Continuous biocatalytic synthesis of (R)-2-octanol with integrated product separation†
Christina
Kohlmann
a,
Susanne
Leuchs
ab,
Lasse
Greiner
*ab and
Walter
Leitner
ac
aInstitut für Technische und Makromolekulare Chemie, RWTH Aachen University, Worringer Weg 1, 52074, Aachen, Germany. E-mail: greiner@dechema.de
bDECHEMA e.V. Karl-Winnacker-Institut, Theodor-Heuss-Allee 25, 60486, Frankfurt am Main, Germany
cMax-Planck-Institut für Kohlenforschung, Mülheim a.d.R., Germany
First published on 15th February 2011
Abstract
The continuous biocatalytic synthesis of (R)-2-octanol carried out in an enzyme membrane reactor showed an excellent process stability of the biocatalysts, resulting in a turnover number of 15 million for the applied alcohol dehydrogenase. Utilisation of the ionic liquid AMMOENG™ 101 as a feasible cosolvent increased substrate concentration, and improved space time yields and turnover numbers of the cofactor by factors of 3 and 6, respectively. Moreover, 80% less waste was generated, while producing the same amount of product. Integrated product separation was realized viasolid phase extraction. Extraction of the applied solid phase with supercritical carbon dioxide allowed more than 30 reuses of the solid phase.
Introduction
The biocatalytic production of enantiopure alcohols from the reduction of prochiral ketones by alcohol dehydrogenases (ADHs) has numerous advantages compared to chemical reaction routes. In comparison to transfer hydrogenations with chiral catalysts, there is no need for transition metals, high pressures or high temperatures,1 whereas, in contrast to asymmetric chemical methods like CBS reduction, the usage of harmful borane and costly chiral oxazaborolidine can be avoided.2 As ADHs are highly selective, while working under mild reaction conditions (e.g. room temperature and ambient pressure), their application could enable environmentally friendly and competitive synthesis routes. Unfortunately, bioprocess engineering faces two main challenges: very often, industrially attractive products result from the conversion of hardly water soluble ketones, causing low productivities. Furthermore, the separation of the alcohol from the aqueous reaction medium is challenging when aiming for a sustainable process. Frequently, products are extracted by an organic solvent and separated from it viadistillation. However, this approach implicates high energy and material consumptions, as well as contamination of both the product and the reaction medium with at least traces of the organic solvent. The application of an organic solubiliser leads to even more complicated mixtures.The application of non-conventional media could contribute to overcome the limitations implied by aqueous reaction environments. For example, ionic liquids (ILs) are powerful solubilisers of various substances and beyond that in biocatalytic reactions improve activities, stabilities and selectivities of biocatalysts, as well as having positive influences on enzyme kinetics and reaction rates.3 Furthermore, supercritical fluids (scFs) could become alternative extracting agents.4 Prevalent supercritical carbon dioxide (scCO2) is used in biocatalysis, as it reaches critical conditions at moderate temperatures and pressures (>31 °C; >7.83 MPa).5 In addition, it is non-flammable, non-toxic, environmentally benign and available in reasonable quantities and at reasonable cost.6 Notably, it is very promising for the extraction of reaction media containing ILs, as it is able to dissolve in ILs, whereas ILs cannot be dissolved in scCO2.7 Unfortunately, CO2 reacts with water to form carbonic acid, resulting in a pH drop in unsuitable reaction media.8 Moreover, CO2 tends to form carbamides with the amino residues of proteins, leading to a destabilised biocatalyst.9 Thus, either the contact of scCO2 with sensitive biocatalysts needs to be avoided or different scFs should be applied.6 A potential approach to achieve separation of the synthesis reaction and scCO2 extraction is its combination with solid phase extraction (SPE). SPE can be beneficial for process engineering, as it is simple and safe in handling, and can be easily automated.10 Besides, when using an appropriate eluent, the SPE material can be recycled effectively.
