Antiproliferative mechanisms of quercetin in rat activated hepatic stellate cells
Li-chen
Wu
*ab,
In-wei
Lu
a,
Chi-Fu
Chung
b,
Hsing-Yu
Wu
b and
Yi-Ting
Liu
b
aDepartment of Applied Chemistry, National Chi Nan University, Puli, Nantou 545, Taiwan. E-mail: lw25@ncnu.edu.tw; Fax: +886-49-2917956; Tel: +886-49-2910960
bGraduate Institute of Biomedicine and Biomedical technology, National Chi Nan University, Puli, Nantou 545, Taiwan
First published on 21st March 2011
Abstract
Quercetin, rich in fruits and vegetables, has been used as a nutritional supplement because of its anti-inflammatory and antioxidative properties. Its positive effects on anti-hepatic fibrosis have also been suggested. However the anti-hepatofibrotic mechanisms upon which quercetin acts have yet to be well characterized. In the present study, we investigated the anti-proliferative effect of quercetin on activated hepatic stellate cells (aHSCs), the central role of hepatofibrosis, and evaluated the proteins involved in growth inhibition by a 2D gel electrophoretic analysis. Activated HSCs were isolated from Sprague Dawley rats and were spontaneously activated in vitro. Quercetin restrained the proliferation of aHSCs rather than quiescent HSCs and heptotcytes by inducing a G1 arrest as examined by cell cycle analysis and evidenced by increased levels of p53, p21CIP1/WAF1, as well as p27KIP1, and decreased abundance of cyclins (D1, D2, A, E). An apoptosis through extrinsic pathway as demonstrated by elevated expression of Fas/Fas ligand (FasL), annexin V labeling, chromatin condensation, sub-G1 fraction (7.39%), caspase-3 activity, was also observed. The 2D electrophoresis analysis revealed that quercetin negatively regulated protein molecules associated with metabolism (α-enolase, phosphoglycerate kinase), survival, cytokinesis (tubulin), and protein folding (protein disulfide isomerase A3) leading to cell growth retardation. Furthermore, quercetin might restrain HSC activation through reducing the levels of inflammatory cytokines (CXCL10, Midkine). Taken together, quercetin exerted diverse mechanisms to inhibit the growth of aHSCs. Proper consumption of quercetin could be beneficial to control the progression of heptofibrosis.
Introduction
Quercetin, a flavonol distributed widely in foods such as citrus fruits, onions and buckwheat, is claimed to be associated with several health benefits, including cardiovascular disease prevention, anti-ulcer, anti-viral, anti-cancer and anti-inflammatory activity.1 This flavonol has been determined to be safe to humans and is classified as IARC group 3, inadequate evidence of carcinogenicity in humans. Obtaining sufficient amounts of quercetin is usually achieved by consumption of dietary supplements that contain higher amounts of quercetin than would typically be found in food sources. The dose of quercetin and rutin (quercetin-3-rutinoside) in dietary supplements is approximately 1200–1500 mg per day (400–500 mg, 3 times a day),2 which could cover the biopreventative doses at 500 and 730 mg per day for sarcoidosis (for 4 successive days)3 and hypertension (for 28 successive days),4 respectively. In addition, an animal study suggested that the consumption of approximately 600 mg of quercetin and rutin potentially reduced precancerous lesions in the large intestine.2Significant recovery of hepatofibrosis through treatment with quercetin has also been indicated. A study revealed that consumption of quercetin at 70 mg kg−1 for 8 weeks significantly reduced CCl4-induced liver fibrosis.5 It was also reported that quercetin behaves as a potent aHSC (activated hepatic stellate cells) inactivator as evidenced by the significant reduction of TGF-β expression after quercetin administration to dimethylnitrosamine-induced liver damage.6 Additionally, quercetin has been noted to inhibit aHSCs proliferation in vitro through its interaction with PDGF receptors/receptortyrosine kinase and the downstream MAPK pathway.7
HSCs, the core of hepatofibrosis, transform from their quiescent phenotype to activated states (myofibroblast-like cells) under the effects of fibrogenic stimuli to perpetuate fibrosis through the amplification of the extracellular matrix (ECM). Reduced levels of aHSCs were usually observed during the resolution of hepatofibrosis in acute human liver injuries due to their eradication by NK cells,8 the proceeding of apoptosis through enhanced expression of CD95L (Fas ligand) and p53,9 and the restrained TIMP-1 expression.10 Accordingly, the induction of growth inhibition or cell death of aHSCs becomes a potential strategy to treat hepatofibrosis.
The search for novel compounds to ameliorate hepatofibrosis is essential in treating liver diseases, and thus the discovery of the underlying mechanisms appears equally critical. The efficacy of quercetin on hepatofibrosis has been indicated; however the associated mechanisms of quercetin-induced antiproliferative effect on aHSCs are not well-characterized. In the present study, we investigated the factors that contributed to this effect. The cell cycle analysis and apoptotic-associated protein molecules were examined to reveal possible mechanisms. The protein profiles of quercetin-treated aHSCs were also analyzed to explore more factors related to the induced cell growth retardation and bioactivities modulated by quercetin. Results show that quercetin restrains aHSC proliferation by arresting cell cycle at G1 phase and enhancing Fas/FasL expression to lead to apoptosis through extrinsic pathway. Additionally, quercetin modulated several bioactivities including metabolism, cytokinesis, protein folding, oxido-reduction, and inflammation. Taken together, our findings suggested that quercetin acts as a multiple-function modulator that not only regulates the growth of aHSCs, but also mediates the inflammatory responses to reduce the progression of hepatofibrosis.
