 Open Access Article
Open Access ArticleBimetallic catalysts for the Fischer–Tropsch reaction†
V. Roberto
Calderone
a,
N. Raveendran
Shiju
a,
Daniel Curulla
Ferré
b and
Gadi
Rothenberg
a
aVan't Hoff Institute for Molecular Sciences, University of Amsterdam, Science Park 904, 1098 XH, Amsterdam, The Netherlands
bGaz & Energies Nouvelles, Total S.A. Paris La Défense 6, France
First published on 26th April 2011
Abstract
This short critical review summarises and analyses the developments in Fischer–Tropsch catalysis using bimetallic alloys. We introduce a simple notation for such catalysts, and monitor the reports of synergistic effects and composition/performance relationships. Special attention is given to CoFe alloys on a variety of supports, and to the effects of catalyst preparation methods and pre-treatment conditions. The key drawbacks in comparing the large amount of data available on Fischer–Tropsch catalysis are the high dimensionality of the problem and the lack of long time-on-stream studies. Based on the new understanding coming from characterisation studies of supported bimetallic particles, we propose a structured approach for effectively studying Fischer–Tropsch catalysis.
 Vincenzo Roberto Calderone | Vincenzo Roberto Calderone earned his Master and PhD at University of Genoa. His research is based since on applying colloidal chemistry methods to the synthesis of advanced materials. After his PhD, his interests shifted into catalysis and, specifically, to the Fischer-Tropsch process. He joined Prof. Albert Philipseʼs group in Utrecht taking on a project regarding the synthesis of monodispersed cobalt-based catalyst for F-T. He currently works at Prof. Rothenbergʼs group in Amsterdam managing a research line which, in collaboration with Total Gaz & Energies Nouvelles, focuses on the preparation of newly developed F-T catalysts. |
 N. Raveendran Shiju | N. Raveendran Shiju obtained his PhD in heterogeneous catalysis from National Chemical Laboratory, Pune. He then worked at the Royal Institution of Great Britain, the University of Cincinnati and the University of Huddersfield. In 2009 he accepted an Assistant Professor position at the University of Amsterdam, in the Heterogeneous Catalysis and Sustainable Chemistry group. Shiju now leads a team of PhD students and post-docs, working on catalyst design for green technology. His research interests range from materials science to reaction engineering. In 2010 he was awarded the Amsterdam Science Park Ideas Prize for the discovery of a new catalyst for making nylons. |
 Daniel Curulla Ferré | Daniel Curulla holds a PhD in Chemistry (2001) from the University Rovira i Virgili (Tarragona, Spain). He worked as research associate at the Technology University of Eindhoven (2001-2007), in the group of Prof Niemantsverdriet. In 2004, he got with the Vernieuwingsimpuls Veni grant. Since 2008, he works for Total S.A. at the R&D department of the branch Gaz & Energies Nouvelles. His main research interests are in nanocatalysis, synthetic fuels, carbon dioxide valorization and photocatalytic water splitting. |
 Gadi Rothenberg | Gadi Rothenberg is Professor and Chair of Heterogeneous Catalysis and Sustainable Chemistry at the University of Amsterdam. He has published over 100 papers in peer- reviewed journals and discovered two catalysts, for which he received the Marie Curie Excellence Award in 2004 and the Paul N. Rylander Award in 2006. He also invented a method for monitoring pollutants in water, and co-founded the companies Sorbisense and Yellow Diesel. In 2007 he was voted “teacher of the year” by the chemistry students. His most recent invention is a biodegradable non-toxic resin, made from 100% plant-based materials. |
It’s commonly accepted that you can't always get what you want. Unfortunately for the petrochemical industry, this pragmatism does not apply to Fischer–Tropsch (F–T) synthesis. Here, you can always get what you want, but unluckily you often also get a plethora of products that you don't want. For over a century, chemists have struggled with optimising F–T catalysts. The task, which seems easy at first glance (just add something to cobalt), is daunting. Catalyst performance depends on precursor composition, preparation methods and treatment parameters. And on top of it all, the catalyst keeps changing in the first three weeks on-stream. Still, whenever crude oil prices peak, so does interest in F–T catalysis, because this is the one process that can make nearly anything out of nearly everything.
Introduction
Efforts to synthesise hydrocarbons by the catalytic hydrogenation of carbon monoxide date back to 1902, when Sabatier and Senderens synthesised hydrocarbons from CO and H2. Later, in 1922, Hans Fischer and Franz Tropsch proposed the Synthol process, in which aliphatic oxygenated compounds were produced by the reaction of CO with H2.1 They used cobalt and/or iron as the first catalysts for syngas conversion. There are several excellent reviews on different aspects of using these catalysts for the F–T reaction.2–4 The most important limitation to the industrialization of F–T processes is the availability of cheap oil. Industrial interest in F–T reactions always peaks during oil crises (Fig. 1).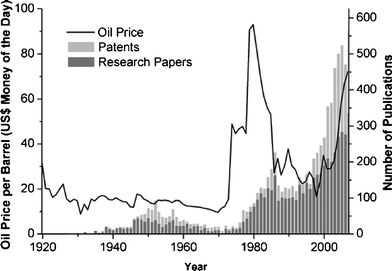 | ||
| Fig. 1 The oil price (line) related to the output of peer reviewed F–T synthesis research papers and patents (bars) in 1925–2007 (from ref. 5). | ||
Increasing regulations on CO2 emissions can deter the development of F–T technologies, especially coal-to-liquid (CTL) technology, where part of the CO is converted to CO2 to re-establish the desired H2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CO stoichiometric ratio, ∼ 2:1 (eqn. 1). F–T may, however, play an important role in the future development of biomass-to-liquid (BTL) technology, which is supposed to be almost CO2 neutral. The Kyoto protocol recognizes the importance of developing and deploying new technologies with less impact on the environment. While F–T is no new technology, it is versatile enough to be adapted for converting a variety of feedstocks to fuels. F–T can convert biomass to a clean fuel (so-called green diesel). It is also an environmentally benign way to convert biosyngas, produced by the gasification of organic residues related to human activities, to liquid fuels. Residue upgrading is comparatively easy compared to conventional crude oil refineries. Synthetic liquid fuels from F–T generally have low contents of sulphur, nitrogen and aromatic compounds compared to gasoline or diesel from crude oil. If effective controls are established, there will be no water effluent from an F–T complex. Hence, F–T fuels are considered as more environmentally friendly than those produced from crude oil.
:CO stoichiometric ratio, ∼ 2:1 (eqn. 1). F–T may, however, play an important role in the future development of biomass-to-liquid (BTL) technology, which is supposed to be almost CO2 neutral. The Kyoto protocol recognizes the importance of developing and deploying new technologies with less impact on the environment. While F–T is no new technology, it is versatile enough to be adapted for converting a variety of feedstocks to fuels. F–T can convert biomass to a clean fuel (so-called green diesel). It is also an environmentally benign way to convert biosyngas, produced by the gasification of organic residues related to human activities, to liquid fuels. Residue upgrading is comparatively easy compared to conventional crude oil refineries. Synthetic liquid fuels from F–T generally have low contents of sulphur, nitrogen and aromatic compounds compared to gasoline or diesel from crude oil. If effective controls are established, there will be no water effluent from an F–T complex. Hence, F–T fuels are considered as more environmentally friendly than those produced from crude oil.