Within our work, we have focused on the Lactobacillus brevisalcohol dehydrogenase (LbADH)-catalysed reduction of 2-octanone with a limiting solubility of 7.8 mmol L−1 in aqueous phosphate buffer (Fig. 1). The enantioselective reduction of 2-octanone was used as a model reaction for membrane mediated biphasic reactors11 or in biphasic systems with LbADH.12 The water miscible IL AMMOENG™ 101 (Fig. 2) was applied as a performance additive, since it has previously been described as a potential solubiliser.3a For cofactor regeneration, the glucose dehydrogenase (GDH)-catalysed oxidation of β-D-glucose was chosen, as previous findings have shown it to be superior to substrate coupled regeneration. Reactions with or without the addition of IL were carried out continuously by applying an enzyme membrane reactor (EMR). To achieve integrated product separation, a stainless steel column filled with an appropriate solid phase extraction material was integrated into the product stream. Afterwards, the SPE column was recycled viascCO2 extraction. The setup used for this approach can be seen in Fig. 3.
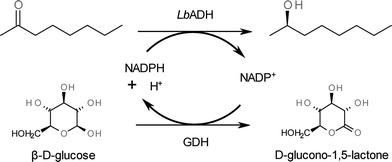 | ||
| Fig. 1 LbADH-catalysed reduction of prochiral ketones to the corresponding (R)-alcohols with GDH-catalysed cofactor regeneration. | ||
 | ||
| Fig. 2 Structure of the ionic liquid AMMOENG™ 101. | ||
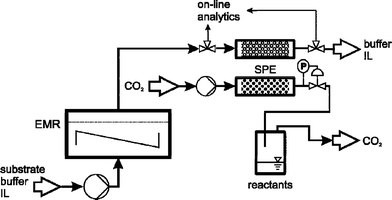 | ||
| Fig. 3 Flow scheme for continuous synthesis in an enzyme membrane reactor (EMR), and subsequent SPE of the loaded column downstream and extraction with carbon dioxide (P: pressure transducer, IL: ionic liquid). | ||
Results and discussion
To choose the appropriate reactor concept, the influence of the different reactants on both biocatalysts has to be considered. Hence, photometric measurements with varied concentrations of substrate, cofactor and product, as well as with varied concentrations of the respective cosubstrate and coproduct, were performed. As the solubility of 2-octanone in aqueous phosphate buffer is low (7.8 mmol L−1), experiments with the addition of 50 g L−1AMMOENG™ 101 were also conducted (resulting in a maximal solubility of 50 mmol L−1). The outcomes of the experiments for LbADH and GDH are shown in Fig. 4 and Fig. 5, respectively.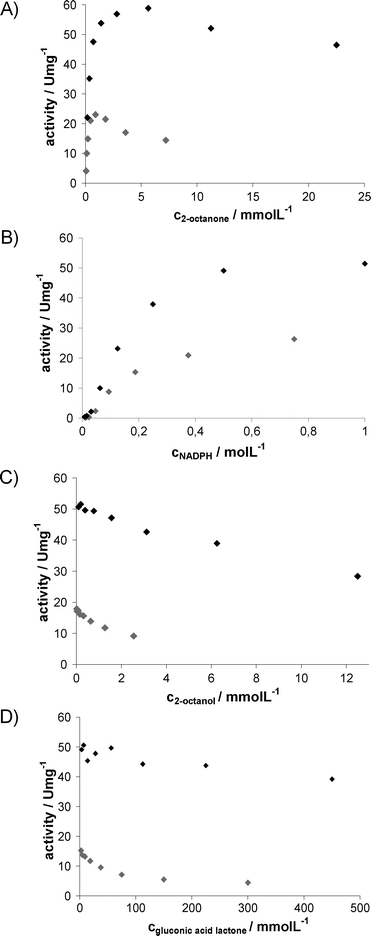 | ||
Fig. 4 Activity of LbADH as a function of reactant concentrations with and without IL;  : buffer, ♦: addition of 50 g L−1AMMOENG™ 101. (A) 0.5 mmol L−1NADPH. (B) 3.6 mmol L−1 or 11.25 mmol L−12-octanone, respectively. (C) 3.4 mmol L−1 or 10 mmol L−12-octanone, respectively; 0.5 mmol L−1NADPH. (D) 2.4 mmol L−1 or 11.25 mmol L−12-octanone, respectively; 0.5 mmol L−1NADPH. : buffer, ♦: addition of 50 g L−1AMMOENG™ 101. (A) 0.5 mmol L−1NADPH. (B) 3.6 mmol L−1 or 11.25 mmol L−12-octanone, respectively. (C) 3.4 mmol L−1 or 10 mmol L−12-octanone, respectively; 0.5 mmol L−1NADPH. (D) 2.4 mmol L−1 or 11.25 mmol L−12-octanone, respectively; 0.5 mmol L−1NADPH. | ||
