||
| Fig. 8 Revenue-cost balance for the biochemical production of ethanol, and alkene oligomer production scenario 1 (corn stover) and scenario 2 (loblolly pine). The calculation of the return on investment (ROI) and operating costs as well as electricity sales is based on rigorous ASPEN models, nominal prices for raw materials and utilities, and the methodology followed by Kazi, et al.30Fuel price and hence fuel sales are adjusted so that the total production cost of each strategy (ROI + income tax + operating costs) is equal to the total revenues (electricity sales + fuel sales). The MSP is the price that leads to this “break even” point. a) Absolute values; b) Percentages of the total. | ||
Sensitivity analysis
We conducted sensitivity analysis studies to determine how variations in key economic and processing parameters impact the MSP in the two oligomer strategies. We investigated the impact of feedstock price, total installed equipment cost, levulinic acid reduction catalyst refurbishing cost, and electricity price. To calculate the corresponding sensitivities, we varied the nominal (base case) values by ±20%. In addition, the impact of selling the generated CO2 instead of venting it into the atmosphere (base case) was also considered. The results are shown in Fig. 9 and 10. | ||
| Fig. 9 Sensitivity analysis for the production of alkene oligomers – scenario 1 (corn stover) (high levels = red bars, low levels = blue bars). | ||
 | ||
| Fig. 10 Sensitivity analysis for the production of alkene oligomers – scenario 2 (loblolly pine) (high levels = red bars, low levels = blue bars). | ||
For scenario 1, the feedstock price and the installed equipment cost have the largest impact; a 20% variation in the former leads to a 9.5% change in the MSP, while a similar variation in the latter leads to a 9.0% change. We note that the feedstock cost accounts for 65% of the total operating cost, or 40% of the total production cost (see Fig. 7). For scenario 2, we note that the two aforementioned parameters remain the most important, but the impact of the equipment cost is higher; increasing it by 20% leads to a 12% change in the MSP, while a similar increase in feedstock price results in a MSP that is only 8% higher. This difference is because the capital cost for scenario 2, although lower than in scenario 1, is a larger fraction of the total production cost, as shown in Fig. 8b. At the same time, the feedstock cost in scenario 2 is 32% of the production cost (60% of the total operating cost) as opposed to 40% in scenario 1. The sensitivity of the MSP with respect to the price of electricity in scenario 1 is 3% and in scenario 2 is 5%, despite the fact that more electricity is produced and sold in scenario 1. This result can be explained if we recognize that the lower MSP of oligomers in scenario 2 leads to substantially smaller fuel sales, which means that electricity sales in scenario 2 are a larger fraction of total sales than in scenario 1 (see Fig. 8b). The cost of catalyst refurbishing had essentially negligible effect (<1% for both scenarios). Finally, using a CO2 price of $36 ton−1, based on an amine recovery system,36 we calculated that selling the CO2 in the high pressure gas stream coming from the CO2/butene separation had a relatively small effect (2% and 4% variation for scenarios 1 and 2 respectively).
Discussion
Impact of newly developed levulinic acid reduction catalyst systems
Due to their inherently high value, the stability of precious metal catalysts is an important concern. The experimental results described above demonstrate that the RuRe/C catalyst maintains stable activity in the presence of sulfuric acid. The TPR study revealed that the Ru and Re metals are in intimate contact on the RuRe/C catalyst under reducing conditions. The XRD study demonstrated that the Ru and Re particles are well-dispersed on the carbon support even after reaction testing in the presence of sulfuric acid. In addition, ICP analyses of reactor effluent streams (see supporting information†) revealed that under the acidic conditions used during reaction kinetics studies in the presence of sulfuric acid, the catalyst is exposed to relatively high concentrations (e.g., 100 ppm) of metal ions that are leached from the reactor walls (i.e., Fe, Ni, Cr, Co, Mo). While the reaction kinetics results presented here demonstrate that the RuRe/C bimetallic catalyst shows good stability and selectivity in the presence of these metal ions from the reactor walls, it is recommended that additional reaction kinetic studies be conducted using various biomass-synthesized feeds to determine the long term effects of soluble humins or other soluble residues on the performance of this catalyst.The application of the newly developed bimetallic RuRe/C catalyst for the reduction of levulinic acid in the presence of sulfuric acid could lead to several advantages for an integrated biofuel production scheme. We have demonstrated that the levulinic acid reduction step can be operated at high conversion with good selectivity to GVL, thus minimizing concerns related to levulinic acid recycle. The proposed GVL extraction process, utilizing butyl acetate, enables the recycling of most of the sulfuric acid catalyst, eliminating costly neutralization and disposal steps. The simultaneous conversion of formic acid provides an internal hydrogen source that can be used to minimize external hydrogen requirements. In addition, the carbon removed from the carbohydrate starting material as CO2 could potentially be sequestered in relatively pure streams from the GVL production and the alkene oligomer production steps.
