A simple and green procedure to prepare poly(ethylene glycol) networks: Synthesis and properties†
Luis Carlos
Cesteros
*
Grupo de Nuevos materiales y Espectroscopia Supramolecular. Departamento de Química Física. Facultad de Ciencia y Tecnología. Universidad del País Vasco, Apartado 644, Bilbao, 48900, Spain. E-mail: c.cesteros@ehu.es; Fax: +34 946013500; Tel: +34 946015966
First published on 1st December 2010
Abstract
Poly(ethylene glycol) networks have been prepared by reaction between dihydroxy-terminated poly(ethylene glycol) (PEG) and diepoxy-terminated poly(ethylene glycol) (DEPEG) (M ≈ 600 g mol−1) in concentrated aqueous NaOH solutions by a process involving alkoxide–epoxy-type reactions. Yields of formation of the networks depend on the concentration of NaOH used, being maximum (>80%) for concentrations of 20% by weight. The PEG/DEPEG molar ratio in the feed plays a decisive role in both the yield of the reaction and the properties of the obtained network, controlling the degree of crosslinking. High PEG/DEPEG ratios lead to hydrogels with a great swelling capacity. The swelling capacity can also be controlled by varying the PEG molecular weight for a same PEG/DEPEG ratio. By employing PEG samples of molecular weights 600, 1500 and 3000 g mol−1, and different PEG/DEPEG ratios, hydrogels with degrees of swelling ranging from 3.4 to 21.6 have been obtained. The thermal transitions in the networks, i.e. the glass transition (Tg) and/or the melting temperature (Tm) show a linear dependence on the PEG/DEPEG ratio.
Introduction
Hydrogels are hydrophilic polymer networks that swell in the presence of water. Such systems have many applications in different fields, but are receiving special attention because in the swollen state these materials resemble living tissues. Thus, hydrogels have important applications in biomedicine, drug delivery and tissue regeneration.1,2Possibly, the most important property of a hydrogel is its ability to swell, mainly controlled by its chemical structure. The chemical structure of the network also plays a decisive role in biomedical applications, since it regulates its biocompatibility. In this regard, poly(ethylene oxide) (PEO) or poly(ethylene glycol) (a name commonly used for poly(ethylene oxide) with a molecular weight less than about 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) is a polymer of choice for these applications.3Poly(ethylene glycol) (PEG) is a linear polyoxyalkylene which contains a hydroxyl group on each end of its chain and is generally represented by the formula: HO(CH2CH2O)nH. PEG has remarkable water solubility, chain flexibility, chemical stability, lack of toxicity and biocompatibility.4 It also features the ability to control the bioactivity of surfaces and bulk materials: when attached to a surface, PEG decreases the adsorption of proteins and the adhesion of cells. PEG is one of the best biocompatible polymers to be combined with a biologically active compound (such as a drug, a protein, a peptide or an enzyme) to produce a conjugate with improved properties such as compatible solubility characteristics, reduced toxicity, enhanced surface compatibility, increased circulation time and reduced immunogenicity.5
000 g mol−1) is a polymer of choice for these applications.3Poly(ethylene glycol) (PEG) is a linear polyoxyalkylene which contains a hydroxyl group on each end of its chain and is generally represented by the formula: HO(CH2CH2O)nH. PEG has remarkable water solubility, chain flexibility, chemical stability, lack of toxicity and biocompatibility.4 It also features the ability to control the bioactivity of surfaces and bulk materials: when attached to a surface, PEG decreases the adsorption of proteins and the adhesion of cells. PEG is one of the best biocompatible polymers to be combined with a biologically active compound (such as a drug, a protein, a peptide or an enzyme) to produce a conjugate with improved properties such as compatible solubility characteristics, reduced toxicity, enhanced surface compatibility, increased circulation time and reduced immunogenicity.5
It is therefore not surprising that there are a large number of investigations devoted to the preparation of hydrogels based on PEG. The most common methods to yield covalently crosslinked PEG hydrogels are gamma or electron radiating of high molecular weight PEO aqueous solutions6,7 and crosslinking by reaction based on the end groups of pre-existing PEG chains. The second method allows for a greater control of the network structure, and takes advantage of the relatively simple preparation of PEG derivatives by modification of their terminal hydroxyl groups.8
Thus, the synthesis of hydrogels from dihydroxy-terminated PEG has been reported,9,10 but also from diacid-PEG,11diisocyanate-PEG,12 diacid chloride-PEG,13diamino-PEG,14 and diepoxy-terminated PEG,15 as well as using various vinyl terminal groups,16–18 with different crosslinking reagents.
Recently, Laine et al. have published two papers19–20 regarding the synthesis and characterization of PEO networks prepared by anionic ring-opening polymerization of diepoxy-terminated PEG (DEPEG) chains. In the first paper, the authors developed a synthetic method to functionalize PEG chains with glycidyl units in a rather simple way, by reaction of PEG with epichlorohydrin. The modified PEG is then crosslinked by ring-opening polymerization using potassium tert-butoxide as initiator, resulting in a highly hydrophilic network (hydrogel) where PEG chains are linked by glycerol units. By this method, and depending on the molar mass of the DEPEG chains, crosslinked materials with an exceptional swelling behavior are obtained. For instance, by employing PEG with a molecular weight of 8000 g mol−1 materials with a degree of swelling of 30 can be obtained. In their second paper, new PEO hydrogels were prepared from DEPEG but this time employing dianionic glycerol and glycolic acid as initiators. The main goal of these works was the development of new drug delivery systems based on hydrogels with innocuous components.