Nevertheless, despite the ongoing research efforts, controlling product selectivity and overcoming catalyst deactivation remain the major obstacles for commercialising F–T technology. F–T catalysts must be selective towards desirable products (i.e., gasoline, diesel fuel, or α-olefins) for the synthesis to have any commercial viability (eqn 1). Recently, several groups have explored using bimetallic catalysts to control selectivity and suppress deactivation. Here, we give an overview of the recent literature on bimetallic F–T catalysts, and discuss the pros and cons of working with bimetallic systems.
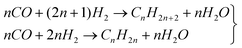 | (1) |
Most studies using bimetallic catalysts have focused on a combination of conventional F–T transition metals, namely Co, Ni, Fe and Ru. Among these, Co and Fe are the most widely used. Ni is relatively inexpensive but also gives more lower alkanes. Ru shows good catalytic properties but its annual world supply cannot even fulfil the requirements of an average plant. We therefore focus first on Co and Fe, followed by a number of examples of other bimetallic catalyst systems.
As with any solid-catalysed reaction, F–T synthesis proceeds on surface sites. Hence, it requires precise control of several parameters to maximize the surface active species. These parameters may include reducibility of the active metal, dispersion of the metal, phase composition and the type of metal–support interaction. Often, these parameters are interdependent. In the case of bimetallic catalysts, the question then becomes how these parameters change in the presence of the second metallic species. Unfortunately, many of the studies on F–T synthesis do not report these parameters. We have thus restricted ourselves to those publications where sufficient parameters were reported for conclusions to be reached on the effect of these parameters on the activity, selectivity and stability of the bimetallic catalysts.
Catalyst notation
The major parameter determining the intrinsic properties of the catalyst is clearly the ratio between the two metals. First, lacking a general and accepted notation for describing F–T catalysts, we proposed the following (eqn 2),| n%(xM1yM2)/support | (2) |
Preparation methods and activity
Several methods such as coprecipitation, impregnation and deposition–precipitation were used to prepare bimetallic catalysts. In one of the studies, (CoFe)/TiO2 catalysts prepared by coprecipitation and impregnation were compared. In the former method, a co-solution of the Fe(NO3)3 and Co(NO3)2 was precipitated with a Na2CO3 solution. TiO2 powder was then stirred into the hot precipitate and mixed thoroughly until a homogeneous mixture was obtained. In the latter case, the catalysts were prepared by a single co-impregnation of a hot solution of iron and cobalt nitrates onto pre-calcined TiO2 by incipient wetness procedures. CoFe/TiO2 prepared by impregnation was more active than that prepared by co-precipitation. The results of (CoFe)/TiO2 catalysts prepared using co-precipitation also showed a lower activity than the equivalent Co/TiO2 catalyst.6 Selectivity patterns of the bimetallic catalysts differed from those of monometallic ones. The Fe/TiO2 catalyst produced olefins and oxygenates, and also showed a much higher WGS activity compared to Co/TiO2. The latter exhibited better hydrocarbon chain growth probabilities and a very low selectivity towards methane.Duvenhage et al. also reported a bimetallic system prepared by dual impregnation. Here, cobalt was first deposited on titania by incipient wetness impregnation and then calcined. Then, the Co/TiO2 system was impregnated with iron and re-calcined. This catalyst was even less active than the corresponding co-impregnated system. It behaved like an iron-rich catalyst, though the Fe content was much lower than the Co content. Clearly, Fe segregates from CoFe systems in the iron-rich part of the phase diagram (Scheme 1). Other studies on CoFe catalysts, reported by Nakamura and co-workers, also showed that the product selectivity was correlated with the most abundant metal.7
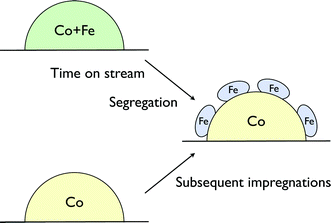 | ||
| Scheme 1 Co–Fe catalysts in the iron-rich part of the phase diagram resemble the structures of the catalyst prepared by subsequent impregnation. | ||
Deng et al. reported recently that silica-supported (CuCo)/SiO2 catalysts, prepared by impregnation of nitrates, gave a >40% selectivity towards higher alcohols.8 The structure and performance of catalysts were significantly affected by the impregnation sequence and synthesis conditions. The (CuCo)/SiO2 catalyst prepared by co-impregnation gave higher alcohol yields.
Another method uses metal carbonyl complexes as precursors of bimetallic active species. Chen et al. prepared carbon-supported Fe, Co and FeCo catalysts from metal carbonyl clusters using Co2(CO)8 and Fe3(CO)12.9 These catalysts do not require a high temperature reduction step because of the initial zero-valent state of the metal atoms and the absence of oxygen functional groups on the carbon surface. The Co/C catalyst derived from Co2(CO)8 produced only paraffins and maintained a steady activity. The Fe/C catalyst had much lower turnover frequencies but a much higher olefin:paraffin ratio. The major product was always methane, with the lowest selectivity (44 mol%) exhibited by iron and the highest selectivity (91.6 mol%) by Co. The mixed-metal clusters showed activity and selectivity behaviours intermediate between these two extremes, and the unpromoted samples displayed more Fe-like behaviour after a high temperature treatment. Adding potassium markedly decreased both activity and methane selectivity, yet enhanced olefin selectivity. The activity of the monometallic catalysts was increased after a high temperature reduction, while that of bimetallic catalysts either remained constant or decreased.
A lower activity was also observed for (CoFe)/TiO2 compared to Co/TiO2 prepared by the impregnation of metal carbonyl complexes.6,10 The catalysts were prepared using either a dimer complex of Fe and Co as the precursor or a mixture of monometallic complexes. The former produced more high molecular weight hydrocarbons with a better olefin selectivity, while the latter yielded more gasoline. Both carbonyl-derived CoFe systems showed a superior selectivity towards olefin products than that found for any of the incipient wetness and precipitated bimetallic systems. This high olefin selectivity and lower methane formation are in marked contrast to the observations of Chen et al.9 However, no advantage with respect to activity was observed when mixing Fe and Co compared to pure Co/TiO2 for either series. Another study using a (FeCo)/Al2O3 catalyst prepared by the impregnation of bimetallic carbonyl complexes also gave a lower activity compared to monometallic cobalt.11 The bimetallic catalyst produced more olefins. Conversely, the catalyst prepared from a mixture of Fe(CO)5 and Co2(CO)8 gave more methane and a higher CO conversion. The difference was attributed to an intermetallic CoFe compound on the surface in the former case.
Metal carbonyl clusters were also used to prepare (CoRu)/SiO2 and (CoRh)/SiO2 catalysts. The selectivity for hydrocarbons was over 90% with the former catalyst. High selectivity for C5–C13hydrocarbons (45–65%) and also selectivity for methane (< 20%) was exhibited by all the catalysts, except for Ru/SiO2 (prepared from H4Ru4(CO)12), which gave preferentially C8+ products (> 75%). Conversely, (CoRh)/SiO2 catalysts, with the exception of Co4/SiO2, gave less CO conversion and tended to produce methane and light hydrocarbons. The chain growth probability was related to the number of cobalt atoms in the precursor. The catalysts prepared from bimetallic clusters produced more oxygenates than those prepared from homometallic ones. However, when the selectivity for oxygenates increased, the activity decreased.12
A different activity pattern was observed for 1%(CoRu)/SiO2 catalysts prepared from carbonyl cluster precursors using a reflux method.13,14 The activity of the different precursors decreased in the order: Ru4/SiO2 ≫ (Co4 + Ru4)/SiO2 > CoRu3/SiO2 > Co3Ru/SiO2 > Co2Ru2(I)/SiO2 > Co2Ru2(II)/SiO2, Co4/SiO2, where the subscripts denote the molar content of each metal in the precursor complex and (I) refers to the hydridic carbonyl precursor. Here, using a CoRu(II) precursor gave an inactive catalyst, but when the same precursor was used with the impregnation method, the resulting catalyst was highly active, surpassing that prepared from the corresponding hydrido-carbonyl CoRu(I) complex. The catalyst prepared using a mixture of two homometallic clusters (Co + Ru) was the least active among those prepared by impregnation, but when using the reflux method it was second only to monometallic Ru/SiO2. Refluxed catalysts showed a generally higher activity per metal atom compared with the corresponding impregnated catalysts.