Process integration
Large capital investment is potentially a barrier for the implementation of new technologies such as those presented here. As discussed above, the single largest capital cost component in all strategies is the boiler/turbogenerator. In this respect, it would be advantageous to couple a biofuels facility with an existing plant that utilizes similar unit operations (i.e., boiler/turbogenerator, wastewater treatment) or to retrofit an existing facility. In addition to equipment cost considerations, the facility should be located in close proximity to a supply of feedstock. For these reasons, a paper mill would appear to be potentially suitable for effective implementation of a biofuels process. Since 1970, the number of paper mills has decreased and the average mill production capacity has increased (from 93,000 to 216,000 MT yr−1).37 This decrease is caused by the closure of smaller mills (<100,000 MT yr−1 production capacity) and their replacement with larger mills. Hence, the integration of a biofuels production process with a relatively smaller scale paper mill may have promising potential.To assess this potential, we estimated how the total installed equipment cost for the loblolly pine processing strategy varies as a function of the softwood processing capacity (see Fig. 11). The results presented above were based on a total processing capacity of 1365 dry MT of loblolly pine per day (478,000 MT yr−1), corresponding to a total installed equipment cost of $149 million. However, if an alkene oligomer production process was developed in a retrofitted paper mill for a lower processing capacity of 200 MT dry softwood per day (70,000 MT yr−1), then the total installed equipment cost would decrease to approximately $44 million. Furthermore, if the facility already included a boiler/turbogenerator and wastewater treatment equipment, then the additional required installed equipment cost could potentially be decreased to as low as $22 million. At the same time, the projected MSP of $4.31 GGE−1 should still be a valid approximation for plant capacities as low as 200 dry MT of loblolly pine per day, because the design for our process is composed of 3 identical processing trains each containing a number of parallel units.
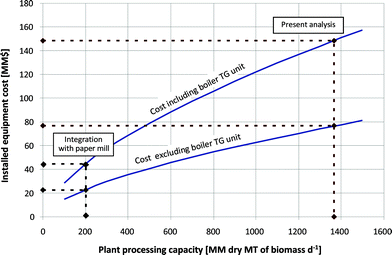 | ||
| Fig. 11 Total capital cost as a function of biomass processing capacity for the conversion of a softwood (loblolly pine) feedstock into liquid alkene oligomers (calculations according to eqn (1)). | ||
Potential for future improvements
The focus of the techno-economic analysis in this study was centered on assessing the feasibility of the alkene oligomer production strategy in comparison with the production of ethanol, the most prevalent biomass-derived fuel in the world today. This analysis revealed that the proposed chemical-catalytic strategy compares favorably, especially when feedstocks rich in C6sugars are used. Importantly, the MSP value of $4.31 GGE−1 can be achieved using the proven experimental yields for levulinic acid production from ligno-cellulose (55% yield), GVL production from levulinic acid (>99% yield), and the production of alkene oligomers (C8–C24) from GVL (99% yield). It should be noted that, in comparison with the ethanol-based process (85% yield of ethanol from cellulose, 76% yield of ethanol from xylose), the overall yield of butene oligomers is relatively low for the base case studied (∼50% yield of oligomer monomers per cellulose monomer processed) with the least selective conversion step being the production of levulinic acid (55% yield). However, the production of levulinic acid from cellulose (and glucose) is not thermodynamically limited, and preliminary results have suggested that levulinic acid yields in excess of 70% may be attainable, leaving significant room for improvement, and ultimately leading to a further significant reduction in the fuel MSP.The