Identification and characterization of five new classes of chlorogenic acids in burdock (Arctium lappa L.) roots by liquid chromatography/tandem mass spectrometry†‡
Rakesh
Jaiswal
and
Nikolai
Kuhnert
*
Chemistry, Jacobs University Bremen, Campus Ring 1, 28759, Bremen, Germany. Tel: +49 421 200 3120; Fax: +49 421 200 3229; E-mail: n.kuhnert@jacobs-university.de
First published on 6th December 2010
Abstract
Burdock (Arcticum lappa L.) roots are used in folk medicine and also as a vegetable in Asian countries especially Japan, Korea, and Thailand. We have used LC-MSn (n = 2–4) to detect and characterize in burdock roots 15 quantitatively minor fumaric, succinic, and malic acid-containing chlorogenic acids, 11 of them not previously reported in nature. These comprise 3-succinoyl-4,5-dicaffeoyl or 1-succinoyl-3,4-dicaffeoylquinic acid, 1,5-dicaffeoyl-3-succinoylquinic acid, 1,5-dicaffeoyl-4-succinoylquinic acid, and 3,4-dicaffeoyl-5-succinoylquinic acid (Mr 616); 1,3-dicaffeoyl-5-fumaroylquinic acid and 1,5-dicaffeoyl-4-fumaroylquinic acid (Mr 614); 1,5-dicaffeoyl-3-maloylquinic acid, 1,4-dicaffeoyl-3-maloylquinic acid, and 1,5-dicaffeoyl-4-maloylquinic acid (Mr 632); 1,3,5-tricaffeoyl-4-succinoylquinic acid (Mr 778); 1,5-dicaffeoyl-3,4-disuccinoylquinic acid (Mr 716); 1,5-dicaffeoyl-3-fumaroyl-4-succinoylquinic acid and 1-fumaroyl-3,5-dicaffeoyl-4-succinoylquinic acid (Mr 714); dicaffeoyl-dimaloylquinic acid (Mr 748); and 1,5-dicaffeoyl-3-succinoyl-4-dimaloylquinic acid (Mr 732). All the structures have been assigned on the basis of LC-MSn patterns of fragmentation, relative hydrophobicity, and analogy of fragmentation patterns if compared to caffeoylquinic acids.
Introduction
Classically, chlorogenic acids (CGAs) are a family of esters formed between quinic acid and certain trans-cinnamic acids, most commonly caffeic, p-coumaric, and ferulic acids1,2 and sometimes dimethoxycinnamic, trimethoxycinnamic, and sinapic acids.3 Representative structures are shown in Fig. 1. In the IUPAC system (−)-quinic acid is defined as 1L-1(OH),3,4/5-tetrahydroxycyclohexane carboxylic acid, but Eliel and Ramirez4 recommend 1α,3R,4α,5R-tetrahydroxycyclohexane carboxylic acid. | ||
| Fig. 1 Structures of burdock roots chlorogenic acids (IUPAC numbering).1 | ||
Several pharmacological activities of CGAs including antioxidant activity,5–7 ability to increase hepatic glucose utilization8–12 inhibition of the HIV-1 integrase,13,14 antispasmodic activity,15 and inhibition of the mutagenicity of carcinogenic compounds16 have been revealed by in vitro, in vivo and human intervention studies so far.
Burdock belongs to the botanical family of the Asteraceae and grows in most temperate climate zones. Burdock roots have been used as a vegetable in Europe (e.g. as dandelion and burdock softdrink) and North America, have however, fallen out of fashion recently. In Asia, in particular in Japan (named gobo), Korea (named u-eong), Taiwan and Thailand burdock roots are still consumed in larger quantities as a popular vegetable. It has also been used in folk medicine as diuretic, antipyretic, and blood purifier. Burdock roots show pharmacological activities such as desmutagenetic activity,17 antioxidant activity,18–20 anti-inflammatory activity,21 and hepatoprotective activity.22 Due to the broad range of biological activities and use in folk medicine burdock roots have been analyzed on several occasions for their chemical constituents.18,19,23
Recently, LC-MSn has been used to characterize cinnamoyl-amino acid conjugates2 and to discrimate between individual regioisomers of monoacyl, diacyl, and triacyl chlorogenic acids.2,3 The MS fragmentation patterns, found to be significantly different for all regioisomers of CGAs, based on fragmentation preferences induced by characteristic hydrogen bonding arrays within the gas phase ions of CGAs in tandem MS spectra, UV spectrum, retention times, and relative hydrophobicity have been utilized to develop structure-diagnostic hierarchical keys for the identification of chlorogenic acids. In this study, we expand on methods developed previously for the structure elucidation of CGAs to the qualitative profiling of chlorogenic acids in burdock root extract. In previous work we have shown that our structure diagnostic hierarchical key can be applied to cinnamoyl and aryl substituted esters of quinic acid. In this study we report on its extension to quinic acid esters bearing a functionalized aliphatic side chain such as succinic acid, malic acid or fumaric acid. Since these side chains carry further substituents such as carboxylic acids or alcohols, which are able to form further hydrogen bonds within the gas phase ions, we address the question on whether our previous observation can after critical evaluation be applied to these new classes of compounds.