Both methods displayed a similar correlation between the ratio of the two metals in the precursor cluster and the catalytic activity, with a minimum at 1:1 Co:Ru. The Co2Ru2(II) precursor was again exceptional: for the impregnated catalysts, Co2Ru2(II)/SiO2 gave a much higher activity than Co2Ru2(I)/SiO2, but the opposite was observed with the reflux method.
The main products here were also straight-chained hydrocarbons (>90 C%). Generally, the products over refluxed catalysts were not as heavy as over impregnated catalysts. The selectivity for C5–C8hydrocarbons increased with respect to the heaviest C8+ products. The catalysts showed a remarkably low activity for the hydrogenation of olefins. Among the C3–C7hydrocarbons, α-olefins predominated over the corresponding paraffins. The heavy hydrocarbon fraction increased with the amount of ruthenium. Higher amounts of CO2 formed with all the refluxed catalysts, indicating that these favour the water-gas shift (WGS).
Studying (CoRu)/SiO2 catalysts prepared via ion-exchange, Reinikainen et al. found that these were more active than ones prepared from tetranuclear carbonyl complexes by reflux techniques.13 Moreover, the amounts of metal analysed after the reaction were better retained for the catalysts prepared by the ion-exchange method. The hydrogen and CO uptake of (CoRu)/SiO2 were much higher than for a physical mixture of Co/SiO2 and Ru/SiO2. Hence, ruthenium aids the reduction of cobalt in the bimetallic system. However, the Ru/SiO2 catalyst gave the highest activity in CO hydrogenation.14 The CO hydrogenation activity of (CoRu)/SiO2 was lower than expected according to the activities of Co and Ru, and that of the Co + Ru mixture was also lower than the average of its components. Regarding selectivity, (CoRu)/SiO2, Ru/SiO2 and the Co + Ru mixture all produced less methane than monometallic cobalt. However, (CoRu)/SiO2 gave less C5+ hydrocarbons than expected, and was relatively more selective for methanol (15.7%). This indicates that neither metal was present in its pure form.
These contradictory results affirm that the performance of F–T catalysts based on cluster precursors is governed by several factors: the structure and composition of the precursor cluster, the method of deposition of the precursor on the support, and the preparation process conditions are all of crucial importance. Additionally, calcination, drying rate and atmospheric composition all influence catalyst performance.
The homogeneous deposition–precipitation of complex nickel-iron cyanides was used in preparing (NiFe)/TiO2.15Catalysts prepared from K3Fe(CN)6 and Na2Fe(CN)5NO precursors exhibited a high activity and selectivity, whereas those prepared from K4Fe(CN)6 were inactive. This lack of activity was attributed to the high potassium concentrations. However, no influence of the nickel-iron ratio on the performance was observed. Comparing the precipitated catalysts with those prepared by the impregnation of carbonyl complexes showed that the latter produce more olefins and long-chain hydrocarbons.6 The methane level produced with the most active carbonyl bimetallic catalyst was 10%, while higher methane levels were obtained with precipitated catalysts.
This work also shows the role of counter ions in determining the properties of the final catalyst. This is an important parameter in co-precipitation methods as well. Oxidic phases are commonly precipitated using bases such as NaOH and KOH. These methods are prone to the introduction of alkali metals in the final catalyst, which thereby decrease its activity. Methods employing non-residue pH regulators (such as urea or ammonium carbonate) can avoid this problem. De Jong and co-workers16 used ammonium carbonate to prepare Co supported on carbon nanotubes. A basic solution containing CoCO3 and (NH4)2CO3 was heated, resulting in Co phase precipitation. NH3 removal from the solution and a consequent decrease in pH was responsible for the precipitation. The catalyst thus prepared was highly active and selective towards C5+ hydrocarbons. We could not find any examples of bimetallic catalysts prepared by this method. The use of easily removable precipitants and pH regulators should be preferred for bimetallic catalysts, as well to avoid unintended metal dopants.
Elsewhere, F–T synthesis was performed with a series of alumina-supported copper-iron catalysts, prepared via the deposition–precipitation of cyanide complexes.17 The initial activity of these catalysts was substantially higher than that of an analogous monometallic iron catalyst. However, the difference decreased with time-on-stream. Interestingly, here, the production of C5+, CO2 and olefins increased with the copper content of the catalyst. A rapid segregation of the iron as iron carbide was observed in this work, also after the exposure of reduced bimetallic CuFe catalysts to CO/H2 mixtures at 275 °C. The deactivation of the catalysts was assigned to the formation of χ-Fe5C2 carbide, which is fairly stable against reaction of carbidic carbon with hydrogen.
Bimetallic catalysts were also prepared using a plasma-spraying technique. In this approach, catalyst precursors were directly plasma-sprayed on the inner surface of the reactor. The CoFe catalyst thus prepared exhibited a higher CO conversion and hydrocarbon selectivity than a cobalt catalyst under similar process conditions. The plasma-sprayed CoFe catalyst was also more active than a powdered CoFe catalyst.18
Another approach used for preparing bimetallic catalysts is anchoring hydridic complexes, such as HFeCo3(CO)12, onto basic supports, such as silica modified by amino donor functions.19 At 20% conversion (1 atm and 240 °C), a maximum selectivity was observed for C6, as well as a high olefin content (∼95% in the C3–C5 range). The selectivity for methane was 12.5%. Increasing the pressure led to improved conversions, higher methane selectivity and less olefins. However, no comparative data for monometallic catalysts were reported.
Elsewhere, using partially degraded CoFe complexes, a normal selectivity pattern was observed with iron-cobalt alloy catalysts under steady-state conditions and elevated pressures.20 Interestingly, the unsupported catalysts maintained a remarkably high activity and selectivity, particularly in view of the hydrogen-poor conditions.