present study has compared the production of alkene oligomers with the production of ethanol in terms of the MSP based on GGE (gallon gasoline equivalent). However, MSP should not be the only comparison criterion. First, the proposed strategy results in a high pressure stream of highly concentrated CO2, which is ideal for carbon capture and sequestration. In addition to leading to a carbon negative process, this also means that if emission regulations or emission trading schemes are introduced, then CO2 consumers will receive credits, which would further improve the economics of the proposed route. Second, a mix of alkene oligomers is a better liquid fuel than ethanol, because it has higher energy density, it is easier to handle, and it does not require modifications of the existing infrastructure. If desired, the alkenes can be converted to alkanes by reduction over a metal catalyst, and only one equivalent of H2 is required to convert one alkene molecule to its corresponding alkane. In this respect, the final fuel components produced from the alkene oligomer strategy do not suffer from blending limitations, in contrast to ethanol and other oxygenated fuel additives. Also, the proposed process has the potential to produce oligomer products that contain more than 93% of the energy content contained within the C6sugar monomers and only 31% of the initial carbohydrate weight (for the stoichiometric conversion of glucose to alkenes plus CO2 and H2O). Thus, the production of alkene oligomers from ligno-cellulosic biomass appears to be an advantageous and economically attractive approach for producing renewable liquid hydrocarbon transportation fuels from non-fossil fuel sources.
Conclusions
GVL has been identified as a viable biomass-derived platform chemical from which liquid hydrocarbon fuels can be derived; however, its efficient and cost effective production remains a challenge. Our studies of the conversion of levulinic acid into GVL over a traditional ruthenium-based catalyst have identified a more active RuRe/C catalyst. This RuRe/C catalyst is selective for the production of GVL from levulinic acid while exhibiting a high activity for the simultaneous decomposition of formic acid. Additionally, this bimetallic catalyst shows good stability in the presence of sulfuric acid, a common homogeneous catalyst used in the conversion of cellulose into levulinic acid.Techno-economic analyses reveal that the production of liquid hydrocarbonsvia our levulinic-acid-based approach is economically competitive with a state-of-the-art ligno-cellulosic ethanol production strategy. The most sensitive process parameters are the feedstock and total installed equipment costs. In addition to cost, it was determined that the feedstock composition plays an important role in the process economics, such that biomass sources rich in C6sugars allow for more cost effective process designs. While the proposed process appears to be economically competitive, there exists ample room for improvement in product yields (i.e., levulinic acid production) and catalyst activities (i.e., levulinic acid conversion) that could further improve the process economics.
Materials and Methods
Ru/C and RuRe/C catalyst preparation and pretreatment
The 5 wt% Ru/C catalyst used in the levulinic acid reduction studies was purchased from Sigma-Aldrich. The 15 wt% RuRe(3:4)/C catalyst was prepared by incipient wetness impregnation of the 5 wt% Ru/C catalyst using an aqueous solution of perrhenic acid (HReO4, 53 wt% Re). Prior to reaction kinetics measurements, each catalyst was reduced at 673 K (0.5 K min−1 ramp, 2 h hold) under flowing H2 (100 cm3 min−1 (STP)). For reactions using feeds containing sulfuric acid, the catalyst was pretreated in situ for 24 h with a 0.5 mol L−1 solution of sulfuric acid at a flow rate of 0.1 cm3 min−1 at the reaction temperature in the presence of flowing H2 (25 cm3 min−1 (STP)).