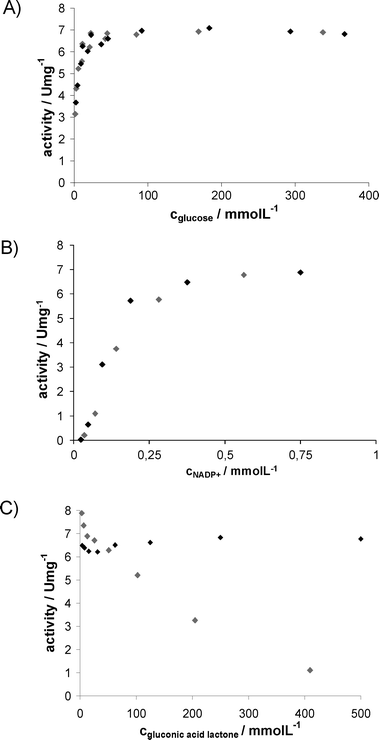 | ||
Fig. 5 Activity of GDH as function of reactant concentrations with and without IL;  : buffer, ♦: addition of 50 g L−1AMMOENG™ 101. (A) 0.5 mmol L−1NADP+. (B) 100 mmol L−1glucose. (C) 100 mmol L−1glucose, 0.5 mmol L−1NADP+. : buffer, ♦: addition of 50 g L−1AMMOENG™ 101. (A) 0.5 mmol L−1NADP+. (B) 100 mmol L−1glucose. (C) 100 mmol L−1glucose, 0.5 mmol L−1NADP+. | ||
All kinetic experiments concerning LbADH show activation by AMMOENG™ 101, therefore the IL is a beneficial solubiliser for this enzyme. Unfortunately, LbADH is inhibited by both the substrate and the product, though these inhibitions are less pronounced in the presence of the IL. As expected, ADH is not affected by glucose (data not shown), while its activity is decreased by the coupled product from cofactor regeneration. The coupled product gluconic acid δ-lactone is in equilibrium with the corresponding gluconic acid; the given concentrations are based on the added amount as this resembles the reaction conditions. Notably, coproduct inhibition is reduced by the addition of AMMOENG™ 101. This may be explained by interaction of gluconic acid with tertiary ammonium salts, which are used for reactive extraction.13 The activity of GDH is not directly influenced by the addition of AMMOENG™ 101. Also, 2-octanone and 2-octanol do not affect the enzyme. However, inhibition by high concentrations of gluconic acid δ-lactone (or the corresponding gluconic acid) is reduced as well.
With regard to kinetics in buffer, high concentrations of all the reactants decrease biocatalyst activity, whereas in the presence of the IL, not only increased substrate concentrations are feasible, but also all types of inhibitions are reduced or, as for gluconic acid δ-lactone, become negligible. To be able to carry out effective syntheses, continuous reactions applying an EMR were chosen as a promising strategy, as only negligible substrate concentrations are present when performed at high conversions.
As the kinetic investigations advised continuous reactions, where the long term performance of the biocatalyst is crucial, the storage stabilities of both enzymes were also determined. The experimental values for ADH stability were perfectly in line with previously published results;3a in pure buffer, LbADH showed a half-life of 41 h, whereas with the addition of 50 g L−1AMMOENG™ 101, improvement by a factor 1.5 to 61 h was possible. Unfortunately, the GDH had a half-life of 7.9 h in buffer that dropped to merely 2.5 h in the presence of the IL. Nevertheless, as storage stabilities are not directly comparable to process stabilities and might show similar trends, these findings do not preclude continuous reactions.
A wide range of SPE materials for different purposes are commercially available. Therefore, we selected different materials from manufacturer instruction guides and tested their ability to extract 2-octanone and 2-octanol from aqueous potassium phosphate buffer.