Materials and methods
Chemicals and reagents
Dulbecco's modified Eagle medium (DMEM), fetal bovine serum (FBS), penicillin-streptomycin and trypsin-EDTA were purchased from Hyclone Inc. (Logam, UT, USA). Quercetin, DMSO, MTT, DTT, and propidium iodide were from Sigma (St. louis, MO, USA). Culture plates were from NUNC (Roskidle, Denmark). Annexin V-FITC apoptosis detection kit I, anti-Fas and anti-Fas ligand (FasL) were purchased from BD Biosciences (San Jose, CA, USA). Anti-β-actin, anti-cyclin D1, anti-cyclin D2, anti-α-tubulin, anti-β-tubulin, anti-α-enolase, anti-Prx-V, anti-p-IκBα, anti-nuclear factor κB (anti-NFκB, p65) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Protein assay kit was from Bio-Rad protein assay (Hercules, CA, USA). Caspase-3 colorimetric assay kit was from R&D system (Minneapolis, MN, USA). Nuclear extraction kit was from Panomic (Fermont, CA, USA).Primary hepatic and hepatic stellate cells isolation and culture
Primary hepatic cells were prepared as described by Seglen (1976).11Hepatic stellate cells (HSCs) were prepared from Sprague Dawley rat liver as described by Kawada et al..7 The liver was perfused, digested with pronase and collagenase, and then excised. After further digestion with pronase and collagenase, the resulting suspension was filtered and centrifuged on a 11% (v/v) Nycodenz cushion (Sigma, St. Louis, MO, USA), which produced a stellate cell-enriched fraction in the upper whitish layer. These cells were cultured in DMEM supplemented with 10% FBS and antibiotics (70 mg L−1penicillin and 100 mg L−1streptomycin) at 37 °C with a humidified atmosphere of 5% CO2. HSCs were activated for 6 passages and used throughout the study.Cell viability, proliferation and cell morphology monitoring
Cell viability was detected by the MTT assay (3,4,5-dimethylthiazol-2-yl-2,5-diphenyl- tetrazolium bromide, MTT; 5 mg mL−1 in PBS). Activated HSCs were previously plated in 12-well plates 5 × 104cells/well). After stabilization for 12 h, aHSCs were co-cultured without or with a series of quercetin (25, 50, 75 and 100 μM) for 24 h. The control group is vehicle-treated (dimethyl sulfoxide, DMSO) cells. The results were measured at absorbance 570 nm and expressed as a percentage (%) of the control. The anti-proliferative effect was measured using BrdU kit (Roche Diagnostic, Germany). Activated stellate cells (1 × 104cells/well) were cultured in 96-well plates for 12 h, followed by the treatment of quercetin at 25, 50, 75, and 100 μM for 24 h. Cells were labeled with 10 μM BrdU for another 2 h. The absorbance of the samples was measured with ELISA reader at 450 nm.For cell morphology monitoring, aHSCs were plated in 6-well plates at a cell density of 2 × 105cells/well. After stabilization for 12 h, the medium were changed to fresh conditioned medium containing 75 μM quercetin for 24 h. The morphologic changes in aHSCs were observed with a phase-contrast microscope (Olympus, Tokyo, Japan).
Apoptosis
Two-dimensional (2D) gel electrophoresis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g) for 15 min at 4 °C. The cytosolic proteins in the supernatant were collected and dialyzed to remove salts. The protein was stored at −80 °C until use. Protein concentration was determined by the Bio-Rad protein assay kit (Hercules, CA, USA).
000 × g) for 15 min at 4 °C. The cytosolic proteins in the supernatant were collected and dialyzed to remove salts. The protein was stored at −80 °C until use. Protein concentration was determined by the Bio-Rad protein assay kit (Hercules, CA, USA).
Reverse transcription polymerase chain reaction (RT-PCR) analysis
Total cellular RNA was prepared from 6 × 106cells/sample according to the manufacturer's instructions provided for Trizol reagents (Invitrogen, Carlsbad, CA, USA). The RT-PCR reaction was conducted with 500 ng of total RNA, reverse transcriptase (Super Script III, Invitrogen), and oligo dT18 primers. The primers used for specific genes were listed as following; protein disulfide-isomerase A3 (PDIA3): forward primer 5′-GATGGTGGCAAAG- ACATTCC-3′ and reverse primer 5′-CACGGCCACCTTCATACTTC-3′; cytokine B10 (CXCL-10): forward primer 5′-GCATCGACTTCCATGAACAGAC-3′ and reverse primer 5′-TAGGTTCCTGTGAGTATTCTGAGTTATG-3′; Midkine: forward primer 5′-GTTGCCCTCTTGGCTGTCAC-3′ and reverse primer 5′-TGGTCTCCTGGCA- CTGGGCA-3′; GADPH: forward primer 5′-AGCCCAGAACATCATCCCTGC-3′ and reverse primer 5′-TAGCCCAGGATGCCCTTTAGT-3′.Immunoblot analysis
aHSCs were cultured in 10-cm dishes (2 × 106cells/dish) with medium containing 75 μM quercetin for 24 h. Cells were lysed in RIPA buffer (50 mmol L−1 Tris-HCl, pH 7.4, 150 mmol L−1NaCl, 1% (v/v) NP-40, 5 mmol L−1EDTA, 1 mmol L−1DTT, 1 mmol L−1Na3VO4 amd 1 mmol L−1 PMSF) and centrifuged at 12000 × g for 15 min at 4 °C. The cytosolic proteins in supernatant were collected and their concentrations were determined before SDS-PAGE separation. Proteins were then transferred onto a PVDF membrane for immunoblotting. Protein blots were exposed to X-ray film (Amersham Pharmacia Biotech, Uppsala, Sweden) by ECL detection reagents.