These investigations open a new field in the synthesis of PEO hydrogels with a controlled chemical structure, being very similar to that of the precursor polymer. However, the anionic synthesis of these hydrogels involves a long process of purification for the reagents and an extremely careful cleaning of the reaction vessels. The question raised in this paper is: would it be possible to prepare PEG networks from DEPEG by using non-toxic reagents and a simpler and greener procedure? Some recently published experimental results suggest that a network could be prepared from DEPEG in an aqueous basic environment using PEG as the crosslinking agent.
Star-shaped highly branched polymers have been prepared, via anionic synthesis, by reacting monomethoxy poly(ethylene glycol) alkoxide anions and diepoxides.21,22 In this method, by anionic synthesis, a PEG-alkoxide anion reacts with the diepoxide (diglycidyl ether of ethylene or neopentyl glycol) yielding secondary alkoxide anions from the primary ones present in the PEG. Any subsequent reactions of diepoxide with the first generation of the secondary alkoxide anions reproduce mostly secondary alkoxide anions in the consecutive generations. Thus there is a sequence of parallel consecutive reactions leading to hyperbranching, and a network formation was observed at a certain conversion of epoxy groups.
In a recently published patent23, non-toxic and biocompatible hydrogels are prepared by combining polymers possessing hydroxyl groups and a molecule containing at least two carbon–carbon double bonds, in an aqueous basic environment. The crosslinking takes place through the formation of ether bonds between the hydroxyl groups of the polymer and the alkene group of the other molecule. The reaction environment consists on the aqueous concentrated solution of a strong base (20 wt% NaOH). Under those aqueous basic conditions, the authors claim that at least a part of hydroxyl groups of the polymer are transformed into alkoxides, which quickly and quantitatively react with the alkene groups of the molecule used as crosslinking agent. The formation of alkoxides in concentrated solutions of KOH has also been observed in phase transfer catalytic systems.24,25
In short, the PEG-based alkoxides behave as strong bases capable of reacting with epoxides by ring opening reactions, and it is possible to obtain, at least partially, PEG alkoxides using concentrated aqueous NaOH solutions. Under these conditions, the reaction between DEPEG and PEG would be possible. Moreover, since the alkoxide-epoxide reaction generates a new hydroxyl function, capable of forming an additional alkoxy group in the basic medium, it can react with another epoxide group, acting as a potential crosslinking point. Consequently, a network could be obtained using only PEG, DEPEG, water and NaOH.
This paper shows that it is possible to obtain such networks in a very simple way, working in a basic aqueous environment. The synthesis were performed with a commercial DEPEG sample (600 g mol−1) and PEG of three different molecular weights: 600, 1500 and 3000 g mol−1. Using different PEG/DEPEG molar ratios and varying the molecular weight of the PEG, it is possible to control the swelling capacity of these networks.
Results and discussion
The aim of this work is the preparation of PEG networks from mixtures of PEG and DEPEG using a simple reaction process based on the formation of PEG alkoxides in concentrated aqueous NaOH solutions.Fig. 1 presents a simplified outline of the chemical processes that could lead to the formation of the network: In step 1 the reaction between DEPEG and PEG-alkoxide leads to the formation of a new hydroxyl group. Further reactions of this type can occur at both ends of the growing chain. In a strongly basic medium some of the hydroxyl groups generated in the previous reactions can be transformed into alkoxides (as is represented in the figure), that in step 2 would react with another DEPEG molecule, creating a branching point. In step 3 the reaction of an epoxy group at the end of a branch with an alkoxy group derived from a previous PEG–DEPEG reaction causes crosslinking between both chains. It is obvious that once a branch is formed, chain growth processes may occur before crosslinking. These can be accomplished through any reaction involving epoxy and alkoxide groups of different chains. In order to simplify Fig. 1, all hydroxyl groups are shown in the form of alkoxides, though as mentioned previously they can be found in both forms. Note that the network thus obtained has a chemical structure almost identical to that of PEG.
 | ||
| Fig. 1 Chemical processes involved in the network formation. | ||
According to the crosslinking mechanism proposed in Fig. 1, the ratio DEPEG/PEG in the reaction medium must play an important role. The presence of PEG in large quantities favours chain growth. By contrast, a low PEG content favours the reactions that lead to branching and, eventually, produce crosslinking.
With these ideas in mind, the first trials were conducted using a DEPEG/PEG600 molar ratio of 4/1 and working with aqueous NaOH solutions with concentrations ranging from 5 to 30% by weight. In order to promote the formation of alkoxide groups, PEG600 samples were maintained under stirring at room temperature for 24 h in NaOH solutions before adding DEPEG. When using NaOH with a concentration above 14%, PEG is not longer soluble,26 and an aqueous biphasic system is formed, although both phases are intimately dispersed under stirring.
In all cases, it was observed that after some time (shorter for higher NaOH concentrations), solved or dispersed PEG600 takes on an orange–yellow colour, which progressively intensifies. The appearance of this colour is probably caused by the formation of PEG–Na+ complexes. Neumann and Sasson27 in their study on the base-catalyzed reaction of isomerization of allylanisole with PEG as phase-transfer catalysts discovered that, as the aqueous phase approached saturation with potassium hydroxide (KOH), a yellow–brown PEG–KOH complex phase would form. In fact, PEG has the ability to serve as a phase-transfer catalysts since the poly(ethylene glycol) chains can form complexes with metal cations, similar to crown ethers, as has been reviewed.28
After the addition of DEPEG, and for those experiments performed with a NaOH concentration above 12%, the formation of a yellowish solid mass was observed in the reactor over a period of time that ranged from 18 to 48 h.
The obtained solid masses were immersed in distilled water, showing a marked swelling. The yellow–orange colour slowly disappears after successive washing steps using water (several weeks). However, when 1 M HCl is used for the washing process, the disappearance of the coloration is much faster, and can be completed within 24–72 h. Finally, a transparent and stable hydrogel is obtained.