Results and discussion
Preliminary assessment of data
All the data for chlorogenic acids presented in this paper uses the recommended IUPAC numbering system,1 structures are presented in Fig. 1. When necessary, previously published data has been amended to ensure consistency and avoid ambiguity.In general, new CGAs can be identified in an All MSn EIC (extracted ion chromatogram) by their unique fragments at m/z 173 and 191 and UV spectra at 320 nm (λmax). Selected ion monitoring at m/z 353 (CQA), 515 (diCQA), 613 (FudiCQA), 615 (SucdiCQA), 631 (MdiCQA), 715 (diSucdiCQA), 731 (SucMdiCQA), 747 (diMdiCQA), and 777 (SuctriCQA) immediately located 19 chromatographic peaks eluting between 23–55 min, each with a UV spectrum typical of chlorogenic acids (λmax 320 nm). Compounds displaying m/z values at 353 and 515 have been reported earlier, whereas compounds with m/z values of 613, 615, 631, 715, 731, 747, and 777 are novel CGA derivatives containing caffeic acid and aliphatic acid substituents as ester groups.
Five CGAs were identified showing an m/z value in the negative ion mode at 631 (MdiCQA). All of these produced MSn peaks by the loss of an acyl residue of mass unit 114 Da corresponding to loss of a maloyl moiety. Three CGAs produced MSn peaks by the loss of an acyl residue of mass unit 98 Da corresponding to a fumaroyl moiety and nine CGAs produced a loss of 100 corresponding to a succinoyl moiety. In nature only fumaric acid (trans-isomer) and S-malic acid (L-isomer) are present. In the case of malic acid there are two possible acylation positions and for the confirmation of these positions targeted MSn experiments were performed. The elemental composition of all compounds at the m/z values above was determined by high resolution LC-ESI-TOF mass spectrometry. All compounds identified displayed mass errors below 5 ppm.
The chemical nature of the non-caffeoyl side chains was established firstly using high resolution in source fragmentation experiments in negative ion mode showing fragment ions with the neutral losses of 100.0154, 97.9998 and 116.0104 Da for the succinoyl, fumaroyl and maloyl residues, respectively. Furthermore a hydrolysis experiment, in which the burdock extract was treated for 1 h with 1 N NaOH, followed by HPLC analysis, revealed the presence of fumaric acid (rather than its isomer maleic acid), (S)-malic acid and succinic acid by comparison to authentic standards.
For all previously investigated chlorogenic acids the mass loss leading to the formation of the MS2 base peak has always correspond to a single acyl residue, accompanied by water when the compound is acylated at C4.2,3 In this study, only five of the 15 novel CGAs fragmented in this manner (9, 10, 16, 17, and 19). Seven novel CGAs lost simultaneously a cinnamoyl and an alkoyl residue (5–8, 11, 12, and 18), one lost a cinnamoyl and two alkoyl residues (15), and the ninth lost two cinnamoyl residues and an alkoyl residue (14). This observation indicates that an internal hydrogen bond from the free side chain carboxylic acid moiety activates the aliphatic side chain for more facile fragmentation if compared to the cinnamoyl moieties. Hence an uncritical adaptation of our previous structure assignment tools is not possible here and requires modification. We propose here to adopt the following general regiochemistry assignment strategy: Targeted MS3 experiments of the ion at m/z 515 and MS4 experiments of the ions at m/z 353 allow unambiguous determination of the regiochemistry of the ions corresponding to dicaffeoyl quinic acid, where ions observed are identical to ions formed from the compounds originally reported thus allowing the definition of the caffeoyl regiochemistry. Definition of dicaffeoyl regiochemistry is possible for any combination of regiochemistry, except for 1,3 dicaffeoyl and 3,5 dicaffeoyl, which both yield in their tandem MS spectra 3-caffeoyl quinic acid. Hence for this combination no unambiguous assignment of regiochemistry is possible and two structural alternatives are left open. Once the two positions of caffeoyl regiochemistry are defined the further substituents must form an ester bond to any one of the two remaining quinic acid alcohol groups. Should the substitution pattern remaining correspond to n,4 (n being one of the free alcohol and the 4-OH being the other one) we apply our previous observation that a 4-substututed derivative loses its acyl side chain most reluctantly producing a dehydrated quinic acid fragment ion at m/z 173 in the process. Should this feature be observed in the spectra the third substituent occupies position 4 