As emphasised by the maximal loadings of the different materials shown in Fig. 6, especially the polystyrenedivinylbenzene copolymer-based materials, HR-X and HR-P are very effective in binding 2-octanol and 2-octanone. With these materials, loadings of up to 0.70 g per gram of material was possible, while with the different silica based material, at best 0.35 g per gram of material could be extracted. For cost effectiveness, we chose HR-P over HR-X for integrated product separation.
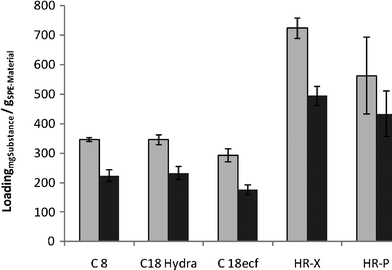 | ||
| Fig. 6 Maximum loading of adsorption materials (light grey: 2-octanone, dark grey: 2-octanol). | ||
Continuous synthesis of (R)-2-octanol
Continuous synthesis was carried out in buffer and with the addition of 50 g L−1AMMOENG™ 101, and compared on basis of space time yields (STY = amount of product produced per litre of reaction volume per day), turnover numbers (TON = amount of product per amount of catalyst or cofactor, respectively) and environmental factor (E factor = mass of waste per mass of product) (see Table 1). TONs of the enzymes were calculated, assuming the protein content of the preparation to be pure enzyme. To ensure that cofactor regeneration with GDH is not limiting, a 17-fold excess of GDH was chosen. For this ratio of LbADH to GDH, no further increase in overall rate was found in batch experiments.| Reaction medium | Pure buffer | 50 g IL L−1 |
|---|---|---|
| a Calculated for the time with more than 90% conversion. | ||
| c 2-octanone /mmol L−1 | 6.0 | 25.0 |
| m ADH /mg | 0.6 | 1.2 |
| m GDH /mg | 10.0 | 20.0 |
| τ/h | 2.4 | 8.0 |
| STY/mmol L−1d−1 a | 56.1 | 72.0 |
| ee (%) | >99.9 | >99.9 |
| TONADHa | 11.0 × 106 | 15.2 × 106 |
| TONGDHa | 1.5 × 105 | 2.1 × 105 |
| TONNADP+a | 58 | 245 |
| E factora | 1420 | 380 |
In comparison to the continuous reduction to (S)-2-octanol in a membrane-mediated biphasic system carried out by Liese et al., 11 an 18-fold higher cofactor utilisation could be achieved, whereas the STY is about 2-fold lower. Eckstein et al.12 reported turnovers for the cofactor higher than 1000 in biphasic batches by recycling of the aqueous reactive phase, but at a lower STY of 20 mmol L−1d−1.
Surprisingly, the reaction progress of the continuous reaction runs demonstrate an excellent process stability of the biocatalyst system. While, in the beginning, full conversion was obtained for both reaction media, in the buffer, the conversion did not drop below 90% for 100 residence times (10 days, Fig. 7). Meanwhile, with the IL buffer mixture, 90% conversion was reached after 72 residence times (24 days, Fig. 8). Apparently, storage and process stability are not comparable. Also the TONs of both biocatalysts showed similar results in both reaction media. The selectivity of LbADH does not change in the presence of 50 g L−1AMMOENG™ 101, resulting in an enantiomeric excess of more than 99.9% in both cases. The addition of the IL allowed for an increase in substrate concentration. This gave a number of improvements that were mirrored in the key performance indicators (Table 1). Firstly, the STY was 1.3-fold increased. Secondly, the TONNADP+ was improved by a factor of 4.2. Finally, the E-factor as an indicator for produced waste per unit of product could be reduced 3.7 times, denoting that 70% less waste is generated.