After establishing the range of NaOH concentrations that lead to the formation of a network, the yield of the crosslinking reaction as function of the NaOH solution concentration and the reaction time was analyzed. The yield of the reaction was calculated after the process of purification and drying of the gels described in the experimental section. In Table 1 the yields for samples prepared with different PEG/DEPEG ratios, NaOH concentrations and reaction times are shown. The obtained results show a very marked dependence of the reaction yield with the NaOH concentration.
| PEG molecular weight (g mol−1) | PEG (%) | DEPEG (%) | NaOH (%) | Reaction time (in hours) | Yield (%) | Degree of swelling | T g (°C) | T m (°C) | ΔHm (J g−1) |
|---|---|---|---|---|---|---|---|---|---|
| a Crystallinity develops during the heating of the sample. | |||||||||
| 600 | 20 | 80 | 14 | 72 | 33.5 | 24.1 | −50.6 | 4.0a | 1.9 |
| 600 | 20 | 80 | 16 | 48 | 65.8 | 13.8 | −51.6 | — | — |
| 600 | 5 | 95 | 16 | 72 | 66.7 | 5.1 | −47.2 | — | — |
| 600 | 10 | 90 | 16 | 72 | 68.9 | 6.0 | −47.9 | — | — |
| 600 | 15 | 85 | 16 | 72 | 68.0 | 7.5 | −48.8 | — | — |
| 600 | 20 | 80 | 16 | 72 | 64.9 | 10.0 | −48.5 | 3.5a | 0.6 |
| 600 | 25 | 75 | 16 | 72 | 57.5 | 13.9 | −50.1 | 7.3a | 29.0 |
| 600 | 0 | 100 | 20 | 48 | 47.4 | 10.2 | −47.0 | — | — |
| 600 | 0 | 100 | 20 | 72 | 73.9 | 3.6 | −46.0 | — | — |
| 600 | 5 | 95 | 20 | 72 | 77.3 | 3.4 | −46.4 | — | — |
| 600 | 10 | 90 | 20 | 72 | 81.8 | 3.5 | −47.1 | — | — |
| 600 | 15 | 85 | 20 | 72 | 82.7 | 3.8 | −48.1 | — | — |
| 600 | 20 | 80 | 20 | 72 | 83.8 | 4.3 | −49.5 | — | — |
| 600 | 25 | 75 | 20 | 72 | 80.9 | 5.2 | −50.2 | 1.5a | 7.0 |
| 600 | 30 | 70 | 20 | 72 | 76.3 | 6.8 | −51.0 | 5.3a | 32.3 |
| 600 | 35 | 65 | 20 | 72 | 66.8 | 11.1 | −51.2 | 9.2a | 35.6 |
| 600 | 40 | 60 | 20 | 72 | 51.6 | 21.6 | −52.5 | 9.5a | 40.1 |
| 600 | 20 | 80 | 20 | 48 | 82.9 | 4.8 | −50.2 | — | — |
| 600 | 20 | 80 | 24 | 72 | 80.3 | 4.7 | −50.5 | — | — |
| 600 | 20 | 80 | 28 | 72 | 70.8 | 6.9 | −50.4 | — | — |
| 1500 | 5 | 95 | 20 | 72 | 79.9 | 4.4 | −49.4 | 1.3a | 0.9 |
| 1500 | 10 | 90 | 20 | 72 | 80.8 | 4.9 | −50.8 | 10.3a | 28.9 |
| 1500 | 15 | 85 | 20 | 72 | 82.4 | 5.2 | −52.6 | 14.1a | 36.0 |
| 1500 | 20 | 80 | 20 | 72 | 81.8 | 5.7 | −45.0 | 22.2 | 48.8 |
| 3000 | 5 | 95 | 20 | 72 | 87.6 | 4.9 | −49.2 | 25.8a | 28.3 |
| 3000 | 10 | 90 | 20 | 72 | 89.7 | 6.8 | −45.3 | 35.3 | 38.9 |
| 3000 | 15 | 85 | 20 | 72 | 83.0 | 8.1 | −45.6 | 44.3 | 56.1 |
| 3000 | 20 | 80 | 20 | 72 | 73.9 | 11.5 | −45.0 | 50.7 | 65.6 |
Thus, setting the reaction time at 48 h, the yield passes through a maximum (ca. 83%) when 20% NaOH is used. On both sides of the maximum the yield drops quickly (see Fig. 2). Increasing the reaction time to 72 h, the yield is also maximum for 20% NaOH and no significant changes are observed for lower NaOH concentrations. On the other hand, notice (Fig. 2) that the reaction time significantly affects the reaction yields when using higher NaOH concentrations (24 and 28%), when the process slows down considerably. In the latter two cases, the experiments carried out at times over 72 h of reaction showed no significant changes in the yields.
 | ||
| Fig. 2 Yield of gel formation as a function of NaOH concentration for samples prepared with a PEG600/DEPEG molar ratio of 20% at different reaction times: (○) 48 h; (●) 72 h. | ||
The results displayed in Fig. 2 lead to the question of what is the mechanism for the alkoxy anion formation. Two distinct scenarios can be envisaged: either the formation of the alkoxy anions takes place at the interface between the two phases, or a phase transfer catalysis process occurs, involving first the transfer of NaOH inside the PEG phase, followed by the formation of the alkoxy anions. In both cases the concentration of the NaOH solution plays an important role. Nevertheless, the high yields obtained in these crosslinking reactions seem to be more compatible with a phase transfer mechanism, in which PEG is very efficient.28 In this case the solubility of PEG in the aqueous phase, which is controlled by the NaOH concentration, is an important parameter and can explain the results presented in Fig. 2. At higher concentrations of NaOH the decrease of PEG content in the aqueous phase reduces the phase transfer process, slowing down the crosslinking kinetics and the yield. The 20% aqueous NaOH solution seems to offer the best compromise between the solubility of PEG and the NaOH concentration required for the formation of an adequate amount of alkoxy anions.