of the quinic acid, if not it must occupy the alternative position n. If positions 1,3-, 1,5- or 3,5 are unoccupied after definition of caffeoyl regiochemistry assignment of aliphatic side chain regiochemistry is more difficult and is only reliable if tandem mass spectra of both remaining regioisomers are available that allow a direct comparison of the ease of fragmentation of the aliphatic side chain, where we apply our previous rule that fragmentation occurs in the order of 1 similar to 5 after 3 after 4 position. Energy resolved mass spectra show that the 1-substituent fragments easier if compared to the 5-substituent, however this effect cannot be readily extracted from MSn spectra obtained in ramped auto MSn scans as applied here, again resulting in two alternative structures possible. Should experimental tandem mass spectra for both regioisomers not be available, further supporting arguments including relative hydrophobicity, comparison of fragmentation patterns of chemically similar side chains (e.g. succinoyl and maloyl) or phytochemical arguments (e.g. Asteraceae are rarely acylating at C1 of the quinic acid) are employed in a more tentative assignment of regiochemistry. Tetraacyl CGAs containing three caffeoyl residues can be investigated by targeting parent ion +677.
These procedures are pursued in the following discussion. Targeted MS3 experiments for m/z 631 + 573 (arises by the loss of 58 amu which corresponds to the CH2COOH group), 631 + 469 (arises by the loss of the caffeoyl residure), and 631 + 307 (arises by the loss of two caffeoyl residues) produced MS3 secondary peaks at m/z 249 ([maloylquinic acid-CH2COOH–H+]−) by the loss of a CH2COOH group (58 Da) which suggested that the maloyl residue is attached by its C1 position as opposed to its C4 position to the quinic acid moiety. This finding represents another nice example, demonstrating the ability of tandem mass spectrometry to act as a reliable tool in structure assignment of regioisomeric compounds.
Characterisation of caffeoylquinic acid (Mr 354) and dicaffeoylquinic acids (Mr 516)
One caffeoylquinic acid and three dicaffeoylquinic acids eluting between ∼18–40 min were easily located and assigned using the hierarchical keys previously developed2,3,24 as the well-known 5-caffeoylquinic acid (1), 1,4-dicaffeoylquinic acid (2), 1,5-dicaffeoylquinic acid (3), and 3,5-dicaffeoylquinic acid (4). Detailed mass spectra have been published previously2 and are not repeated here.Characterisation of putative dicaffeoyl-succinoylquinic acids (Mr 616)
Four isomers (5–8) were detected at m/z 615 eluting between ∼43–47 min and were assigned as dicaffeoyl-succinoylquinic acids. In theory a total of 12 different regioisomers of dicaffeoyl-succinoyl quinic acid are expected with the eight missing isomers either not being present at all, present in too low concentrations or co-eluting with the detected isomers. The most hydrophilic isomer 5 produced the MS2 base peak at m/z 353 by the loss of a caffeoyl residue and a succinoyl residue. Its MS2 at m/z 515 and MS3 spectra at m/z 353 are identical to the MS2 and the MS3 spectra of 3,4-dicaffeoylquinic acid (Fig. 2).2 This suggested that isomer 5 is one of two possible 3,4-dicaffeoyl-succinoylquinic acids. The secondary peak at m/z 453 in the MS2 spectrum is of very low intensity; this suggested that the loss of a caffeoyl residue is less favorable if compared to the loss of the succinoyl residue secondary peak at m/z 515. On the basis of the above arguments isomer 5 was tentatively assigned as 3,4-dicaffeoyl-5-succinoylquinic acid or 3,4-dicaffeoyl-1-succinoylquinic acid.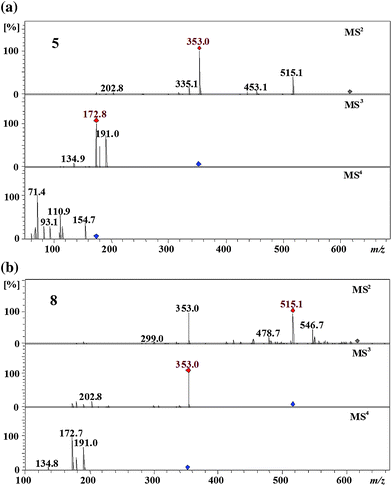 | ||
| Fig. 2 MS4 spectra of 3,4-dicaffeoyl-5-succinoylquinic acid (5) and 3-succinoyl-4,5-dicaffeoylquinic acid (8) (parent ion at m/z 615 in negative ion mode). | ||
The most hydrophobic isomer 8 produced the MS2 base peak at m/z 515 ([dicaffeoylquinic acid-H+]−) by the loss of a succinoyl residue and secondary peaks at m/z 353 ([caffeoylquinic acid-H+]−), by the loss of a succinoyl residue and a caffeoyl residue, and at m/z 453 ([caffeoyl-succinoylquinic acid-H+]−), by the loss of a caffeoyl residue (Fig. 2). Its MS3 and MS4 spectra were identical to the MS2 and the MS3 spectra of 4,5-dicaffeoylquinic acid,2 which suggested that isomer 8 is 4,5-dicaffeoyl-succinoylquinic acid. It loses the succinoyl residue preferentially to the caffeoyl residue which suggested that the succinoyl residue is found at the C3 position of the quinic acid moiety. Accordingly, isomer 8 was tentatively assigned as 3-succinoyl-4,5-dicaffeoylquinic acid.