Statistical analysis
Data were presented as mean ± standard deviation. Statistical significance was analyzed by one-way ANOVA. Values of P < 0.05 were considered significant.Results
Anti-proliferative effect and morphological alterations of quercetin on rat hepatic cells (HCs) and activated quiescent hepatic stellate cells (aHSCs and qHSCs)
The anti-proliferative effect of quercetin on rat HCs, qHSCs and aHSCs is revealed in Fig. 1A, B. Several concentrations of quercetin (0, 25, 50, 75, and 100 μM) were co-cultured with HCs, qHSCs, and aHSCs for 24 h. Significant (P < 0.01) growth inhibitory effects on aHSC appeared dose-dependently, and reached 61.0% at 75 μM, whereas no significant inhibition was shown at the same quercetin level for HCs (91.0%) and qHSCs (95.3%) (Fig. 1A). Additionally, as shown in Fig. 1B, quercetin dose-dependently suppressed the BrdU incorporation, indicating that quercetin is a potential growth inhibitor for aHSCs. Moreover, blebbed aHSCs increased upon increasing the quercetin concentration (at 75μM), indicating that quercetin is cytotoxic toward rat aHSCs (Fig. 2B). According to the cytotoxicity results of these cell types, quercetin level at 75 μM was selected for the following study. Furthermore, based on the results demonstrated in Fig. 1A, B, the growth inhibitory effect of quercetin was found specifically on aHSCs, but not significantly in normal hepatocytes and quiescent HSCs. Therefore normal hepatocytes and quiescent HSCs were excluded from the follow-up studies; only the aHSC was subjected to the investigation of mechanism of action at the molecular level on quercetin-induced cell death.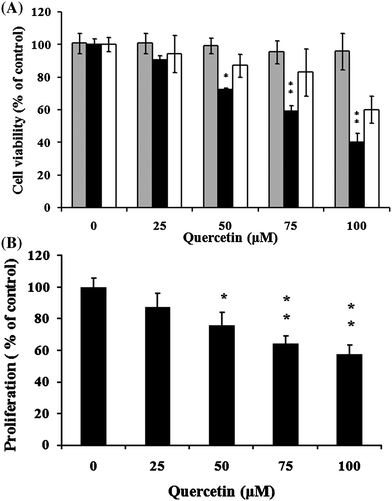 | ||
Fig. 1 Effects of cell viability and antiproliferation of quercetin on primary cells. Cell viability of qHSC( ), aHSCs (■) and hepatic cells (□) (A); Antiproliferative effect of aHSCs (■) (B). Cells were cultured with quercetin at 25, 50, 75, and 100 μM for 24 h. Data were expressed at mean ± SD from three different experiments. The asterisk (*) indicates a significant difference from the control group (*, P < 0.05; **, P < 0.01). ), aHSCs (■) and hepatic cells (□) (A); Antiproliferative effect of aHSCs (■) (B). Cells were cultured with quercetin at 25, 50, 75, and 100 μM for 24 h. Data were expressed at mean ± SD from three different experiments. The asterisk (*) indicates a significant difference from the control group (*, P < 0.05; **, P < 0.01). | ||
 | ||
| Fig. 2 Determination of quercetin induced apoptosis. Activated HSCs (2 × 105cells/well in 6-well plate) were incubated with 10, 30, 50, 75 μM of quercetin for 24 h. Cells were then harvested and incubated with FITC-conjugated annexin-V and propidium iodide (PI). FITC-positive/PI-negative cells were measured by flow cytometry (A). HSCs were stained with DAPI (magnification, ×200) after co-incubation with or without 75 μM quercetin for 24 h. Arrows indicate the condensation of chromatin (B). A sub-G1 peak appeared in 48 h with (right) or without (left) 75 μM quercetin treated histogram (C). The distribution of cells in DNA content was determined by ModFit LT 3.0 software. Data was expressed at mean ± SD from three different experiments. The asterisk (*) indicates a significant difference from control group (*, P < 0.05; **, P < 0.01). | ||
Quercetin arrests the cell cycle in aHSCs at G1 stage