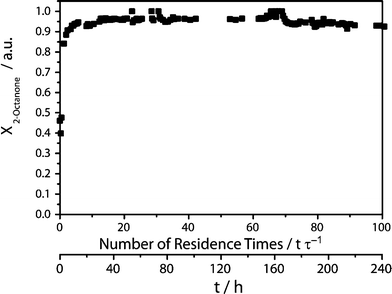 | ||
| Fig. 7 Conversion X of 2-octanonevs. residence time or time, respectively. | ||
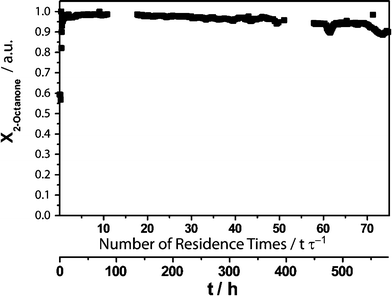 | ||
| Fig. 8 Conversion X of 2-octanonevs. residence time or time, respectively with the addition of 50 g L−1AMMOENG™ 101. | ||
Integrated product separation
To enable integrated product separation, a stainless steel column filled with the previously selected polystyrenedivinylbenzene copolymer HR-P was integrated into the permeate stream of the EMR. The outlet of the column was monitored online viaGC before and after the column. After reaching its full capacity, the SPE column was replaced by a second column and extracted using scCO2. Conditions for scCO2 extraction were optimised from experiments with varied temperature and pressure, with extraction at 8.0 MPa and 45 °C with 0.1 mL (liquid) min−1 showing best results. Results for different reuses for two columns applied in continuous syntheses in pure buffer and buffer containing 50 g L−1AMMOENG™ 101 are shown in Fig. 9.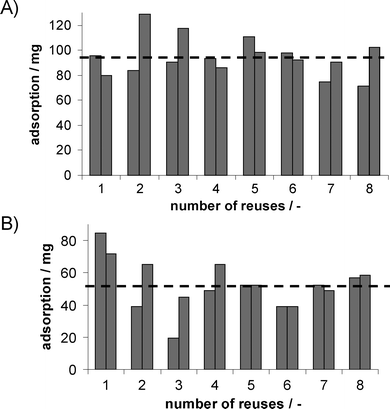 | ||
| Fig. 9 Achieved adsorptions for two SPE columns loaded with HRP-Pvs. reuses applied in continuous reactions in (A) buffer and (B) with the addition of 50 g L−1AMMOENG™ 101. The dashed line represents the average loading of 95 mg in buffer and 51 mg with the addition of 50 g L−1AMMOENG™ 101 (an additional washing step with water was performed after the third reuse, see text). | ||
In buffer, the adsorption reached was relatively stable, whereas in buffer containing 50 g L−1AMMOENG™ 101, a loss in capacity was observed (Fig. 9). As a consequence, average adsorptions in buffer (90 mg for column 1 and 100 mg for column 2) were much higher than for buffer containing 50 g L−1AMMOENG™ 101 (50 mg for column 1 and 53 mg for column 2). Nevertheless, this trend is not due to ageing of the SPE material, but due to binding of the IL, as washing with water after the third reuse resulted in higher adsorptions. Furthermore, when changing the reaction medium, the respective average adsorption can be reached. In total, more than 30 reuses were carried out under the same conditions without an apparent loss of capacity. Moreover, recovery rates for scCO2 extraction of an average 75% in buffer and 62% with the addition AMMOENG™ 101 were achieved.
Conclusion
The application of non-conventional media in biocatalysis enables novel process strategies, overcoming limitations of low aqueous solubilities of reactants or downstream processing. Ionic liquids have proved to be promising performance additives, being biocompatible and powerful solubilisers at the same time. By applying a reaction medium containing 50 g L−1AMMOENG™ 101 in the continuous ADH catalysed synthesis of (R)-2-octanol, STY and TONNADP+ could be improved by factors 1.3 and 4.2, respectively. Furthermore, an excellent selectivity with >99.9% ee in both cases was observed. By applying higher IL contents, it might be possible to further enhance the results. Moreover, 70% less waste is generated, while producing the same amount of product. Combining a continuous reaction, and solid phase extraction and regeneration of the SPE material with scCO2 not only prevented deactivation of the biocatalysts by scCO2, but also enabled processing of the solvent-free product. Furthermore, scCO2 seems to be an effective extracting agent for the used HR-P material, as more than 30 reuses were possible without an apparent loss in capacity.Experimental section
Material