Cabet et al. prepared cobalt-iron alloy/cobalt-doped magnetite catalysts (spinel structure), in which the Co and Fe are in a metallic state without any reducing treatment. The preparation was based upon the coprecipitation of hydroxides in a boiling basic medium. Depending on the catalyst treatment, products ranged between hydrocarbons and CO2. When the spinel was preserved, a selectivity for hydrocarbons of 56.4% (31.1% C2–C4) was obtained at a CO conversion of 5%.21
Composition and activity
Several studies have reported an enhanced activity by the mixing of two metals. Regarding the Fe–Co system, Arai et al. observed higher activity for FeCo/TiO2 bimetallic catalysts compared to the pure metal catalysts (Table 1, ESI†).22 Moreover, bimetallic catalysts were more selective towards higher molecular weight hydrocarbons. Among the compositions studied, catalysts with equal amounts of both metals, e.g. 10%(50Fe50Co)/TiO2, showed the highest activity, the highest yield of C6+hydrocarbons and the lowest methane selectivity. Similar results were obtained with CoNi/TiO2 and NiFe/TiO2. Another study also reported an enhanced CO conversion for FeCo/silica bimetallic catalysts.10 Adding iron to the cobalt catalyst increased the CO conversion compared to Co/silica. The 15%(33Fe67Co)/SiO2 catalyst was the most active at 533K. However, the major products were C1–C4hydrocarbons. The selectivity shifted towards heavier hydrocarbons at higher temperatures.Higher CO conversions were observed for a 12% (30Fe70Co)/ZrO2 catalyst compared to 11%Fe/ZrO2. The bimetallic catalyst showed a lower methanation activity, and higher C4 and C2–C4olefin selectivities.23 Another study has reported that the characteristics of “bimetallic” FeCo on Y zeolite are dominated by Fe. The low molecular weight olefin:paraffin ratios were similar for FeCo/HY and Fe/HY.24 Ishihara et al. have reported an enhanced activity for CoNi/SiO2 catalysts compared to monometallic Co and Ni on silica.25 10%(50Ni50Co)/SiO2 gave a six-fold higher conversion than Co and/or Ni. The CoNi alloy catalysts showed a high selectivity for higher hydrocarbons, particularly gasoline compounds, compared to Ni alone. Comparing catalysts on different supports showed that electron-donating oxides such as MgO, PbO and ZnO lowered the overall activity of the 50Co50Ni alloy.26 The CO conversion, as well as the chain growth probability, was higher over 10%(50Ni50Co)/TiO2 and 10%(50Ni50Co)/MnO2.
Iglesia et al. have reported enhanced F–T reaction rates for bimetallic 12%(99.4Co0.6Ru)/TiO2. The yield (normalised per second and cobalt content) doubled for the bimetallic precursor after reduction in H2 compared to Co/TiO2, and the selectivity to C5+ products also increased. The rate and C5+ selectivity were increased further when the catalyst was calcined prior to reduction. Bimetallic catalysts also increased the molecular weight and paraffin content of the F–T products. A similar bimetallic synergy was observed for 23%(99.3Co0.7Ru)/SiO2. In this case, calcination was required to induce the bimetallic synergy. The deactivation of bimetallic catalysts was qualitatively similar to that of Co. However, a hydrogen treatment at 483 K for 14 h restored rates and selectivities of the bimetallic catalysts to values similar to those observed during the initial 24 h of catalyst operation (this was not observed for monometallic Co catalysts).
Xiao et al. have reported an increase in the activity of bimetallic catalysts with increasing molar Co/Ru ratio. Catalysts made from homometallic Co or Ru carbonyl precursors were less active than any bimetallic combinations.27
While some publications have reported an enhanced activity for bimetallic catalysts compared to monometallic catalysts, many others reported an opposite trend (Fig. 2). The contradictory results on the selectivity also becomes clear when C5+ selectivity is plotted against conversion (Fig. 3). Kiviaho and co-workers prepared silica-supported bimetallic catalysts Co4 −nRun and Co4 −nRhn by impregnating tetranuclear cobalt, CoRu, CoRh and rhodium carbonyl clusters as catalyst precursors.28 Notably, the catalytic activities were lowest for 1:1 Co:Ru and Co:Rh ratios. The authors attributed this difference to the different pre-treatments of the catalysts. Similarly, Ma et al. observed a decrease in CO conversion with increasing Fe content for FeCo/SiO2 catalysts.29 The results, obtained at a steady-state after stabilizing for over 50 h, showed that Fe-rich catalysts needed higher reaction temperatures to reach a given CO conversion compared to Co-rich examples. Moreover, the C2–C5 product fraction increased at the expense of C5+. Duvenhage's group have also reported a decrease in the overall activity of CoFe/TiO2 bimetallic systems compared to that of pure Co/TiO2.30 The CO conversion using 10%(50Fe50Co)TiO2 was less than half that obtained with 10%Co/TiO2, but higher than that of 10%Fe/TiO2. The increase in iron content shifted the products to lower hydrocarbons, LPG and petrol fractions. Methane selectivities for the bimetallic catalysts ranged between 15–20 wt%, depending on the metal ratios. A lower CO TOF was reported for 5%(80Fe20Co)/silica compared to monometallic catalysts.31
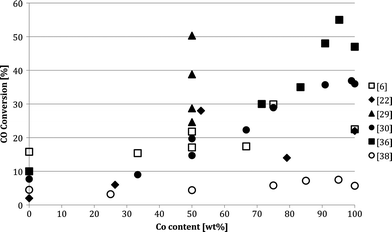 | ||
| Fig. 2 Conversion as a function of Co:Fe relative ratio, as reported by different authors. | ||
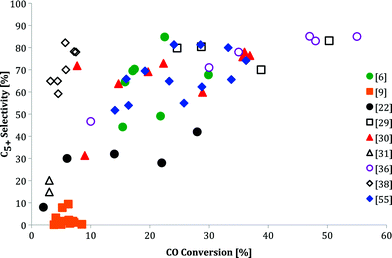 | ||
| Fig. 3 C5+ selectivity as a function of CO conversion, as reported in nine different F–T studies over Co–Fe catalysts. No clear relationship between the two value sets can be recognized. | ||
Amelse et al. have reported a lower activity for NiFe/SiO2 than for either single metal.32 The results of Arai et al., however, show a higher activity, higher selectivity for C6+hydrocarbons and a lower selectivity for methane with NiFe/TiO2 than with the individual components.22 Here, 10%(75Ni25Fe)/TiO2 was the most active, giving more C2–C5olefins than methane. Once again, however, different studies report different trends for NiFe catalysts. Ishihara et al. have reported a maximum activity for a 10%(75Ni25Fe)/TiO2 sample, but the selectivity for C2+hydrocarbons did not depend on the Ni:Fe ratio.33 Conversely, Udrea et al. found a composition-dependent behaviour for NiFe/Al2O3 catalysts.34 The highest activity and selectivity for C2+ hydrocarbons were obtained with 10%(80Ni20Fe)/Al2O3 and 10%(50Ni50Fe)/Al2O3. The difference in activity and selectivity was explained by two competing processes: the formation of C2–C5hydrocarbons and their hydrogenolysis. The increase in methane at higher Ni loadings was attributed to C–C bond scission.16
In another study using equimolar (50Ni50Fe)/SiO2, the product distributions were similar to those of 100Ni/SiO2.35 Both the NiFe alloy phase and an unreduced ferrous phase were identified by post-reaction, room temperature Mossbauer spectroscopy. Notably, no bulk carbide formation was detected in the alloy phase after 6 h using a feed of 3.3:1 H2:CO at 250 °C.
Many of the CoFe catalyst systems show a non-linear activity–composition relationship. For example, Tavasoli et al. have reported a higher CO conversion with increasing Fe content up to 0.5 wt% for CoFe supported on carbon nanotubes.36 However, higher Fe loadings decreased the activity. The monometallic cobalt catalyst gave preferentially C5+ hydrocarbons, while monometallic iron was selective for C1–C5 products. Adding small amounts of Fe to the Co did not change the selectivity. However, higher amounts of iron gave more methane and less C5+ products. Notably, monometallic iron gave 3.5 times as much olefin compared to monometallic cobalt. Using bimetallic catalysts, the olefin:paraffin product ratio increased with the addition of iron. A reduced hydrogen mobility, as well as lower hydrogen adsorption rates, could explain the decrease in olefin hydrogenation for the iron-containing catalysts.37 Similarly, the alcohol selectivity of monometallic iron was about 4 times higher than that of monometallic cobalt. Once again, adding iron to the cobalt yielded more alcohols.