We further examined the cell cycle profile of aHSCs to delineate quercetin-induced anti-proliferative effect. The cell cycle profiles were determined at different time points (0, 6, 12, and 24 h) after quercetin treatment. Cells were synchronized by serum starvation in a medium containing 0.5% serum. After incubation for 24 h, cells re-entered the cell cycle through an exchange of 5% FBS medium in the presence or absence of quercetin at 75 μM. The percentage of DNA contents at G1 phase were 87.0% and 72.5% with and without quercetin-treatment for 24 h, respectively (Table 1), indicating an arrest at G1 phase.| Time (h) | Control | Quercetin | ||||
|---|---|---|---|---|---|---|
| G0/G1 (%) | S (%) | G2/M (%) | G0/G1 (%) | S (%) | G2/M (%) | |
| 0 | 90.9 | 4.2 | 4.9 | 89.3 | 4.2 | 6.5 |
| 6 | 88.3 | 6.2 | 5.5 | 86.4 | 5.5 | 8.1 |
| 12 | 83.2 | 7.5 | 9.3 | 87.3 | 7.9 | 4.8 |
| 24 | 72.5 | 19.3 | 7.6 | 87.0 | 4.8 | 8.2 |
Quercetin induces HSC apoptosis
Apoptotic analysis was performed to delineate quercetin-induced cell death (Fig. 2A–C). Results of annexin V staining showed that significant fluorescent intensity (P < 0.01) showed in a dose-dependent manner ranging from 15% to 58% upon increasing quercetin concentration (from 10 to 75 μM) in quercetin-treated aHSCs (Fig. 2A). Additionally, chromatin condensation and cell blebbing also increased as revealed by DAPI staining (Fig. 2B). The induced apoptosis was further characterized by the existence of sub-G1 phase (Fig. 2C, 7.39% at 24 h, 75 μM quercetin); elevated caspase-3 activity (Fig. 3A, a 1.5-fold increase, with quercetinvs. without, 0.37 vs. 0.25 a.u., respectively, 75 μM, 24 h, P < 0.05). Moreover, treating with caspase inhibitor, z-VAD-fmk, protected aHSCs from death dose-dependently (Fig. 3B).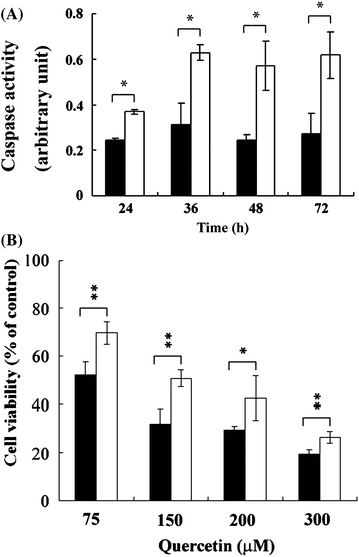 | ||
| Fig. 3 Quercetin-induced caspase-dependent cell death in activated HSCs. Activated HSCs were co-cultured with (□) or without (■) 75 μM quercetin for 24, 36, 48, and 72 h. Caspase activity was evaluated (A). Cells were co-cultured with quercetin (75, 150, 200, and 300 μM) and with (□) or without (■) z-VAD-fmk (50 μM) for 24 h (B). Cell viability was determined. The asterisk (*) indicates a significant difference between treated and untreated groups (*, P < 0.05; **, P < 0.01). | ||
To further explore molecules associating with quercetin-induced apoptosis, investigation on the expression of cell cycle-, apoptosis pathway-, and cell survival-related molecules was performed. Results of immunoblot analysis (Fig. 4A, B) reveals that quercetin (75 μM, 24 h) significantly elevated the abundance of cell cycle regulatory proteins p27KIP1, p21CIP1/WAF1, and p53, but suppressed the expression of cyclins D1, D2, A, and E, insinuating a classic G1 arrest. The up-regulation of apoptosis-associated molecules such as cytochrome c, Fas, and FasL suggested the involvement of extrinsic apoptotic pathway. Moreover, the suppressed survival signals including phosphorylated Akt (p-Akt; Fig. 4A), phosphorylated IκBα (p-IκBα) (Fig. 7A) and nuclear NFκB (Fig. 7A) proteins, accompanied with the induced apoptosis. The hindered nuclear entry of NFκB was further examined by analyzing the nuclear NFκB protein-DNA binding activity. As seen in Fig. 5, the binding activity significantly (P < 0.05) decreased with the treatment (75 μM), suggesting a decreased abundance of nuclear NFκB proteins.