Having established that the maximum yield of gel formation is obtained using NaOH with a concentration of 20%, further investigations were carried out in order to determine the influence of the PEG600/DEPEG feed molar ratio. As previously indicated, for high molar PEG/DEPEG ratios, either branched or linear polymers should be formed instead of networks. The results obtained using 20% NaOH and varying the PEG600/DEPEG ratio are shown in Fig. 3. The conversion goes through a maximum at about 20% of PEG600. Notice that, as expected, the yield significantly declines for higher PEG600 contents and no gel formation is observed for PEG600 contents above 40%. Fig. 3 also shows that it is feasible to prepare the gel without the direct involvement of pure PEG, using DEPEG only (yield 73.9%). This is not surprising, since the partially modified DEPEG chains present in the sample can start the reaction process through their hydroxyl end groups.
 | ||
| Fig. 3 Yield of gel formation as a function of the mol% of PEG600 in the feed mixture PEG600/DEPEG, for samples prepared with 20% NaOH solutions. | ||
In short, the most suitable conditions for the synthesis of PEG600 gels by this method are achieved using an aqueous solution of NaOH with a concentration of 20 wt%, a mixture PEG600/DEPEG with a content of 20 mol% of PEG600, and a reaction time of 72 h.
According to the proposed mechanism for the formation of the gel, PEG chains of any length could be used to build the networks. By varying the molecular weight of PEG the chain lengths of the networks could be controlled and, therefore, their swelling properties.
Several synthesis have been carried out using PEG samples with molecular weights 1500 and 3000 g mol−1 (PEG1500 and PEG3000), in addition to PEG600. At 30 °C these samples are crystalline solids insoluble in the NaOH solution. It was therefore necessary to carry out the synthesis at a temperature of 60 °C, above the melting point of PEG, which permits good mixing of the PEG and DEPEG polymers. In these experiments, 20% NaOH was used, and a gel was produced in all cases. Table 1 shows the yields of different crosslinking reactions using PEG1500 and PEG3000 in the feed.
When using PEG of different molecular weights in the synthesis, it can be noticed that the yellowish colour observed when adding NaOH is less intense the higher the molecular weight of PEG. This would support the hypothesis of the formation of a PEG–Na+ crown-ether type complex at the polymer chain end.24 PEG samples with a high molecular weight possess a lower ratio of end groups and produce a less coloured material.
The resulting gels were characterized by infrared spectroscopy. Fig. 4 shows the IR spectra obtained for some gels and their precursors. As can be seen, the spectra of the gels are similar to that of pure PEG, in agreement with the proposed mechanism. In some of them a small band around 740 cm−1 can be detected (particularly in those obtained from pure DEPEG) which could indicate the presence of a number of unreacted epoxy groups. Consequently, a network with a chemical structure quasi-similar to PEG is produced by this simple method.
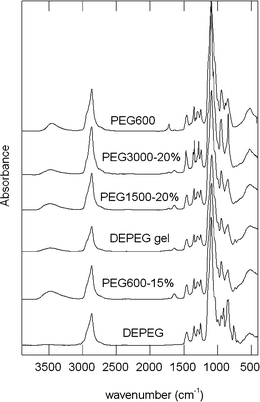 | ||
| Fig. 4 ATR infrared spectra of PEG600, DEPEG and some of the synthesized networks. | ||
Solid-state 13C NMR spectra provide more information about the network structure. It is possible to resolve the spectral contributions attributed to the groups involved in branching and chain growth processes: tertiary carbon atoms (δ = 79.2 ppm) and carbon atoms located between an ether group and a hydroxyl group (δ = 73.4 ppm). As can be seen in Fig. 1, the first contribution is only produced by branching, and the second one is produced by chain growth (two carbon atoms) as well as branching (one carbon atom). Both signals are displayed in Fig. 5 for PEG600/DEPEG samples prepared with different amounts of PEG600 ranging from 5 to 35%. As can be seen, and in agreement with the proposed reaction mechanism, the branching contribution progressively diminishes as the PEG content in the feed increases, in contrast to the increasing chain growth signal.
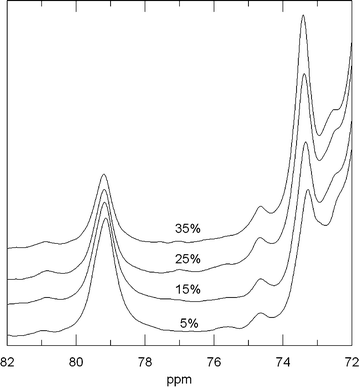 | ||
| Fig. 5 Solid-state 13C spectra for networks prepared from different PEG600/DEPEG molar ratios. | ||
Swelling behavior
The swelling behavior can also give us important information about the network structure. In the case of the hydrogels prepared from PEG600, the highest degree of swelling is found for that with a lower reaction yield (Table 1). This sample prepared with a 14% NaOH solution and a reaction time of 72 h has a swelling ratio of 24.1. It is evident that, at this low NaOH concentration, the formation of alkoxide groups is not sufficiently facilitated, leading to a reduction of the cross-linking reactions, which produces a very open network with a high swelling capacity.The concentration of NaOH solutions plays an important role on the swelling capacity (network structure) of the hydrogels. Comparing the networks prepared from PEG600 with the same PEG600/DEPEG molar ratio, but using 16% or 20% NaOH solutions, significant differences in their degrees of swelling are observed (Fig. 6). For any PEG600/DEPEG ratio, hydrogels prepared with 20% NaOH show a lower swelling capacity, i.e. denser networks are forming for similar feed compositions. It seems clear that the attachment of epoxy and enolate groups is more favored working with 20% NaOH solutions that in the case of 14% NaOH. The effect of NaOH concentration seems to have a certain limit; if we use 24% NaOH the degree of swelling becomes slightly higher, and results significantly higher when using 28% NaOH (see Table 1). This effect is similar to that observed for the yield of gel formation as a function of the NaOH concentration (Fig. 2), previously discussed.