Levulinic acid reduction experiments
Reaction kinetics measurements for the reduction of levulinic acid were carried out using a packed bed reactor. A schematic of the flow reactor system used for reaction kinetics studies is shown in the supporting information in Fig. S1.† A packed-bed reactor consisting of a half-inch or quarter-inch tube (Hastelloy C327) was used for the levulinic acid reduction experiments. Quartz wool was used above and below the catalyst bed to keep the catalyst in place. The reactor was heated with an aluminium block heated externally by a well-insulated furnace (Applied Test Systems). Type-K thermocouples (Omega) were used to measure the reaction temperature outside the reactor wall. The reaction temperature was maintained by PID temperature controllers (Love controls) that were connected to a variable transformer (Tesco). Mass flow controllers (Brooks 5850E) were used to regulate the flow of H2, He and other gases during the experiments. The liquid feed was pumped from a graduated cylinder by an HPLC pump (Lab Alliance series 1) to a needle located at the entrance of the catalyst bed. A back-pressure regulator (GO model BP-60), located downstream from the reactor, was used to control the total pressure, which was measured by three gauges located downstream from the liquid pump, at the inlet of the reactor and downstream of the reactor. A gas-liquid separator (Pemburthy) at room temperature was used to collect the liquid phase for analysis. A bubble flow meter, located downstream from the back pressure regulator, measured the gas flow rate out of the reaction system.The aqueous product distributions were measured by HPLC analysis (Waters, using a Biorad Reztec column) of the liquid effluent drained from the separator located downstream of the flow reactor. Non-UV active species (e.g., sugars) were quantified using a refractive index detector (Waters 410), and UV-active species were quantified using a UV detector (Waters PDA 990) at appropriate absorbance wavelengths. Total organic carbon analysis (TOC) was used to determine the total organic carbon in solution. An HPLC-MS (Agilent), equipped with an ion trap, was used to qualitatively identify species in solution.
Catalyst characterization
Adsorption isotherms for CO on Ru/C and RuRe/C catalysts were collected using a standard volumetric gas handling system employing capacitance manometers for precision pressure measurement (±0.5 × 10−5 mbar). Following reduction pretreatment at 673 K (0.5 K min−1 ramp, 2 h hold), adsorbed hydrogen was desorbed by vacuum treatment (1 × 10−7 mbar) for 1 h, and the sample was subsequently cooled to 300 K. For chemisorption studies, small amounts of CO (1–10 μmol) were dosed onto the catalyst until saturation of the surface sites was achieved. The amount of gas adsorbed was determined volumetrically from the dose and equilibrium pressures and system volumes and temperatures. Times of 20–30 min were allowed for each dose to reach equilibrium. After saturation, the catalyst was evacuated at 300 K for 1 h to remove weakly bound probe gas molecules and a second isotherm was collected. The irreversible CO uptake was calculated by subtracting the two isotherms.TPR studies were carried out using a continuous flow apparatus consisting of a mass flow controller (Teledyne-Hastings) and tube furnace connected to a variable power-supply and PID temperature controller (Love Controls) with a K-type thermocouple (Omega), shown in the supporting information in Fig. S2.† The effluent was monitored by a mass spectrometer system consisting of a quadruple residual gas analyzer (Stanford Instruments RGA 200) inside a vacuum chamber. Vacuum was provided by a diffusion pump connected in series to a rotary pump. The effluent was introduced into the vacuum chamber via a constricted quartz capillary, resulting in a pressure of 5 × 10−5 mbar inside the chamber. Dried, unreduced catalyst samples (100–300 mg) were loaded into a 12.6 mm (0.5 inch) outer diameter, fritted quartz tube and reduced in situ. A temperature ramp of 10 K min−1 was used to heat the catalyst from room temperature to 873 K where it was held for 20 min.
A Scintag PAD V X-ray diffractometer with monochromated Cu-Kα X-ray tube was used in XRD studies. The tube voltage and current were 45 kV and 40 mA, respectively. Diffraction patterns for were collected in the 5° to 40° 2θ range, with 0.02° intervals and a dwell time of 2 s. Prior to diffraction studies, the catalyst samples were reduced in flowing hydrogen (200 cm3 min−1) at 673 K and passivated in a flowing 2% O2/He mixture (150 cm3 min−1).
Process synthesis and techno-economic analysis
The process synthesis studies were based on detailed process simulation models developed in ASPEN Plus, incorporating experimental data and other published information to specify raw materials compositions, reactor operational conditions, overall reactor conversions and yields, separation recoveries, etc. Conversion reactor models coupled with rigorous separation models (e.g., multi-stage distillation and liquid–liquid extraction units) were used to obtain accurate values. The solution of the corresponding material and energy balances provided the necessary information to conduct equipment sizing.38 Finally, detailed capital cost calculations based on unit capacities, operational conditions, and selected materials of construction were performed using ASPEN Process Economic Analyzer. In order to have a common basis, we adopted the methodology which was used for the economic analysis by NREL for the evaluation of their ethanol process, accounting for all sources of operational revenue (e.g., liquid fuels and electricity), operational costs (e.g. raw material and utility consumption) capital depreciation, taxes, internal rate of return, etc.Acknowledgements
This work was supported by the Department of Energy through the Great Lakes Bioenergy Research Center.Notes and references
- D. Martin-Alonzo, J. Q. Bond and J. A. Dumesic, Catalytic conversion of biomass to biofuels, Green Chem., 2010, 12, 1493–1513 RSC.