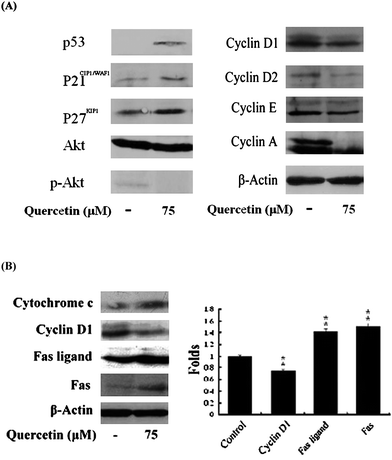 | ||
| Fig. 4 Regulation of cell cycle- and apoptotic pathway-associated proteins by quercetin. Activated HSCs were treated with or without quercetin (75 μM) for 24 h. Cell cycle-associated-, survival- (A), and apoptotic pathway-associated proteins (B) were determined. The levels of proteins were analyzed by immunoblot analysis. The asterisk (*) indicates a significant difference from control group (**, P < 0.01). | ||
 | ||
| Fig. 5 Decrease of nuclear NFκB—DNA binding activity by quercetin treatment in activated HSCs. Cultures were treated with 75 μM for 24 h. Activated NFκB p65 DNA binding activity was determined by enzyme-linked immunosorbent assay according to the manufacturer's instructions. Data were mean ± SD of triplicates. The asterisk (*) indicates a significant difference from control group (*, P < 0.05). | ||
Identification of proteins modulated by quercetin and associated with quercetin-induced antiproliferative effect on aHSCs
A 2D gel electrophoresis analysis was performed to study the proteins that were regulated by quercetin and were associated with quercetin-induced antiproliferative effect (Fig. 6). Extracts of aHSCs were loaded onto an 18 cm IPG strip (pH 3–10) and then run on a second-dimension SDS-PAGE apparatus (12%). Proteins were visualized using colloidal Coomassie Blue. After computer analysis of the protein profiles of quercetin-treated (75 μM, 24 h) and untreated groups, a total of 512 spots were detected. Among them, 74 spots were regulated by quercetin, and 10 spots were identified by MS analysis and fingerprint matching (Fig. 6, Table 2). The down-regulated proteins included α-enolase, protein disulfide isomerase A3 (PDIA3), α-smooth muscle actin (α-SMA), tropomyosin 4, midkine (MK), cytokine B10 (CXCL-10), tubulin, and phosphoglycerate kinase (PGK). The up-regulated proteins were peroxiredoxin-V (Prx-V) and NFκB inhibitor (IκB). Some of these spots might be used as biomarkers for hepatofibrosis.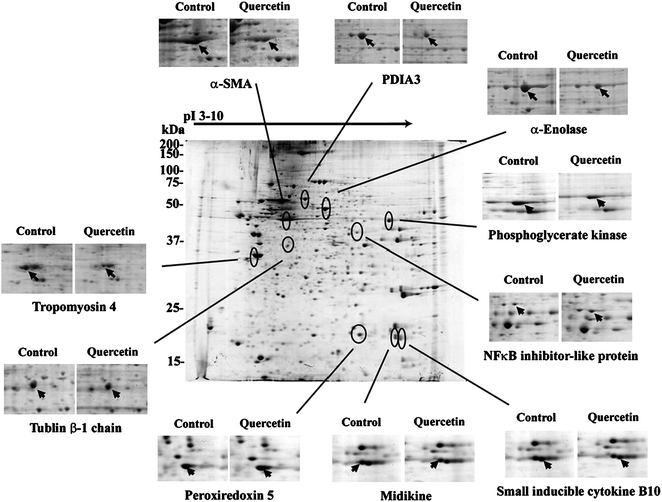 | ||
| Fig. 6 Two-dimensional electrophoresis of proteins from cellular fractions of activated HSCs. Isoelectric focusing was performed using immobilized pH gradients (IPG) 3–10. A total of 350 μg protein was applied per IPG gel. HSCs were derived from normal liver and were cultured for 9 days (in vitroactivation, control). The procedures are referred to in “Materials and methods”. Activated HSCs were treated with 75 μM quercetin for 24 h. The changes of down- and up-regulated proteins were displayed. The pointed spots were identified by MS analysis and peptide fingerprinting. Each result was from one of three different experiments with similar pattern. | ||
| No. | Accession No. | Name | Function | Coverage (%) | Matches |
|---|---|---|---|---|---|
| 1 | P04764 | α-Enolase | Glycolysis | 32% | 10 |
| 2 | P11598 | Protein disulfide-isomerase A3 precursor (PDIA3) | Protein folding | 23% | 12 |
| 3 | P68136 | α-Smooth muscle actin (α-SMA) | Contractile | 29% | 10 |
| 4 | P09495 | Tropomyosin α 4 chain | Contractile | 36% | 11 |
| 5 | Q9R1S9 | Midkine precursor (MK) | Inflammation | 55% | 5 |
| 6 | P48973 | Small inducible cytokine B10 (CXCL-10) | Inflammation | 78% | 5 |
| 7 | P04691 | Tubulin β chain | Cytokinesis | 21% | 6 |
| 8 | P16617 | Phosphoglycerate kinase (PGK) | Glycolysis | 36% | 11 |
| 9 | Q9R063 | Peroxiredoxin-V (Prx-V) | Oxidoreduction | 50% | 8 |
| 10 | Q8R2H1 | NFκB inhibitor-like protein (IκB) | NFκB inhibitor | 24% | 5 |
Validation of identified proteins
The identified proteins from 2D gel electrophoresis analysis were validated by immunoblotting (α-enolase, Prx-V, tubulin α/β, nuclear NFκB and p-IκBα) or RT-PCR (CXCL-10, PDIA3, and MK). Fig. 7A, B reveals that quercetin significantly inhibited the expression of α-enolase and tubulin α/β, whereas it enhanced the expression of Prx-V at protein level. It also decreased the expression of CXCL-10, PDIA3, and MK. Additionally, the abundance of pIκBα and nuclear NFκB were negatively modulated. | ||
| Fig. 7 Validation of 2D electrophoresis analysis by immunoblot analysis or by RT-PCR (A). Activated HSCs were treated with or without quercetin (75 μM) for 24 h. Procedures of immunoblot analysis and RT-PCR were described in the section of Materials and Methods. The levels of immunoblot analysis and RT-PCR products were quantified by densitometer (B). The asterisk (*) indicates a significant difference from control group (*, P < 0.05; **, P < 0.01). | ||
Discussion