 | ||
| Fig. 6 Dependence of the degree of swelling with PEG content for hydrogels prepared using 16% NaOH and PEG600 (●); 20% NaOH and PEG600 (○), PEG1500 (■) and PEG3000 (□). In all cases the reaction time was 72 h. | ||
Fig. 6 also highlights the influence of the PEG/DEPEG ratio on the swelling capacity when the NaOH concentration is fixed. A higher PEG content leads to a gradual increase in the degree of swelling of the network. As previously discussed, and confirmed by NMR, a larger amount of PEG favors PEG–DEPEG chain growth as opposed to branching (responsible for crosslinking) and, ultimately, leads to a network with greater chain lengths between crosslinking points, and therefore a higher swelling capacity. As we will see below, the PEG/DEPEG feed ratio also has important consequences on the thermal behavior of these materials.
The same effect occurs if instead of PEG600, samples of PEG1500 or PEG3000 were used. Again, there is an increase of the degree of swelling in the order: PEG3000 > PEG1500 > PEG600, related to the increase in the chain length. This effect is also evident in Fig. 6, for networks prepared from PEG600, PEG1500 and PEG3000 in 20% NaOH at different PEG/DEPEG ratios.
The degrees of swelling reported by Laine20 for hydrogels prepared from PEG3400 ranged between 6.6 and 22, depending on the type of initiator used. The value of 11.5 reported here for the sample prepared with a 20% of PEG3000 is similar to the reported by Laine using glycolic acid as initiator.
Swelling experiments also permit the evaluation of the average molecular weight between crosslinks. As has been pointed out,29 gels formed by the condensation of terminally modified polymers fit well to the classic model developed by Flory & Rehner30 to describe the conditions of swelling equilibrium:
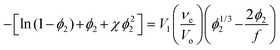 | (1) |
Taking into account that νe is related to the average molecular weight between crosslinks, Mc, by: νe = mp/Mc, where mp is the total mass of PEG in the gel; eqn (1) becomes:
 | (2) |
Table 2 shows the experimental results obtained for the density of the xerogels and the polymer volume fractions in swelling equilibrium conditions at 25 °C for all the hydrogels prepared from PEG600 and 20% NaOH solutions. The average molecular weights between crosslinks obtained from these experimental values, using eqn (2) with f = 3, are also included. The reported values fall within a reasonable range taking into account the methodology of synthesis, the molecular weights of the precursors and the proposed mechanism (Fig. 1). Taking into account that the starting materials are PEG600 and DEPEG (also ca. 600 g mol−1), it can be seen that the Mc values are close to this value for samples prepared with lower PEG600 content. This indicates that branching is highly favoured over chain growth, leading to a very compact network, with chain lengths similar to those of precursors. As PEG600 content in the feed increases, so does Mc. As was shown before, an increase in the PEG/DEPEG ratio favours chain growth, resulting in much more open networks, with greater swelling capacities. The highest value obtained for Mc is close to 12000 g mol−1, which represents around 19 linear chain growth reactions for each crosslinking one. After that, higher PEG/DEPEG ratios prevent the formation of the gel.
| PEG600/DEPEG molar ratio | ρ (g cm−3) | ϕ 2 | M c (g mol−1) |
|---|---|---|---|
| 5 | 1.162 ± 0.003 | 0.268 ± 0.004 | 736 ± 44 |
| 10 | 1.165 ± 0.002 | 0.257 ± 0.003 | 819 ± 18 |
| 15 | 1,177 ± 0.005 | 0.227 ± 0.009 | 1140 ± 114 |
| 20 | 1.166 ± 0.001 | 0.207 ± 0.002 | 1421 ± 27 |
| 25 | 1.171 ± 0.004 | 0.167 ± 0.002 | 2400 ± 64 |
| 30 | 1.173 ± 0.005 | 0.127 ± 0.003 | 4569 ± 343 |
| 35 | 1.167 ± 0.010 | 0.081 ± 0.001 | 11754 ± 77 |
Thermal behavior
With respect to thermal behavior of these materials, the glass transitions, the crystallization and fusion processes by DSC, and also their thermogravimetric behavior have been analyzed. In Table 1 some of these data are displayed.For all networks, the glass transition can be detected by DSC, and Tg is low, below −45 °C. Most of these materials exhibit an elastomeric behavior at room temperature. Some of them present crystallinity and behave like rigid materials.