- J. J. Bozell and G. R. Petersen, Technology development for the production of biobased products from biorefinery carbohydrates - the US Department of Energy's “Top 10” revisited, Green Chem., 2010, 12, 539–554 RSC.
- D. J. Hayes, F. Fitzpatrick, M. H. B. Hayes, J. R. H. Ross, Biorefineries: Industrial Processes and Products, Vol. 1 (Wiley-VCH, Weinheim), ( 2006) Search PubMed.
- B. Girisuta, L. P. B. M. Janssen and H. J. Heeres, A kinetic study on the conversion of glucose to levulinic acid, Chem. Eng. Res. Des., 2006, 84, 339–349 CrossRef CAS.
- B. Girisuta, L. P. B. M. Janssen and H. J. Heeres, Kinetic study on the acid-catalyzed hydrolysis of cellulose to levulinic acid, Ind. Eng. Chem. Res., 2007, 46, 1696–1708 CrossRef CAS.
- J. C. Serrano-Ruiz, D. J. Braden, R. M. West and J. A. Dumesic, Conversion of cellulose to hydrocarbon fuels by progressive removal of oxygen, Appl. Catal., B, 2010, 100, 184–189 CrossRef CAS.
- I. T. Horvath, H. Mehdi, V. Fabos, L. Boda and L. T. Mika, γ-Valerolactone - a sustainable liquid for energy and carbon-based chemicals, Green Chem., 2008, 10, 238–242 RSC.
- H. Mehdi, et al., Integration of homogeneous and heterogeneous catalytic processes for a multi-step conversion of biomass: from sucrose to levulinic acid, γ-valerolactone, 1,4-pentanediol, 2-methyltetrahydrofuran, and alkanes, Top. Catal., 2008, 48, 49–54 CrossRef CAS.
- J. P. Lange, et al., Valeric Biofuels: A Platform of Cellulosic Transportation Fuels, Angew Chem, 2010, 122, 4581–4585 Search PubMed.
- J. Q. Bond, D. M. Alonso, D. Wang, R. M. West and J. A. Dumesic, Integrated Catalytic Conversion of gamma-Valerolactone to Liquid Alkenes for Transportation Fuels, Science, 2010, 327, 1110–1114 CrossRef CAS.
- L. E. Manzer, Production of 5-methylbutyrolactone from levulinic acid, Int. Patent 6617464 B2, ( 2003).
- L. E. Manzer, Catalytic synthesis of alpha-methylene-gamma-valerolactone: a biomass-derived acrylic monomer, Appl. Catal., A, 2004, 272, 249–256 CrossRef CAS.
- Z.-p. Yan. and L. Lin, Synthesis of γ-valerolactone from biomass-based levulinic acid over Ru/C catalyst in, Energy Fuels, 2009, 23, 3853–3858 CrossRef CAS.
- R. J. Haan, J. P. Lange, L. Petrus, C. J. M. Petrus-Hoogenbosch, Hydrogenation process for the conversion of a carboxylic acid or an ester having a carbonyl group, US Patent 2007208183 ( 2007).
- A. Corma, S. Iborra and A. Velty, Chemical routes for the transformation of biomass into chemicals, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS.
- M. Osada, N. Hiyoshi, O. Sato, K. Arai and M. Shirai, Subcritical water regeneration of supported ruthenium catalyst poisoned by sulfur, Energy Fuels, 2008, 22, 845–849 CrossRef CAS.
- T. Miyazawa, S. Koso, K. Kunimori and K. Tomishige, Glycerol hydrogenolysis to 1,2-propanediol catalyzed by a heat-resistant ion-exchange resin combined with Ru/C, Appl. Catal., A, 2007, 329, 30–35 CrossRef CAS.