In the present study, we further characterized the molecules regulated by quercetin and the elements associated with quercetin-induced antiproliferative and apoptotic effects on rat aHSCs. Quercetin significantly restricted the growth of rat aHSCs (Fig. 1), as consistent with a previous study,7 and showed no significant cytotoxicity on HCs and qHSCs. The examination of quercetin-induced mechanism of action on these two cell types at the molecular level is thus excluded. The antiproliferative effect induced by quercetin could be partly resulted from a cell cycle arrest at G1 phase with a suppressed expression of cyclins D1/D2, which guard the early G1 stage; and E/A, which guard the late G1 stage; as well as the activation of p27KIP1, p53 and its downstream effector p21CIP1/WAF1. In addition, we also found that the enhanced expression of extrinsic apoptotic pathway factors, Fas and FasL, might be responsible for quercetin-induced apoptosis. Cell-type-dependent cell cycle arrest induced by quercetin has been suggested such as the G1 phase13 and the G2/M phase.14 Interestingly, some cells, for example, lymphoblastic leukemia cells (HSB-2), are resistant to quercetin.15It is known that antimitogenic signals (e.g. transforming growth factor-β and IL-6) mobilize nuclear p27KIP1 to disturb the function of cyclin E-cdk2 complex.16 This prevents the activation of S-phase specific transcription factor, such as E2F, and results in cell growth inhibition. The loss of this regulatory mechanism may lead to hyperproliferative disorders which are commonly seen in cancer cells. Generally, aHSCs constantly expressed TGF-β receptor (TGF-β-R) to receive the signals from TGF-β, unlike hepatocytes, which express TGF-β-R dependent on local environmental stimuli.17 The secretion of substantial amount of TGF-β to sustain the production of ECM is a feature of aHSCs. However, higher levels of TGF-β might probably lead to marked hypo-proliferation. Presumably, aHSCs prevent TGF-β-induced cell growth inhibition through the activation of PI3K-Akt pathway.18 In the present study, we revealed that quercetin was able to inactivate Akt phosphorylation and hence increase the abundance of p27KIP1 leading to a G1 phase cell cycle arrest.
The toxicity of oxidized quercetin could possibly be associated with the induced apoptosis. It is indicated that the apoptosis of human chronic myeloid (K562) leukemia cells was linked to a similar toxicity.15Quercetin is one of the most prominent antioxidants used commonly to scavenge free radicals, and it may become toxic after oxidation.19 In an antioxidant network,20 oxidized quercetin can be recycled through interactions with other antioxidants, such as ascorbic acid or glutathione (GSH). Nevertheless, the oxidized form of quercetin, ortho-quinone (Q), reacts preferably with thiol groups, rather than with ascorbic acid, to form adducts with GSH such as 6-GSQ or 8-GSQ19 and protein thiols.21 The presence of toxic quercetin-thiol adducts could interfere with enzyme activity such as quercetin-mediated arylation of glutathione-S-transferase-P1-1 at cysteine4722 or remove GSH from the antioxidant networks, leading to growth inhibition or cell death. Therefore, we suspected that the toxic adducts may be involved partially in the apoptotic effect.
To further investigate proteins regulated by quercetin, a 2D electrophoresis study was performed. This analysis revealed that proteins involved in metabolism, cytokinesis, protein folding, oxido-reduction, inflammation, and cell survivability were regulated. In the aspect of metabolism, the expression of glycolytic proteins, such as α-enolase and phosphoglycerate kinase (PGK), was down-regulated (Table 2). Quercetin has been reported to inhibit glycolysis and gluconeogenesis in rat liver via the reduction of oxidative phosphorylation, the inhibition of Na+/K+-ATPase activity, and the suppressed expression of glucokinase and glucose 6-phosphatase.23 The decreased level of α-enolase and PGK caused by quercetin could also be another mechanism to hinder glycolysis. Besides that, elevated PGK levels have been reported in aHSCs.24 Therefore, quercetin could be used to limit the activation of HSCs. The results of 2D gel electrophoresis might reveal the link between glycolysis and apoptosis, which agreed with a previous finding that the two independent systems, glycolysis and apoptosis, maneuver an enzyme complex containing an apoptosis inducer (BAD) and a glycolytic protein (glucokinase) to modulate cell survival through maintaining glucose homeostasis.25
As mentioned above, oxidized quercetin could render cytotoxicity leading to cell death. The 2D gel analysis indicated that a cellular defensive mechanism was activated by promoting the expression of an oxido-reduction enzyme, peroxiredoxin-V (Prx-V), an ubiquitously expressed enzyme which possesses peroxynitrite reductase activity and antioxidant activity equivalent to that of catalase,26 to neutralize the oxidative stress resulted from oxidized quercetin.