Lögdberg et al. also observed that alloying cobalt with small amounts of iron improved the activity of Co/Al2O3, especially when using hydrogen-poor feeds (H2:CO ratio = 1:1).38 This was accomplished without increasing the WGS activity, and is therefore thought to be due to a higher F–T activity of the CoFe mixtures compared to monometallic Co. Conversely, alloying iron with small amounts of Co increased the relative WGS activity and lowered the F–T activity compared to monometallic Fe. The bimetallic catalysts showed essentially no synergy effects with respect to hydrocarbon selectivities and olefin/paraffin ratios. Small amounts of Fe increased the overall activity but reduced the C5+ fraction selectivity. The authors related the results to a dilution effect of Co with Fe. They also observed a more pronounced deactivation effect for catalysts with a higher Fe content.
Copper may facilitate the reduction of iron oxide to metallic iron during hydrogen activation by lowering the reduction temperature which, in turn, reduces the sintering of the metallic iron formed.39 Some early patents claim that adding Cu enhances the selectivity for oxygenated products.40 In one study, more C5+ was obtained using FeCu than with monometallic iron.41 However, another study concluded that copper does not alter activity or selectivity.42 Hence, the effects of alloying Cu and Fe remain a moot point.
Wielers et al. have observed that using 20%(FeCu)/SiO2, the F–T activity passes through a maximum at a Cu content of 10–20%.40 A monometallic copper catalyst was inactive under these conditions.43 The reducibility of 20%(100Fe)/SiO2 catalyst was rather low, and the reduced iron particles were (partly) encapsulated by iron(II) silicate.44Iron present in the bimetallic particles was not encapsulated by iron(II) silicate. This led to a higher F–T activity for 20%(50Fe50Cu)/SiO2. Copper did not affect the probability of chain growth, nor did it prevent iron clusters present in a copper matrix from forming iron carbide. It was observed that CO could induce at 25 °C a significant iron enrichment in the surface of the bimetallic Fe–Cu particles.44 Hence, it was suggested that the iron–iron carbide concentration on the surface is appreciably higher than the iron–iron carbide bulk concentration of the bimetallic particles during the F–T process, leading to islets (or even a complete layer) of this active phase. With increasing copper concentration, these islets will become smaller, gradually decreasing the activity. Changes in selectivity were only observed at higher copper concentrations, supporting the concept that at low copper fractions the copper atoms are covered by iron–iron carbide islets (Scheme 2). Both the CO2 and CH4 selectivities increased with increasing copper concentration, at the expense of olefins. This shows that the WGS reaction and the hydrogenation reaction by exposed copper becomes significant at higher Cu concentrations. Hence, the surface concentrations of individual catalyst components play a major role in determining the activity and selectivity. Filamentary carbon growth was observed on the monometallic iron particles as well as the on iron–iron carbide islets present on the surface of the bimetallic particles. It was suggested that this growth was suppressed at high Cu concentrations and the deactivation rate was reduced.47 However, longer time-on-stream studies are necessary to confirm this hypothesis.
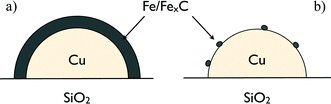 | ||
| Scheme 2 Schematic of two 20%(xFeyCu)/SiO2 catalysts with different x and y ratios. (a) a copper-poor catalyst (x ≫ y); (b) a copper-rich catalyst (x ≪ y). | ||
Reduction behaviour of bimetallic catalysts and F–T activity
In general, mixed CoFe catalysts have a different reduction behaviour to monometallic ones. TPR studies of monometallic cobalt generally show two peaks, attributed to a two-step reduction of Co3O4 (Co3O4→CoO→Co0; Fig. 4, top).45,46 The first reduction step occurs at ∼300 °C and the second at 440 °C. Typical TPR profiles of Fe/SiO2 show three peaks at around 405, 580 and 720 °C, respectively. These are consistent with three consecutive reduction steps: α-Fe2O3→Fe3O4→FeO→Fe (Fig. 4, bottom).47 The reduction behaviour of bimetallic catalysts depends on several factors, such as the preparation method, type of precursors, pre-treatment and support. Some studies have reported a higher reducibility for CoFe than for the individual components. TPR studies of CoFe/SiO2, prepared by impregnation, have shown a shift to lower temperatures with increasing cobalt content.48 Similar results were obtained using TiO2.30 More reduction was observed for 10%(50Fe50Co)/TiO2 than for 5%(100Co)/TiO2 or 10%(100Co)/TiO2.49 However, less reducibility was reported for 10%(25Fe75Co)/TiO2 and 10%(75Fe25Co)/TiO2 catalysts compared to pure (100Co)/TiO2.49 The extent of reduction of monometallic iron catalysts was roughly half that of monometallic Co, with an inverse relationship between reduction and dispersion.49 This shows that dispersed particles are not easily reduced.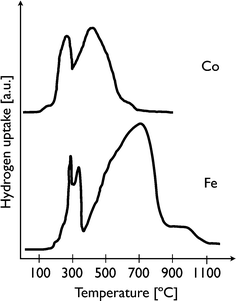 | ||
| Fig. 4 Typical TPR plots of the cobalt (top) and the iron (bottom) reduction. | ||
Similar trends were observed for catalysts prepared by precipitation. Higher reduction levels were reported for 10%(50Fe50Co)/TiO2 compared to monometallic cobalt. TPR studies by Ma et al. showed that reducibility decreases with increasing Fe content.48
Catalysts prepared by precipitation were more easily reduced than those made by impregnation.6 The shift in the two reduction peaks associated with iron and cobalt was lowered by about 100 °C. The catalyst prepared from a mixture of metal carbonyl complexes ([CpFe(CO)2]2/Co(CO)8) reduced at a slightly lower temperature than mixed metal carbonyl dimer (Cp(CO)2FeCo(CO)4). This suggests different Fe/Co interactions with the TiO2 support. The reduction behaviour is further complicated by the formation of mixed phases of Fe/Co and support. For example, an ilmenite (FeTiO3) phase was identified for Fe/TiO2 that was reduced for 16 h at 350 °C.30
Higher reduction was also observed upon adding Cu, Au and Ag to Co. Jacobs et al. showed that cobalt reducibility increased from 50% for monometallic Co/Al2O3 to 93% for 15%(1.63Cu98.37Co)/Al2O3.50 However, the activity in terms of CO conversion and selectivity towards higher hydrocarbons decreased significantly for bimetallic catalysts. The authors also observed a higher reducibility for AgCo and AuCo catalysts. In the case of AgCo, the reducibility correlated linearly with the CO conversion and selectivity towards higher hydrocarbons. For AuCo, the linear relationship was valid when the amount of Au was <5%. Reducibility and dispersion were not correlated.
Guczi et al. have studied the structure and catalytic activity of (ReCo)/Al2O3 and (ReCo)/NaY zeolite, prepared by sol/gel and incipient wetness techniques.51 They established that Re promotes cobalt reduction, but that the formation of bimetallic particles depends on the support: sol/gel prepared and Al2O3-supported samples produced bimetallic particles, whereas using NaY as a support did not. Rhenium promoted the cobalt activity and the selective formation of higher hydrocarbons. The alumina-supported sample was more active than the zeolite one. This was partly due to the higher reducibility of cobalt in the former. However, the selectivity was unchanged.