The glass transition temperature of the gels depends both on the feed composition and on the molecular weight of PEG. As shown in Fig. 7, the analysis of all samples prepared from PEG600 and 20% NaOH shows that the glass transition temperature of the xerogels linearly decreases with the content of PEG600. As shown in the same figure, the gels containing PEG1500 have lower glass transition temperatures. The composition dependence is also linear, with the exception of the sample containing 20% of PEG1500, which has a much higher Tg, clearly attributable to the crystallinity developed at temperatures below its Tg. The latter effect explains that the gels containing PEG3000 are those having a higher Tg (5% excepted).
 | ||
| Fig. 7 Plot of the glass transition temperature of the xerogel as a function of the PEG content in feed for samples prepared from: PEG600 (●) and PEG1500 (■). | ||
With the exception of gels that show crystallinity below their Tg, an inverse relationship between Tg and the network chain length (swelling capacity) is observed for samples prepared under similar conditions. The effect of the crosslinking degree of a polymer network on the glass transition is well established:32,33 the Tg value increases and the rate of increase of Tg accelerates as the average molecular weight Mc between crosslinks decreases. The dependence of the glass transition temperature of the network on the average number of repeat units between crosslinks, n, has been expressed as:33
 | (3) |
As shown in Fig. 8, by plotting the Tg for PEG600 gel samples versus 1/n (determined from the data in Table 2, taking into account a molecular weight of 44 g mol−1 for PEG repeat units) a straight line is obtained (R = 0.9916). The intercept value (T∞g) obtained using the least-squares method is 221.2 K, which satisfactorily agrees with the experimental values close to 218 K that have been published for PEG samples of high molecular weights.34,35 From the slope of the regression line, and taking a value of 3 for Nrot in PEG (according to the rules for the calculation of Nrot33), a value of 1.2 is obtained for the constant c; lower than that reported by Bicerano33 in other networks (5 ± 2).
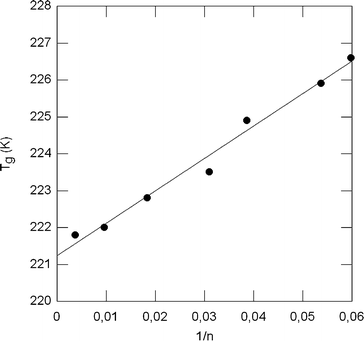 | ||
| Fig. 8 Glass transition temperatures observed for PEG600/DEPEG networks as a function of the inverse of the average number of repeat units between crosslinks. | ||
As mentioned previously, crystallinity is observed in some of the prepared networks. This is not a surprising result because PEG is an easily crystallizable polymer and many of the gels prepared from it are crystalline materials.9,12,15,19,20 To a lesser or greater extent crystallinity is observed for all the networks prepared from PEG1500 or PEG3000. In these samples, crystallinity increases with the molecular weight and the PEG content in the feed. Fig. 9 shows the DSC curves for the whole set of gels prepared from PEG1500.
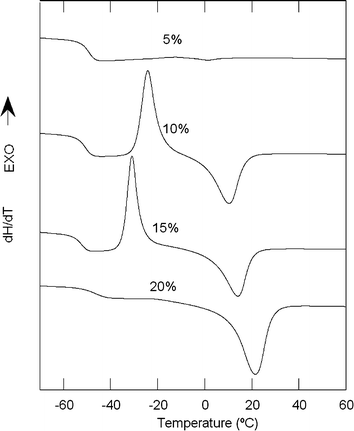 | ||
| Fig. 9 DSC curves for xerogels prepared from PEG1500. | ||
In the gels with lower PEG1500 contents (5, 10 and 15%), the crystallinity develops during the heating of the sample, above the glass transition temperature, and the endothermic melting peak is observed later. The gel with a higher content of PEG1500 (20%) crystallized during the cooling process, showing only the glass transition and the melting peak. As shown in Table 1, the enthalpy of fusion of these samples is higher as its content in PEG1500 increases.
Crystallinity develops more easily in the gels prepared from PEG3000. In this case, only the gel containing 5% of PEG3000 presents crystallization during heating. The enthalpies of fusion are significantly higher than those of PEG1500-based gels, increasing as a function of the PEG3000 content. Fig. 10 depicts the melting temperatures of both types of gels versus the corresponding PEG contents, and a linear increase is observed. A similar behavior is obtained for the enthalpies of fusion of the samples.
 | ||
| Fig. 10 Melting temperatures as a function of PEG% in the feed for xerogels prepared from: PEG1500 (●) and PEG3000 (■). | ||
For gels prepared from PEG600 and 20% NaOH, crystallinity develops at PEG600 compositions higher than 20% (Table 1). When using 16% NaOH crystallinity can be detected in samples with a 20% of PEG600 and for gels with a 25% of PEG600 is significantly higher than that achieved at the same composition for those prepared with 20% NaOH.
The crystallization of polymer networks has been studied by several authors for different systems.36,37 These investigations have shown that this process is very different from that of linear polymers because the diffusion of molten polymer chains participating in the crystallization is greatly restricted due to the presence of crosslinks. Furthermore, the exclusion of the crosslinking points from the crystalline phase leads to a depression in the crystallization degree as well as in the rate of crystallization.
Consequently, the average chain length within the networks is the main factor that controls crystallinity. For example, it has been found that PEG-based networks do not crystallize at all when the molecular weight of PEG is less than 1000 g mol−1.38 The crystallization behavior of the networks presented here agrees well with these ideas, and crystallinity develops more easily in the networks with longer chain lengths.
As we have seen, the chain length increases both with the PEG/DEPEG ratio and with the molecular weight of PEG used. The crystallization behavior of these networks, just described, shows that crystallinity increases at the same time. It has to be noticed that gels with a higher swelling capacity (i.e. longer chain length) are those that crystallize more easily, presenting within a same set of samples higher temperatures and enthalpies of melting. For the samples prepared from PEG600 and 20% NaOH, crystallinity develops in the network for molecular weights between crosslinks higher than 2000 g mol−1 (see Table 1).
The crystallization behavior of the gels containing PEG3000 can be compared to the results reported by Laine20 for gels prepared from DEPEG with a molecular weight of 3400 g mol−1. Depending on the anionic initiator, melting temperatures around the 49–56 °C and melting enthalpies between 59 and 72 J g−1 are reported, i.e. similar values to those found for the gel prepared with a 20% PEG3000 (Table 1).