Similar to the oxidized quercetin that inactivates enzyme activity causing cell death, decreased expression of enzymes that catalyzes protein folding may also lead to cell growth inhibition or death. Our data suggested that a decreased expression of PDIA3, a nucleus- and ER-located enzyme responsible for the catalyzation of protein folding through the formation and breakage of disulfide bonds between cysteine residues, might result in the loss of normal protein functions leading to cell growth inhibition and cell death. Moreover, proteins involved in cytokinesis could be another factor in contributing to cell growth inhibition or cell death. The down-regulated expression of cytokinesis proteins, tublin α/β and tropomyosin 4, has been linked to restrained cell growth.27Tubulin α/β constitutes microtubules to perform several cellular processes including mitosis, cytokinesis, and vesicular transport. The lack of tubulins could likely interfere with various cellular activities. In addition to tubulin, tropomyosin is also involved in cytokinesis. Tropomyosin, an actin-binding protein regulating actin mechanics, participates not only in cytokinesis, but also in cell contraction. Quercetin could likely regulate these two cytoskeleton proteins to inhibit cell growth or cell contraction to reduce portal hypertention caused by vasoactive substances.28
The regulation of cellular survival signaling was also observed by 2D gel analysis. The regulation of IκB and NFκB is critical to the viability of aHSCs. Activation of NFκB enhances the survival of stellate cells in the presence of apoptotic stimuli.29NFκB usually binds with IκB in the cytoplasm prior to its activation. Once IκB is phosphorylated (p-IκB) by IκB kinase (IKK) and degraded by proteasome, NFκB enters the nucleus to regulate a number of genes, including the inhibitors of the caspase cascade.30 Our 2D electrophoresis results revealed an increased IκB level after quercetin treatment, indicating a likely restrained dissociation of NFκB from IκB. This could probably result in impeded NFκB nuclear translocation and reduced phosphorylation of IκB. The above hypothesis was confirmed by immunoblot analysis in which the levels of p-IκB and nuclear NFκB were significantly reduced after quercetin treatment. It was also noted that the activity of nuclear NFκB was suppressed. These results suggested that quercetin could possibly down-regulate the activation of NFκB, thereby resulting in subsequent cell death.
Finally, quercetin may act as an inflammatory modulator. This flavonol was able to down-regulate CXCL-10 and midkine (MK). CXCL-10 is expressed by inflammatory cells, hepatocytes, and non-parenchymal intrahepatic cell populations.31 This chemokine functions as a potent interferon-γ inducible chemo-attractant of neutrophils and T-cells. Increased CXCL-10 level has been observed in human hepatitis C-associated cirrhosis32 and in liver transplant organ acceptance.33 Similar to CXCL-10, the pleiotropic growth factor MK is also associated with inflammatory responses and is active in cell proliferation, migration, and angiogenesis.34 Elevated levels of hepatic inflammation may substantially result in the activation of HSCs leading to hepatofibrosis.35 The down-regulation of inflammatory mediators, CXCL-10 and MK, could thus possibly lessen the degree of inflammation and reduce the activation of HSCs, thereby delaying the progress of hepatofibrosis.
Conclusion
Taking dietary nutritional supplements on a regular basis to achieve beneficial effects on health has become common nowadays. Quercetin, a bioactive compound in the supplement, has gained attention for years because of its effectiveness on several bioactivities. Herein, we revealed that this flavonol may exert several mechanisms to induce the antiproliferative effect on aHSCs, a central role of hepatofibrosis, but not HCs and qHSCs. The progression of hepatofibrosis is likely to be limited by quercetin due to the reduced level of aHSCs either by restrained growth or by induction of apoptosis. Accordingly, it could be concluded that proper consumption of quercetin might be helpful to control the progression of hepatofibrosis.Acknowledgements
This work was supported by NSC96-2627-M-260-002, NSC97-2113-M-260-006-MY2, Taiwan National Science Council; TCVGH-NCNU 977902, TCVGH-NCNU 987901, TCVGH-NCNU 987904, Taichung Veterans General Hospital and National Chi-Nan University.References
- A. W. Boots, G. R. Haenen and A. Bast, Eur. J. Pharmacol., 2008, 585, 325–337 CrossRef CAS.
- S. R. Volate, D. M. Davenport, S. J. Muga and M. J. Wargovich, Carcinogenesis, 2005, 26, 1450–1456 CrossRef CAS.
- A. W. Boots, M. Drent, E. L. Swennen, H. J. Moonen, A. Bast and G. R. Haenen, Respir. Med., 2009, 103, 364–372 CrossRef.
- R. L. Edwards, T. Lyon, S. E. Litwin, A. Rabovsky, J. D. Symons and T. Jalili, J. Nutr., 2007, 137, 2405–2411 CAS.
- A. G. López-Reyes, N. Arroyo-Curras, B. G. Cano, V. J. Lara-Díaz, G. E. Guajardo-Salinas, J. F. Islas, V. Morales-Oyarvide, L. A. Morales-Garza, F. J. Galvez-Gastelum, G. Grijalva and J. E. Moreno-Cuevas, Ann. Hepatol., 2008, 7, 130–135 Search PubMed.
- E. S. Lee, H. E. Lee, J. Y. Shin, S. Yoon and J. O. Moon, J. Pharm. Pharmacol., 2003, 55, 1169–1174 CrossRef CAS.
- N. Kawada, S. Seki, M. Inoue and T. Kuroki, Hepatology, 1998, 27, 1265–1274 CrossRef CAS.