LbADH and GDH are available from X-Zyme GmbH (Düsseldorf, Germany). AMMOENG™ 101 was supplied by Solvent Innovation (Cologne, Germany now Merck KgA). NADPH and NADP+ were provided by Carl Roth GmbH (Karlsruhe, Germany), and N-methyl-N-trimethylsilyl-trifluoracetamide (N-MSTFA) by CS-Chromatographie Service GmbH (Langerwehe, Germany). SPE materials are available from Macherey-Nagel (Düren, Germany). All other reagents were purchased from Sigma-Aldrich (Schnelldorf, Germany) and were at least of analytical grade.Preparation of IL buffer mixtures
For the preparation of 1 L of IL buffer mixture (100 mmol, pH 7), K2HPO4 (12.63 g, 72.5 mmol) and KH2PO4 (3.76 g, 27.6 mmol) were diluted in 500 mL water. Taking the determined water content of the IL into account, the IL (50 g) and MgCl2 (238 mg, 2.5 mmol) were added, and fixed to the required pH by the addition of H3PO4. Afterwards, the mixture was filled to a 1 L volume with water.Determination of IL water content
The water content of the different ILs was determined by Karl Fischer titration. The measurements were carried out by directly injecting IL into the titration solution of a 756 KF coulometer from Metrohm.Enzyme activity
Enzyme kinetics were investigated spectrophotometrically, measuring the difference in absorption of the reduced cofactor NADPH at a wavelength of 340 nm for a 2 min reaction time. The extinction coefficient of NADPH was determined for each reaction medium. To explore possible inhibitions, experiments with varied concentrations of substrate, cofactor and product, as well as cosubstrate and coproduct, were conducted. In a 96-well plate, the substrate solution in buffer containing potential inhibitors (typically 180 μL) and the cofactor solution (typically 10 μL to give a final concentration of 0.5 mmol L−1) were mixed. The reaction was started by the addition of the enzyme solution (10 μL).Stability of enzymes in the reaction media
To determine stabilities, the enzymes were stored in the corresponding IL–buffer mixture at 25 °C; samples were withdrawn and analysed for activity. The half-life was estimated from fitting first order exponential decay of activity vs. time.GC analytics
Aqueous samples were analysed viaGC (column: Chirasil-Dex (25 m × 0.25 mm ID) from Varian GC Capillary Columns, carrier gas: H2, 0.4 bar).To determine the reaction progress, the following method was used: 80 °C (3 min), 10 °C min−1 to 120 °C (6 min), 40 °C min−1 to 180 °C (2 min). Retention times: 2-octanone (7.3 min), 2-octanol (9.4 min), internal standard 1-octanol (11.8 min).
To measure ee, the samples (250 μL) were mixed with N-MSTFA (50 μL), heated to 80 °C for 30 min and analysed by GC: 80 °C (3 min), 1 °C min−1 to 100 °C (5 min), 40 °C min−1 to 180 °C (2 min). Retention times: (R)-2-octanol (14.9 min), (S)-2-octanol (15.2 min).
Selection of SPE material
2-Octanone (8 mL, 6 mmol L−1) or 2-octanol (8 mL, 6 mmol L−1) solutions were added to different SPE materials (10 mg of C8, C18ecf or C18 Hydra; or 5.0 mg of HR-X and HR-P) and shaken at 300 rpm overnight. All experiments were carried out at least in duplicate. Afterwards, the aqueous phase was analysed viaGC. The following materials were under investigation:C8: silica-based, pore size 60 Å, particle size 45 μm, specific area 500 m2 g−1, stable from pH 2 to 8, octyl phase, not end-capped, carbon content 8%.
C18ecf: silica-based, pore size 60 Å, particle size 100 μm, specific area 500 m2 g−1, stable from pH 2 to 8, octadecyl phase, end-capped, carbon content 14%.
C18 Hydra: silica-based, pore size 60 Å, particle size 45 μm, specific area 500 m2 g−1, stable from pH 2 to 8, octadecyl phase for polar analytes, carbon content 15%.
HR-X: hydrophobic polystyrenedivinylbenzene copolymer, pore size 55–60 Å, particle size 85 μm, spherical particles, specific area 1000 m2 g−1.
HR-P: highly porous polystyrenedivinylbenzene copolymer, particle size 50–100 μm, specific area 1200 m2 g−1.