With regard to the thermogravimetric behavior of the networks, all behave in a similar manner. The degradation process occurs in a single step that begins at around 340–350 °C and ends ca. 410 °C. These results are similar to those previously reported by Laine.19
Conclusions
It has been shown that it is possible to prepare, in a very simple and environmentally friendly way, hydrophilic networks from PEG and DEPEG in a basic aqueous medium with yields above 80%. The highest yields are obtained in a 20% aqueous NaOH solution and depend on the amount of PEG in feed. For high values of the PEG/DEPEG molar ratio (>40%) no gel formation is observed.The properties of these materials are controlled by the PEG/DEPEG feed ratio. Long chain lengths and large swelling capacities result from high proportions of PEG in the feed. It is also possible to control the network chain lengths by varying the molecular weight of PEG used in the synthesis. As expected, for the same PEG/DEPEG feed ratio, the higher the molecular weight of PEG the higher the degree of swelling of the obtained material.
The values of chain lengths obtained by the classical theory of swelling, confirm that PEG/DEPEG ratio controls the two basic processes involved in the formation of the network: chain growth and branching. The former predominates as the PEG/DEPEG ratio increases, ultimately controlling the topology of the network and its swelling properties.
Similarly, the thermal transitions in these networks are also controlled by the PEG/DEPEG ratio. The networks prepared from high PEG/DEPEG ratios posses the lower glass transition temperatures and, for very high values of this ratio, crystallinity is observed. This behavior is consistent with the increase of chain length in the network with the PEG/DEPEG ratio. For the same reason, crystallinity is also favored by increasing the molecular weight of the PEG used in the synthesis.
Experimental section
Poly(ethylene glycol) diglycidyl ether (DEPEG) (Aldrich), with a nominal average molecular weight of 526 g mol−1, and poly(ethylene glycol) (PEG) samples (Aldrich) having average molecular weights of 600, 1500 and 3000 g mol−1, determined by size-exclusion chromatography (SEC) in a Knauer system provided with two TSKgel Alpha columns calibrated with PEG samples (Polymer Laboratories) and using water as eluent, were used as received. Henceforth, these samples will be denoted as DEPEG, PEG600, PEG1500 and PEG3000, respectively. Sodium hydroxide in pellets (Panreac, pellets 98%), HCl (Panreac), and Cyclohexane (Merck PA), were employed as received.All the experiments were performed using 2 g of the PEG/DEPEG mixture diluted to 30% by weight in the NaOH solution. The reactions were conducted as follows. In 20 ml glass vial, PEG and the corresponding amount of NaOH solution were weighed. The mixture was magnetically stirred at room temperature for 24 h. During this time the solution changed from colourless to yellow–orange (the higher the concentration of NaOH the more intense its colouring). After this time, the appropriate amount of DEPEG was added and the vial was placed, with stirring, in an oil bath at a temperature of 30 °C. The total amount of polymers in the mixture was 30% by weight. After 24–48 h, the formation of an orange solid floating on the mixture was observed.
After the time programmed for the reaction, the solid was washed in a 1 M HCl solution for 24 h. The solid swelled in the acidic solution and lost its colour, becoming a transparent–translucent mass, with the classic appearance of a hydrogel. The cleaning process continues for three additional days using distilled water, by frequently changing the washing liquid. Finally, the hydrogel was dried at room temperature for 48 h and then dried in a vacuum oven at 50 °C to constant mass. The yield of the reaction was calculated from this value.
The equilibrium degree of swelling of the hydrogels was measured by first equilibrating in Milli-Q water small pieces of gel for 48 h at 25 °C and then weighing the samples removed from the liquid using a Mettler–Toledo AG135 electronic balance with a sensitivity of 10−5 g. Before measuring its weight, the samples were quickly and gently blotted to remove liquid water from their surface. The samples were desiccated at 50 °C under high vacuum to determine the dry polymer weight. The degree of swelling was then expressed as ratio between the weights of the swollen polymer and that of dry polymer. The reported values are the average of three determinations.
The determination of the densities of the swollen and dried gels was performed at 25 °C with a density determination kit from Mettler Toledo on a Mettler Toledo AG135 electronic balance, using cyclohexane as the immersion liquid for the samples. The reported values are the average of at least three determinations.
Infrared spectra of the samples were recorded on a Thermo 520 Fourier transform infrared spectrophotometer equipped with a Smart Orbit diamond ATR (attenuated total reflection) accessory. Spectra were taken with a resolution of 4 cm−1 and were averaged over 64 scans.
Solid-state 13C NMR spectra were recorded on a Bruker DSX300 spectrometer (7.05 T) at the resonance frequency of 75.4878 MHz, using a standard pulse sequence, a time domain of 12 K, a spectral width of 24154 Hz and interpulse delay of 8 s. The gel (200–300 mg) was packed into a cylindrical zirconia rotor using a Bruker 4 mm MAS probe and then spun at an MAS frequency of 10–11 kHz.
Thermal analysis of gels was performed with a DSC (differential scanning calorimeter) TA Instruments (DSC 2920) calibrated with indium. Weight samples were about 15 mg in all cases and sample scans were performed between 193 and 393 K at a speed of 10 K min−1 under nitrogen atmosphere. The glass transition temperatures of the gels were determined with the approach of the half-point of the specific heat jump, while the melting and crystallization temperatures correspond to the peak values. All reported values were determined in the second DSC run.
The thermogravimetric study was performed with a thermobalance TA Instruments SDTQ600 under a nitrogen atmosphere. Sample weights were about 3 mg in all cases and the scan speed was 10 K min−1.
References
- N. A. Peppas, ed., Hydrogels in Medicine and Pharmacy, CRC Press, Boca Raton, Florida, 1987 Search PubMed.
- S. A. Hoffman, Adv. Drug Delivery Rev., 2002, 54, 3–12 CrossRef CAS.