In another study, Reinikainen et al. found that both CO conversion and catalyst reducibility were higher for CoRu catalysts compared to monometallic cobalt.13,14 However, monometallic ruthenium was more active and selective towards higher hydrocarbons compared to CoRu, though the reducibility of the former was lower.
TPR studies of CoFe catalysts typically show that cobalt facilitates the reduction of iron.30,52,53 However, the effect of adding iron on CoFe alloys is much more complex. Confusingly, Tavasoli et al. observed a decrease in reducibility with the addition of high amounts of iron to Co/TiO2, though lower Fe contents in the bimetallic catalysts increased the reducibility.25 When iron is present in high concentrations, the catalyst takes on the properties of this metal. Hence the net result is an increase in the Co reduction temperature.54
Generally, higher reduction levels go hand-in-hand with higher activity. This is the case, for example, for 10%(50Fe50Co)/TiO2.55Methane production levels increased with reduction temperature, while the C5+ fraction diminished. This was also observed for Co/Al2O3 and Co/TiO2, reduced between 380 and 530 °C.56 Increased olefin production was observed for 10%(50Fe50Co)/TiO2 and for Co/TiO2 at higher reduction temperatures.57 The WGS activity was low, observable only at low calcination and reduction temperatures. Tavasoli et al. observed a linear relationship between reducibility and dispersion with the CO conversion, and an inverse relationship with methane formation.36 Ma et al. also observed a linear relationship between CO conversion and reducibility.48
The relationship between activity and reducibility, however, is more complex for bimetallic catalysts. Thus, 10%(50Fe50Co)/TiO2 showed more reduction compared with either 5%(100Co)/TiO2 or 10%(100Co)/TiO2. However, the CO conversion values were 36.3, 42.5 and 63.9%, respectively.49 Thus, adding iron increases the reducibility of cobalt in this case, but does not enhance its catalytic performance.
Moreover, the relationship between reducibility and catalytic performance changes at higher reduction temperatures.55 CoFe catalysts reduced at 400 °C showed a lower activity than those reduced at 300 °C, though the reduction level was high.
The influence of crystallite size
There is a clear size–activity relationship in the case of monometallic cobalt catalysts.3 When using large cobalt particles supported on alumina, silica and titania, the yield and the selectivity are independent of the cobalt particle size.58,59 However, the F–T turnover frequencies and the selectivity of cobalt catalysts supported on carbon nanofibres were much lower when the particles were smaller than 8 nm.60 Decreasing the particle size to 8 nm resulted in more methane and more olefins, at the expense of C5+ products. Notably, no such trend could be discerned from the available literature for bimetallic catalysts. No change in particle size was observed for a series of CoNi and CoRu bimetallic catalysts, though their activity differed significantly.25 However, a study on CoFe catalysts reported a decrease in particle size for bimetallic catalysts compared to monometallic catalysts.Deactivation behaviour of bimetallic F–T catalysts
Readers who have reached this point will not be surprised that several reports on the behaviour of bimetallic catalysts towards deactivation are, to put it mildly, inconsistent. Duvenhage et al. observed qualitatively similar deactivation profiles for monometallic and bimetallic (CoFe)/TiO2 catalysts.30 A loss in CO conversion with continued use was seen, especially over the first 50 h on-stream.30 Similar findings for (CoFe)/SiO2 catalysts were made by Butt and Schwartz.61 They also reported that the deactivation effect is more pronounced when the amount of iron in the catalyst increases. At low conversions, bimetallic systems were typically more stable.61 In another study, Duvenhage et al. observed that the lower the temperature of reduction, the higher the initial activity, but the more unstable was the system over the first 50 h on-stream.55According to Lögdberg et al., bimetallic catalysts were more stable against permanent deactivation compared to the pure Co catalyst investigated for 120 h on-stream.38 Still, this would present no advantage in real F–T conditions, since the bimetallic catalysts suffer from a severe temporary deactivation with respect to both WGS and F–T activity upon exposure to the water pressures prevailing at sufficiently high conversion levels.
Tavasoli et al. studied the deactivation behaviour of the (CoRu)/γ-Al2O3 catalyst along the catalytic bed over 850 h on-stream.62 The reactor bed was divided into four sections and samples of spent catalyst from each section were studied for structural changes (Scheme 3). Rapid deactivation was observed during first 200 h. The physical properties of the catalyst in the first two sections did not change. Interaction of cobalt with alumina and the formation of mixed oxides of the form xCoO·yAl2O3 and CoAl2O4 increased along the catalytic bed. The reducibility and dispersion decreased by 2.4–25.5% and 0.5–8.8% for the catalyst in sections 1 and 4, respectively. Moreover, the particle diameter increased by 0.8–6.1% for the catalyst in sections 1 and 4, respectively, suggesting a higher rate of sintering at last catalytic bed.
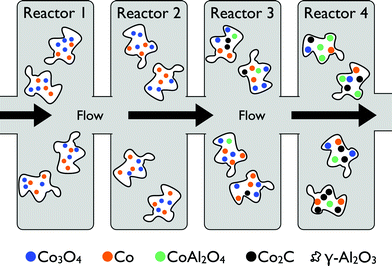 | ||
| Scheme 3 A fluidised-bed reactor concept showing the four sections from which samples were analysed during the first 200 h on-stream (adapted from Tavasoli et al.62). | ||
Factors leading to the lack of a consistent structure–activity relationship
The above analysis of the published literature reveals that several factors complicate the comparative analysis of the performance of bimetallic catalysts. One of these is the diversity of pre-treatment conditions. There are no accepted rules for pre-treatment. The problem is that even small changes in pre-treatment conditions can influence catalytic activity. An example is the study of the effect of calcination and reduction conditions on the activity of (CoFe)/TiO2.55 The activity was almost doubled when the reduction temperature was increased from 250 to 300 °C. The selectivity for C5+ hydrocarbons was also increased at 300 °C. However, reduction at still higher temperatures decreased both the activity and C5+ selectivity. TPO studies of these catalysts showed an increase in the reduction level with increasing reduction temperature, similar to the monometallic catalysts.A related issue is the phases that form during the pre-treatment/reduction stages. Depending on the conditions, the two metals may alloy on the support, exist as separate species or react separately with the support, forming mixed oxides. Often, it is unclear whether alloys really form or not. Duvenhage et al. have suggested that at sufficiently high loadings, CoFe alloys can form on TiO2, Ag2O3 and SiO2.30 According to this, pure Co/TiO2 exists as fcc or hcp, while pure Fe/TiO2 exists as bcc. The 10%(50Fe50Co)/TiO2 catalyst had a bcc alloy of Co and Fe, while in lower Fe content catalysts the Co fcc structure was retained. The possibility of forming a CoFe alloy at some compositions has also been indicated by Lögdberg et al.38 Mossbauer studies conducted on CoFe/SiO2 catalysts also suggested the formation of alloy particles.63 However, another study on silica-supported bimetallic calcined samples showed separate Co3O4 and Fe2O3 phases, with no cobalt–iron mixed oxides.64 After hydrogen reduction at 500 °C, Co0 and Fe0 metallic phases formed in monometallic samples, whilst bimetallic phases comprised mainly two phases: Co0 (major phase, 76%) and a CoFe alloy. The crystallite size of the alloyed phase increased with iron content, whereas that of the Co0 phase remained constant. An improved catalyst performance in this case was attributed to a better dispersion of the active phase.65
The presence of small amounts of CoFe alloys is also related to an increased selectivity for alcohols.36 However, it is unclear whether the alloy phases remain stable during the F–T process, or whether such small alloy amounts are actually causing the change in selectivity. The low amounts of alloy phases could explain the absence of a synergetic behaviour for Co + Fe. This combination does not exhibit the additive properties for activity and selectivity expected from combining the individual metals.