Continuous reaction towards (R)-2-octanol with integrated product separation
Continuous reactions were carried out in an EMR (Jülich Chiral Solutions, Germany) with an overall reaction volume of 16 mL. Previous to insertion of the ultra filtration membrane (NMWL 10000, Millipore) and the supporting disc, the reactor was flushed with water. Afterwards, the reactor volume was exchanged with buffer and bovine serum albumin was introduced to cover the membrane. Then, the reactor was flushed with the substrate solution. To start the experiments, both enzymes were added. To maintain high stability of the cofactor, the substrate solution was stored at 5 °C. EMR experiments were conducted at 27 °C.Continuous reactions in pure buffer (100 mmol L−1potassium phosphate pH 7.0; 2.5 mmol L−1MgCl2) were carried out with 2-octanone (6 mmol L−1), glucose (100 mol L−1), NADP+ (0.1 mmol L−1), LbADH lyophilisate (0.6 mg) and GDH lyophilisate (10.0 mg). To adjust the residence time to 2.4 h, the volumetric flow was fixed at 6.6 mL h−1.
Continuous reactions in IL buffer mixture (100 mmol L−1potassium phosphate pH 7–2; 50 g L−1AMMOENG™ 101, 2.5 mmol L−1MgCl2) were carried out with 2-octanone (25 mmol L−1), glucose (150 mmol L−1), NADP+ (0.1 mmol L−1), LbADH lyophilisate (1.2 mg) and GDH lyophilisate (20.0 mg). To adjust the residence time to 8.0 h, the volumetric flow was fixed at 2.0 mL h−1.
A GC flow cell (mechanical workshop of the institute) was integrated into the product stream to monitor conversion. To enable integrated product separation, the outlet was further connected to a HPLC column (60 mm, ID 3 mm) filled with HR-P material (typically 0.20 g). To control the entire product separation, a second GC flow cell was integrated into the waste stream coming from the SPE column (Fig. 3). When full loading was reached, the column was exchanged and the product extracted using scCO2 (8.0 MPa, 45 °C, 0.1 mL min−1).
Acknowledgements
The authors thank Deutsche Bundesstiftung Umwelt within “ChemBioTec”, Deutsche Forschungsgemeinschaft via the graduate school “scanBioNoCo” (DFG 1166), the Cluster of Excellence “Tailor-Made Fuels from Biomass” by the Excellence Initiative of the German federal and state governments, and the Federal Ministry of Economics and Technology (ZIM) for partial funding. Furthermore, we thank Dr S. Na'amnieh X-Zyme for the kind gift of enzyme preparation.References
- (a) T. Ohkuma, M. Koizumi, K. Muniz, G. Hilt, C. Kabuto and R. Noyori, J. Am. Chem. Soc., 2002, 124, 6508–6509 CrossRef CAS; (b) T. Ohkuma, M. Koizumi, H. Doucet, T. Pham, M. Kozawa, K. Murata, E. Katayama, T. Yokozawa, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1998, 120, 13529–13530 CrossRef CAS; (c) R. Noyori, Angew. Chem., Int. Ed., 2002, 41, 2008–2022 CrossRef CAS; (d) R. Noyori and T. Ohkuma, Angew. Chem., Int. Ed., 2001, 40, 40–73 CrossRef CAS.
- (a) P. Galatsis in Named Reactions for Functional Group Transformations, ed. J. J. C. Li and E. J. John, Wiley & Sons, New Jersey, 2007, pp. 2–21 Search PubMed; (b) A. Hirau, J. Chem. Soc., Chem. Commun., 1981, 315–317 RSC; (c) E. J. Corey, R. K. Bakshi, S. Shibata, C.-P. Chen and V. K. Singh, J. Am. Chem. Soc., 1987, 109, 7925–7926 CrossRef CAS.