- P. Krsko and M. Libera, Mater. Today, 2005, 8, 36–44 CrossRef CAS.
- F. E. Baley and J. V. Koleske, Poly(ethylene oxide), Academic Press, New York, 1976 Search PubMed.
- S. Zalipsky, Bioconjugate Chem., 1995, 6, 150–65 CrossRef CAS.
- R. A. Van Brederode, F. Rodriguez and G. G. Cocks, J. Appl. Polym. Sci., 1968, 12, 2097–2104 CrossRef CAS.
- N. Belcheva, R. Stamenova, C. Tsvetanov, N. Lambov, S. Tsankov and J. Smid, Macromol. Symp., 1996, 103, 193–211 CAS.
- J. M. Harris, E. C. Struck, M. G. Case, M. S. Paley, M. Yalpani, J. M. van Alstine and D. E. Brooks, J. Polym. Sci., Polym. Chem. Ed., 1984, 22, 341–52 CrossRef CAS.
- N. B. Graham and M. Zulfiqar, Polymer, 1989, 30, 2130–35 CrossRef CAS.
- Y. Gnanou, G. Hild and P. Rempp, Macromolecules, 1984, 17, 945–52 CrossRef CAS.
- R. Mincheva, N. Manolova, R. Sabov, G. Kjurkchiev and I. Rashkov, e-Polymers, 2004, no. 058 Search PubMed.
- L. C. Cesteros, C. A. Ramírez, A. Peciña and I. Katime, Macromol. Chem. Phys., 2007, 208, 1764–72 CrossRef CAS.
- L. C. Cesteros, R. González-Teresa and I. Katime, Eur. Polym. J., 2009, 45, 674–79 CrossRef CAS.
- K. L. Moffat and K. G. Marra, J. Biomed. Mater. Res., 2004, 71B, 181–7 Search PubMed.
- M. Teodorescu, B. Cursaru, P. Stanescu, C. Draghici, N. D. Stanciu and D. M. Vuluga, Polym. Adv. Technol., 2009, 20, 907–15 CrossRef CAS.
- M. Dadsetan, J. P. Szatkowski, M. J. Yaszemski and L. Lu, Biomacromolecules, 2007, 8, 1702–9 CrossRef CAS.
- J. Kim, K. Lee, T. E. Hefferan, B. L. Currier, M. J. Yaszemski and L. Lu, Biomacromolecules, 2008, 9, 149–57 CrossRef CAS.
- P. J. Lutz, Polym. Bull., 2007, 58, 161–71 CrossRef CAS.
- R. M. Laine, S. G. Kim, J. Rush, R. Tamaki, E. Wong, M. Mollan, H. J. Sun and M. Lodaya, Macromolecules, 2004, 37, 4525–32 CrossRef CAS.
- R. M. Laine, S. G. Kim, J. Rush, E. Wong, M. Mollan, H. J. Sun and M. Lodaya, Polym. Int., 2007, 56, 1006–15 CrossRef CAS.
- G. L. Lapienis and S. Penczek, Macromolecules, 2000, 33, 6630–32 CrossRef.
- G. L. Lapienis and S. Penczek, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 1576–98 CrossRef CAS.
- G. Balestrieri, Eur. Pat., 2094769, 2009 Search PubMed.
- Y. Kimura and S. L. Regen, J. Org. Chem., 1983, 48, 195–8 CrossRef CAS.
- G. Jin, T. Ido and S. Goto, Catal. Today, 2001, 64, 279–87 CrossRef CAS.
- H. D. Willauer, J. G. Huddleston and R. D. Rogers, Ind. Eng. Chem. Res., 2002, 41, 2591–2601 CrossRef CAS.
- R. Neumann and Y. Sasson, J. Org. Chem., 1984, 49, 3448–51 CrossRef CAS.
- G. E. Totten, N. A. Clinton and P. L. Matlock, J. Macromol. Sci., Part A: Pure Appl. Chem., 1998, 38(1), 77–142 Search PubMed.
- D. L. Elbert, A. B. Pratt, M. P. Lutolf, S. Halstenberg and J. A. Hubbell, J. Controlled Release, 2001, 76, 11–25 CrossRef CAS.
- P. J. Flory, Principles of Polymer Chemistry, Cornell University Press, Ithaca, NY, 1953, pp. 578–579 Search PubMed.
- E. W. Merrill, K. A. Dennison and C. Sun, Biomaterials, 1993, 14, 1117–26 CrossRef CAS.
- T. G. Fox and S. Loshaek, J. Polym. Sci., 1955, 15, 371–90 CrossRef CAS.
- J. Bicerano, R. L. Sammler, C. J. Carriere and J. T. Seitz, J. Polym. Sci., Part B: Polym. Phys., 1996, 34, 2247–59 CrossRef CAS.
- B. E. Read, Polymer, 1962, 3, 529–42 CrossRef CAS.
- J. D. Hoffman and J. I. Lauritzen, J. Res. Natl. Bur. Stand., 1961, 65A, 297–336 Search PubMed.
- P. J. Phillips and W. S. Lambert, Macromolecules, 1990, 23, 2075–81 CrossRef CAS.
- H. Takahashi, M. Shibayama, M. Hashimoto and S. Nomura, Macromolecules, 1995, 28, 5547–53 CrossRef CAS.
- C. Qiao, S. Jiang, D. Dong, X. Ji, L. An and B. Jiang, Macromol. Rapid Commun., 2004, 25, 659–63 CrossRef CAS.
Footnote |
| † Dedicated to Professor Issa A. Katime on the occasion of his 70th birthday and his appointment as Professor Emeritus. |
| This journal is © The Royal Society of Chemistry 2011 |