- W. I. Jeong, O. Park, S. Radaeva and B. Gao, Hepatology, 2006, 44, 1441–1451 CrossRef CAS.
- I. Aprigliano, J. Dudas, G. Ramadori and B. Saile, Liver Int., 2008, 28, 546–557 Search PubMed.
- F. R. Murphy, R. Issa, X. Zhou, S. Ratnarajah, H. Nagase, M. J. Arthur, C. Benyon and J. P. Iredale, J. Biol. Chem., 2002, 277, 11069–11076 CrossRef CAS.
- P. O. Seglen, 1976, Cell Biol., vol. 13, pp. 29–83, Academic Press, New York Search PubMed.
- H. I. Field, D. Fenyö and R. C. Beavis, Prot. Clin. Appl., 2002, 2, 36–47 Search PubMed.
- M. Yoshida, M. Yamamoto and T. Nikaido, Cancer Res., 1992, 52, 6676–6681 CAS.
- S. U. Mertens-Talcott, S. T. Talcott and S. S. Percival, J. Nutr., 2003, 133, 2669–2674 CAS.
- F. Brisdelli, C. Coccia, B. Cinque, M. G. Cifone and A. Bozzi, Mol. Cell. Biochem., 2007, 296, 137–149 CrossRef CAS.
- M. Adachi, Y. Osawa, H. Uchinami, T. Kitamura, D. Accili and D. A. Brenner, Free Radical Bio. Med., 2007, 132, 1434–1446 CAS.
- M. Date, K. Matsuzaki, M. Matsushita, Y. Tahashi, F. Furukawa and K. Inoue, Gut, 2000, 46, 719–724 CrossRef CAS.
- S. Reif, A. Lang, J. N. Lindquist, Y. Yata, E. Gabele, A. Scanga, D. A. Brenner and R. A. Rippe, J. Biol. Chem., 2003, 278, 8083–8090 CrossRef CAS.
- A. Bast and G. R. Haenen, Environ. Toxicol. Pharmacol., 2002, 11, 251–258 CrossRef CAS.
- A. Meister, J. Biol. Chem., 1994, 269, 9397–9400 CAS.
- G. C. Yen, P. D. Duh, H. L. Tsai and S. L. Huang, Biosci., Biotechnol., Biochem., 2003, 67, 1215–1222 CrossRef CAS.
- J. J. van Zanden, O. Ben Hamman, M. L. van Iersel, S. Boeren, N. H. Cnubben, M. Lo Bello, J. Vervoort, P. J. van Bladeren and I. M. Rietjens, Chem.-Biol. Interact., 2003, 145, 139–148 CrossRef CAS.
- F. R. Gasparin, F. L. Spitzner, E. L. Ishii-Iwamoto, A. Bracht and J. Constantin, Xenobiotica, 2003, 33, 903–911 CrossRef CAS.
- D. B. Kristensen, N. Kawada, K. Imamura, Y. Miyamoto, C. Tateno, S. Seki, T. Kuroki and K. Yoshizato, Hepatology, 2000, 32, 268–277 CrossRef CAS.
- N. N. Danial, C. F. Gramm, L. Scorrano, C. Y. Zhang, S. Krauss, A. M. Ranger, S. R. Datta, M. E. Greenberg, L. J. Licklider, B. B. Lowell, S. P. Gygi and S. J. Korsmeyer, Nature, 2003, 424, 952–6 CrossRef CAS.
- B. Knoops, A. Clippe, C. Bogard, K. Arsalane, R. Wattiez, C. Hermans, E. Duconseille, P. Falmagne and A. Bernard, J. Biol. Chem., 1999, 274, 30451–30458 CrossRef CAS.
- R. A. Janssen and J. W. Mier, Mol. Biol. Cell, 1997, 8, 897–908 CAS.
- H. Reynaert, M. G. Thompson, T. Thomas and A. Geerts, Gut, 2002, 50, 571–581 CrossRef CAS.
- M. R. Watson, K. Wallace, R. G. Gieling, D. M. Manas, E. Jaffray, R. T. Hay, D. A. Mann and F. Oakley, J. Hepatol., 2008, 48, 589–597 CrossRef CAS.
- B. Saile, N. Matthes, H. El Armouche, K. Neubauer and G. Ramadori, Eur. J. Cell Biol., 2001, 80, 554–561 CrossRef CAS.
- Y. Zhai, B. Qiao, F. Gao, X. Shen, A. Vardanian, R. W. Busuttil and J. W. Kupiec-Weglinski, Hepatology, 2008, 47, 199–206 CAS.
- M. T. Liu, H. S. Keirstead and T. E. Lane, J. Immunol., 2001, 167, 4091–4097 CAS.
- S. Goddard, A. Williams, C. Morland, S. Qin, R. Gladue, S. G. Hubscher and D. H. Adams, Transplantation, 2001, 72, 1957–1967 CrossRef CAS.
- E. H. van der Horst, B. T. Frank, L. Chinn, A. Coxon, S. Li, F. Polesso, A. Slavin, A. Ruefli-Brasse and H. Wesche, Neoplasia., 2008, 10, 340–347 CAS.
- F. Marra, J. Hepatol., 1999, 31, 1106–1119 CrossRef.
| This journal is © The Royal Society of Chemistry 2011 |