Remarkably, other groups did not observe alloy formation for mixed metal catalysts. X-Ray diffraction of fresh CoFe catalysts by Ma et al. revealed hematite and Co3O4 spinel phases on iron-rich and cobalt-rich catalysts, respectively.48 The spent iron-rich catalyst showed iron carbide reflections, while the cobalt-rich catalyst displayed patterns typical of cobalt oxide (CoO) and metallic cobalt. EXAFS studies on CoCu, CoAg and CoAu revealed only homometallic bonds, suggesting that the metals formed separate phases.50
Another parameter affecting the properties of the catalyst could be the preferential surface segregation of one of the components during pre-treatment, reduction or reaction. An early example was shown for CuNi alloys, where the activity per unit area decreased by a factor of 220 upon alloying.66 No changes in the electronic structure of the nickel atoms were observed. Analysis using low energy Auger electrons (at 100 eV, about 50% of the total signal comes from the first layer) showed a segregation of copper on the surface.67,68
Similarly, surface enrichment of Fe was observed by XPS studies of a Fe–Co/Al2O3 catalyst.38 The surface concentration was similar to the nominal one after 300 s of sputtering for 12%(15Fe85Co)/Al2O3, while 12%(25Fe75Co)/Al2O3 showed an enrichment in surface iron. When this enrichment was higher, the degree of Co reduction was lower. Thus, a thick iron-rich surface layer “protects” the cobalt oxide in the same layer and in the underlying layers from reduction. Surface iron enrichment was also observed by XPS for CoFe catalysts on SiO2, TiO2, MgO and ZrO2.30,69CO and H2 desorption studies showed that the desorption profile of (CoFe)/TiO2 is similar to that of monometallic Fe/SiO2.70 The surface segregation extent and rate depend on several factors, including metal surface energy, pre-treatment conditions and the M1/M2 ratio. Generally speaking, when one metal migrates to the surface, the mixed catalyst will resemble the migrated component. This explains some of the lack of uniformity in reducibility and catalytic performance.
Conclusions
Soberly, the analysis of the literature on bimetallic F–T catalysts as such does not show any direct relationship between structure and catalyst performance. Mostly, this reflects the fact that studies look at F–T catalysts from different perspectives. The many contradictory results are due to a lack of information regarding the actual catalyst. Comparing different catalysts, sharing, for example, a similar nominal composition or initial reaction conditions, can be misleading. Such catalysts may differ in many respects because of the varied preparation or pre-treatment conditions. These differences remain unfortunately hidden due to an incomplete overlap of characterization details. Articles reporting the full structural characterization of the catalyst typically lack details about the particle size of the active phase (or its surface area) and/or the reactor type used. Note that full characterization of a system is a time consuming task indeed. For example, Duvenhage and Coville carried out a comprehensive study on CoFe bimetallic catalysts: the first paper of this series appeared in 1997 and the last one was published in 2005.With rare exceptions, most studies report activity and selectivity values after a few hours of stabilization. In our opinion, since the surface of the catalyst undergoes extensive reconstruction and effects such as surface segregation are important under F–T reaction conditions, studies and characterisation should be done after at least two weeks (∼350 h) on-stream. For researchers, this is an unwelcome conclusion. Nevertheless, it is necessary if we are to really understand and improve on the current F–T catalyst systems. Only a systematic study under carefully controlled conditions for longer times-on-stream will give us useful answers.
References
- F. Fischer and H. Tropsch, Brennst. Chem., 1923, 4, 276 Search PubMed.
- C. H. Bartholomew, in book of abstracts from AIChE Spring Meeting, 2003 pp. 83b Search PubMed.
- A. Y. Khodakov, Catal. Today, 2009, 144, 251–257 CrossRef CAS.
- A. Y. Khodakov, W. Chu and P. Fongarland, Chem. Rev., 2007, 107, 1692–1744 CrossRef CAS.
- E. D. Smit and B. M. Weckhuysen, Chem. Soc. Rev., 2008, 37, 2758–2781 RSC.
- D. Duvenhage and N. Coville, J. Mol. Catal. A: Chem., 2005, 235, 230–239 Search PubMed.
- M. Nakamura, B. J. Wood, P. Y. Hou and B. Wise, Studies in Surface Science and Catalysis, Elsevier, Amsterdam, 1981 Search PubMed.
- S. Deng, W. Chu, H. Xu, L. Shi and L. Huang, J. Nat. Gas Chem., 2008, 17, 369–373 Search PubMed.
- A. Chen, M. Kaminsky, G. Geoffroy and M. Vannice, J. Phys. Chem., 1986, 90, 4810–4819 Search PubMed.
- V. A. de la Peña O'Shea, M. C. A. Alvarez-Galvan, J. M. Campos-Martin and J. L. G. Fierro, Appl. Catal., A, 2007, 326, 65–73 Search PubMed.
- T. Khomenko, A. Kadushin, N. Kutyreva, Y. Maksimov, V. Matveev, A. Slinkin, E. Fedorovskaja and V. Khandozhko, J. Mol. Catal., 1989, 51, L9–L14 CrossRef CAS.
- J. Kiviaho, M. Reinikainen, M. K. Niemela, K. Kataja and S. Jaaskelainen, J. Mol. Catal. A: Chem., 1996, 106, 187–195 Search PubMed.
- M. Reinikainen, J. Kiviaho, M. Kroger, M. Niemela and S. Jaaskelainen, J. Mol. Catal. A: Chem., 1997, 118, 137–144 CrossRef CAS.
- M. Reinikainen, M. K. Niemelä and N. Kakuta, Appl. Catal., A, 1998, 174, 61–75 CrossRef CAS.
- J. van de Loosdrecht, A. J. van Dillen, A. A. Van Der Horst, A. M. Van Der Kraan and J. W. Geus, Top. Catal., 1995, 2, 29–43 Search PubMed.
- G. L. Bezemer, P. B. Radstake, V. Koot, A. J. van Dillen, J. W. Geus and K. P. de Jong, J. Catal., 2006, 237, 291–302 CrossRef CAS.
- E. Boellaard, A. M. Van Der Kraan, A. B. P. Sommen, J. H. B. J. Hoebink, G. B. Marin and J. W. Geus, Appl. Catal., A, 1999, 179, 175–187 Search PubMed.
- A. K. Dalai, N. N. Bakhshi and M. N. Esmail, Fuel Process. Technol., 1997, 51, 219–238 Search PubMed.
- R. Hemmerich, W. Keim and M. Roper, J. Chem. Soc., Chem. Commun., 1983, 428–430 RSC.
- R. Snel, Can. J. Chem. Eng., 1989, 67, 992–998 Search PubMed.
- C. Cabet, A. Roger, A. Kiennemann, S. Lakamp and G. Pourroy, J. Catal., 1998, 173, 64–73 Search PubMed.
- H. Arai, K. Mitsuishi and T. Seiyama, Chem. Lett., 1984, 1291–1294 Search PubMed.
- Y. Bi and A. K. Dalai, Can. J. Chem. Eng., 2008, 81, 230–242 Search PubMed.