- (a) C. Kohlmann, N. Robertz, S. Leuchs, Z. Dogan, S. Lütz, K. Bitzer, S. Na'amnieh and L. Greiner, J. Mol. Catal. B: Enzym., 2011, 68, 147–153 CrossRef CAS; (b) C. Kohlmann, L. Greiner, W. Leitner, C. Wandrey and S. Lütz, Chem.–Eur. J., 2009, 15, 11692–11700 CrossRef CAS; (c) R. A. Sheldon and F. van Rantwijk, Organic Synthesis with Enzymes in Non-Aqueous Media, ed. G. Carrea and S. Riva, Wiley-VCH, Weinheim, 2008, pp. 227–253 Search PubMed; (d) S. Park and R. J. Kazlauskas, Curr. Opin. Biotechnol., 2003, 14, 432–437 CrossRef CAS; (e) R. A. Sheldon, F. van Rantwijk, R. M. Lau, in Ionic Liquids as Green Solvents: Progress and Prospects, ed. R. D. Rogers and K. R. Seddon, Washington, 2003, pp. 192–205 Search PubMed; (f) U. Kragl, M. Eckstein and N. Kaftzik, Curr. Opin. Biotechnol., 2002, 13, 565–571 CrossRef CAS; (g) R. A. Sheldon, R. M. Lau, M. J. Sorgedrager, F. van Rantwijk and K. Seddon, Green Chem., 2002, 4, 147–151 RSC; (h) U. Kragl, N. Kaftzik, S. H. Schöfer, M. Eckstein, P. Wasserscheid and C. Hilgers, Chim. Oggi, 2001, 19, 22–24 CAS; (i) C. Roosen, P. Müller and L. Greiner, Alp. Microbiol. Biotechnol., 2008, 81, 607–614 Search PubMed; (j) F. van Rantwijk and R. A. Sheldon, Chem. Rev., 2007, 107, 2757–2785 CrossRef.
- (a) M. T. Reetz, W. Wiesenhöfer, G. Francio and W. Leitner, Chem. Commun., 2002, 992–993 RSC; (b) M. T. Reetz, W. Wiesenhöfer, G. Franciò and W. Leitner, Adv. Synth. Catal., 2003, 345, 1221–1228 CrossRef CAS; (c) S. Cantone, U. Hanefeld and A. Basso, Green Chem., 2007, 9, 954–971 RSC; (d) P. Lozano, T. de Diego, S. Gmouh, M. Vaultier and J. L. Iborra, Biotechnol. Prog., 2004, 20, 661–669 CrossRef CAS; (e) P. Lozano, T. de Diego, D. Carrie, M. Vaultier and J. L. Iborra, Chem. Commun., 2002, 692–693 RSC.
- A. J. Mesiano, E. J. Beckman and A. J. Russell, Chem. Rev., 1999, 99, 623–633 CrossRef CAS.
- S. K. Karmee, L. Casiraghi and L. Greiner, Biotechnol. J., 2008, 3, 104–111 Search PubMed.
- L. A. Blanchard, D. Hancu, E. J. Beckman and J. F. Brennecke, Nature, 1999, 399, 28–29 CrossRef.
- C. Roosen, M. Ansorge-Schumacher, T. Mang, W. Leitner and L. Greiner, Green Chem., 2007, 9, 455–458 RSC.
- K. Wittmann, W. Wisniewski, R. Mynott, W. Leitner, C. L. Kranemann, T. Rische, P. Eilbracht, S. Kluwer, J. M. Ernsting and C. J. Elsevier, Chem.–Eur. J., 2001, 7, 4584–4589 CrossRef CAS.
- M.-C. Hennion, J. Chromatogr., A, 1999, 856, 3–54 CrossRef CAS.
- A. Liese, T. Zelinski, M.-R. Kula, H. Kierkels, M. Karutz, U. Kragl and C. Wandrey, J. Mol. Catal. B: Enzym., 1998, 4, 91–99 CrossRef CAS.
- (a) M. Eckstein, M. Vilella Filho, A. Liese and U. Kragl, Chem. Commun., 2004, 1084–1085 RSC; (b) M. Eckstein, T. Daußmann and U. Kragl, Biocatal. Biotransform., 2004, 22, 89–96 CrossRef CAS.
- (a) I. Inci, H. Uslu and S. T. Ayhan, J. Chem. Eng. Data, 2005, 50, 961 CrossRef CAS; (b) J. Hirth, M. Bicker and H. Vogel, Chem. Ing. Tech., 2002, 74, 345 CrossRef CAS.
Footnote |
| † This paper was published as part of the themed issue of contributions from the Green Solvents – Alternative Fluids in Science and Application conference held in Berchtesgaden, October 2010. |
| This journal is © The Royal Society of Chemistry 2011 |