- Y. J. Kuo, R. A. Cocco and B. J. Tatarchuk, Hydrogenation and Hydrodesulfurization over Sulfided Ruthenium Catalysts: II. Impact of Surface Phase-Behavior on Activity and Selectivity, J. Catal., 1988, 112, 250–266 CAS.
- Y. J. Kuo and B. J. Tatarchuk, Hydrogenation and Hydrodesulfurization over Sulfided Ruthenium Catalysts, J. Catal., 1988, 112, 229–249 CAS.
- M. Zerella, S. Mukhopadhyay and A. Bell, T. Synthesis of mixed acid anhydrides from methane and carbon dioxide in acid solvents in, Org. Lett., 2003, 5, 3193–3196 CrossRef CAS.
- R. H. Leonard, Levulinic acid as a basic chemical raw material, Ind. Eng. Chem., 1956, 48, 1330–1341 Search PubMed.
- E. L. Kunkes, et al., Catalytic conversion of biomass to monofunctional hydrocarbons and targeted liquid-fuel classes, Science, 2008, 322, 417–421 CrossRef CAS.
- M. Mabry, W. Prichard and S. Ziemecki, in E. I. DuPont, de Nemours and Company US Patent 4,550,185, ( 1985).
- M. Mabry, W. Prichard and S. Ziemecki, in E. I. DuPont, de Nemours and Company US Patent 4,609,636, ( 1986).
- J. T. Schwartz, in E. I. DuPont, de Nemours and Company US Patent 5,478,952, ( 1995).
- V. Pallassana and M. Neurock, Reaction paths in the hydrogenolysis of acetic acid to ethanol over Pd(111), Re(0001), and PdRe alloys, J. Catal., 2002, 209, 289–305 CrossRef CAS.
- A. Aden, Biochemical production of ethanol from corn stover: 2007 state of technology model,NREL/TP-510-43205, 1-10, ( 2008).
- A. Aden and T. Foust, Technoeconomic analysis of the dilute sulfuric acid and enzymatic hydrolysis process for the conversion of corn stover to ethanol, Cellulose, 2009, 16, 535–545 CrossRef CAS.
- A. R. Aden, et al., Lignocellulosic biomass to ethanol process design and economics utilizing co-current dillute acid prehydrolysis and enzymatic hydrolysis for corn stover,NREL/TP-510-32438, ( 2002).
- F. K. Kazi, et al., Techno-economic comparison of process technologies for biochemical ethanol production from corn stover, Fuel, 2010, 89, 520–528 Search PubMed.
- L. d. C. Sousa, S. P. S. Chundawat, V. Balan and B. E. Dale, ‘Cradle-to-grave’ assessment of existing lignocellulose pretreatment technologies, Curr. Opin. Biotechnol., 2009, 20, 339–347 CrossRef CAS.
- M. P. Tucker, K. H. Kim, M. M. Newman and Q. A. Nguyen, Effects of temperature and moisture on dilute-acid steam explosion pretreatment of corn stover and cellulase enzyme digestibility, Appl. Biochem. Biotechnol., 2003, 105, 165–178 Search PubMed.
- T. Marzialetti, et al., Dillute acid hydrolysis of loblolly pine: a comprehensive approach, Ind. Eng. Chem. Res., 2008, 47, 7131–7140 CrossRef CAS.
- R. Smith, Chemical process design and integration, Wiley, Chichester, West Sussex, England, Hoboken, NJ, 2005 Search PubMed.
- 2008 Georgia Timber Report in Georgia Forestry Commision (http://www.gfc.state.ga.us/ForestManagement/TimberSelling.cfm).
- D. Mignard and C. Pritchard, Processes for the synthesis of liquid fuels from CO2 and marine energy, Chem. Eng. Res. Des., 2006, 84, 828–836 CrossRef CAS.
- P. J. Ince, X. Li, M. Zhou, J. Buongiorno and M. Reuter, United States paper, paperboard, and market pulp capacity trends by process and location, 1970-2000, Forest Products Laboratory, US Dept of Agriculture, 2001, 1–36 Search PubMed.
- S. M. Walas, Chemical process equipment : selection and design (Butterworths, Boston), ( 1988) Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details on techno-economic analyses, extraction of GVL using butyl acetate, and the reactor metal leaching study. See DOI: 10.1039/c1gc15047b |
| This journal is © The Royal Society of Chemistry 2011 |