- T. Lin, L. Schwartz and J. B. Butt, J. Catal., 1986, 97, 177–187 Search PubMed.
- T. Ishihara, N. Horiuchi, T. Inoue and K. Eguchi, J. Catal., 1992, 136, 232–241 Search PubMed.
- T. Ishihara, N. Horiuchi, K. Eguchi and H. Arai, J. Catal., 1991, 130, 202–211 Search PubMed.
- F.-S. Xiao, R.-R. Xu, M. Ichikawa, D. F. Shriver, W. Henderson, X.-X. Guo and Q. Xin, Sci. Chin. (Ser. B), 1993, 36, 151 Search PubMed.
- J. Kiviaho, M. Reinikainen, M. K. Niemela, K. Kataja and S. Jaaskelainen, J. Mol. Catal. A: Chem., 1996, 106, 187–195 Search PubMed.
- X. Ma, Q. Sun, F. Cao, W. Ying and D. Fang, J. Nat. Gas Chem., 2006, 15, 335–339 Search PubMed.
- D. Duvenhage and N. Coville, Appl. Catal., A, 1997, 153, 43–67 CrossRef.
- K. Arcuri, L. Schwartz, R. Piotrowsi and J. B. Butt, J. Catal., 1984, 85, 349–361 Search PubMed.
- J. A. Amelse, L. H. Schwartz and J. B. Butt, J. Catal., 1981, 72, 95–110 Search PubMed.
- T. Hishihara, K. Eguchi and H. Arai, Chem. Lett., 1986, 1695–1698 Search PubMed.
- I. Udrea, E. Groner and L. Frunza, React. Kinet. Catal. Lett., 1994, 53, 451–458 Search PubMed.
- G. B. Raupp and W. N. Delgass, J. Catal., 1979, 58, 337–347 CrossRef CAS.
- A. Tavasoli, M. Trépanier, R. M. M. Abbaslou, A. K. Dalai and N. Abatzoglou, Fuel Process. Technol., 2009, 90, 1486–1494 Search PubMed.
- S. Jam, M. Ghalbi Ahangary, A. Tavasoli, K. Sadaghiani and A. Nakhaeipour, React. Kinet. Catal. Lett., 2006, 89, 71–79 Search PubMed.
- S. Lögdberg, D. Tristantini, O. Borg, L. Ilver, B. Gevert, S. Jaras, E. A. Blekkan and A. Holmen, Appl. Catal., B, 2009, 89, 167–182 Search PubMed.
- Catalysis, Science and Technology, ed. M. E. Dry, Springer-Verlag, New York, 1981 Search PubMed.
- A. F. H. Wielers, G. W. Koebrugge and J. W. Geus, J. Catal., 1990, 121, 375–385 CrossRef CAS.
- Y. T. Shah and A. J. Perrotta, Ind. Eng. Chem. Prod. Res. Dev., 1976, 15, 123 CAS.
- I. E. Wachs, D. J. Dwyer and E. Iglesia, Appl. Catal., 1984, 12, 201 Search PubMed.
- Catalysis, Science and Technology, ed. M. A. Vannice, Springer-Verlag, Berlin, 1982 Search PubMed.
- A. F. H. Wielers, A. J. H. M. Kock, C. E. C. A. Hop, J. W. Geus and A. M. Van Der Kraan, J. Catal., 1989, 117, 1 CrossRef CAS.
- J. H. A. Martens, H. E. J. van't Blik and R. Prins, J. Catal., 1986, 97, 200 CAS.
- Y. Okamoto, K. Ngata, T. Adachi, T. Imanaka, K. Inamara and T. Takyu, J. Phys. Chem., 1991, 95, 310 CrossRef CAS.
- R. Brown, M. E. Cooper and D. A. Whan, Appl. Catal., 1982, 3, 177 Search PubMed.
- X. Ma, Q. Sun, W. Ying and D. Fang, J. Nat. Gas Chem., 2009, 18, 232–236 Search PubMed.
- D. Duvenhage and N. Coville, Appl. Catal., A, 2005, 289, 231–239 Search PubMed.
- G. Jacobs, M. C. Ribeiro, W. Ma, Y. Ji, S. Khalid, P. T. A. Sumodjo and B. H. Davis, Appl. Catal., A, 2009, 361, 137–151 Search PubMed.
- L. Guczi, G. Stefler, Z. Koppany and L. Borko, React. Kinet. Catal. Lett., 2001, 74, 259–269 Search PubMed.
- R. J. Kaleficzuk, J. Chem. Technol. Biotechnol., 1994, 59, 73 Search PubMed.
- D. Banerjee and D. K. Chakrabarty, Ind. J. Technol., 1990, 30, 81 Search PubMed.
- M. Rameswaran and C. H. Bartholomew, J. Catal., 1989, 117, 218 CrossRef CAS.
- D. Duvenhage and N. Coville, Appl. Catal., A, 2002, 233, 63–75 Search PubMed.
- R. C. Reuel and C. H. Bartholomew, J. Catal., 1984, 85, 78 CrossRef.
- G. Lohrengel, M. R. Dass and S. Baern, Preparation of Catalysts, Elsevier, Amsterdam, 1979 Search PubMed.
- S. L. Soled, E. Iglesia, R. A. Fiato, J. E. Baumgartner, H. Vroman and S. Miseo, Top. Catal., 2003, 26, 101 CrossRef CAS.
- Ø. Borg, S. Eri, E. A. Blekkan, S. Storsæter, H. Wigum, E. Rytter and A. Holmen, J. Catal., 2007, 248, 89 CrossRef CAS.
- G. L. Bezemer, J. H. Bitter, H. P. C. E. Kuipers, H. Oosterbeek, J. E. Holewijn, X. Xu, F. Kapteijn, A. J. v. Dillen and K. P. d. Jong, J. Am. Chem. Soc., 2006, 128, 3956 CrossRef CAS.
- J. B. Butt, L. H. Schwartz, M. Baerns and R. Malessa, Ind. Eng. Chem., 1984, 23, 51 Search PubMed.
- A. Tavasoli, M. Irani, R. M. M. Abbaslou, M. Trepanier and A. K. Dalai, Can. J. Chem. Eng., 2008, 86, 1070–1080 Search PubMed.
- R. Stanfield and W. Delgass, J. Catal., 1981, 72, 37–50 CrossRef CAS.
- V. A. de la Peña O'Shea, N. N. Menendez, J. D. Tornero and J. L. G. Fierro, Catal. Lett., 2003, 88, 123–128 Search PubMed.
- V. A. de la Peña O'Shea, M. C. A. Ivarez Galvan, J. M. Campos Martın, N. N. Menendez, J. D. Tornero and J. L. G. Fierro, Eur. J. Inorg. Chem., 2006, 5057–5068 Search PubMed.
- V. Ponec, Mater. Sci. Eng., 1980, 42, 135–140 Search PubMed.
- F. J. Kuijers and V. Ponec, Surf. Sci., 1977, 68, 294 Search PubMed.
- K. Watanabe, M. Hashiba and T. Yamashina, Surf. Sci., 1976, 61, 483 Search PubMed.
- F. P. Larkins and A. Z. Khan, Appl. Catal., 1989, 4, 209 Search PubMed.
- T. Ishihara, K. Eguchi and H. Arai, Appl. Catal., 1987, 30, 225 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Tabulated synthesis parameters, testing conditions and testing details of the various CoFe catalysts discussed in this review. See DOI: 10.1039/c0gc00919a |
| This journal is © The Royal Society of Chemistry 2011 |