The isomers 6 and 7 produced almost identical MSn spectra and their MS3 and MS4 spectra were identical to the MS2 and the MS3 spectra of 1,5-dicaffeoylquinic acid (Fig. 3).2 From this it becomes clear that both isomers are 1,5-dicaffeoyl-succinoylquinic acids. There are two possibilities for the succinoyl residue: either at the C3 or at the C4 position of the quinic acid moiety. The isomer 7 produced a dehydrated secondary peak at m/z 335 ([caffeoylquinic acid-H2O–H+]−) in the MS2 spectrum which pointed to the 4-acyl chlorogenic acid.2 On the basis of the above arguments the isomers 6 and 7 were tentatively assigned as 1,5-dicaffeoyl-3-succinoylquinic acid and 1,5-dicaffeoyl-4-succinoylquinic acid, respectively.
 | ||
| Fig. 3 MS4 spectra of 1,5-dicaffeoyl-3-succinoylquinic acid (6) and 1,5-dicaffeoyl-4-succinoylquinic acid (7) (parent ion at m/z 615 in negative ion mode). | ||
Characterisation of putative dicaffeoyl-fumaroylquinic acids (Mr 614)
Two peaks were detected at m/z 613 eluting at 26 min and 37 min. Both isomers 9 and 10 produced an MS2 base peak at m/z 515 ([dicaffeoylquinic acid-H+]−) by the loss of a fumaroyl residue (Fig. 4). Second eluting isomer 10 produced the MS2 secondary peak at m/z 433 by the loss of a caffeoyl residue and a H2O molecule; this was completely absent in the first eluting isomer 9 (Fig. 4). Isomers 9 and 10 have the MS3 and the MS4 spectra identical to the MS2 and the MS3 spectra of 1,3-dicaffeoylquinic acid or 3,5 dicaffeoyl quinic acid and 1,5-dicaffeoylquinic acid 3,2 respectively. Accordingly, isomer 9 is 1,5-dicaffeoyl-fumaroylquinic acid while 10 is 1,3-dicaffeoyl-fumaroylquinic acid or 3, 5-dicaffeoyl-fumaroylquinic. Isomer 10 produced a dehydrated MS2 secondary peak at m/z 433 by the loss of a caffeoyl residue which is characteristic to the vic 3,4- and 4,5-diacyl chlorogenic acids while this peak is absent in first eluting isomer 9. From this it is clear that isomers 9 and 10 have a fumaroyl residue at the C5 and the C4 position, respectively, of the quinic acid moiety. Based on the above arguments, isomers 9 and 10 were tentatively assigned as 1,3-dicaffeoyl-5-fumaroylquinic acid or 3,5 -dicaffeoyl-1-fumaroylquinic and 1,5-dicaffeoyl-4-fumaroylquinic acid, respectively.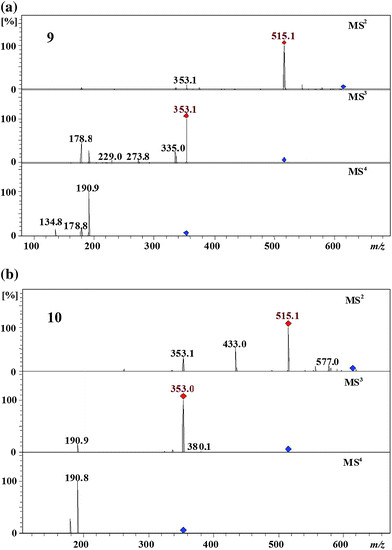 | ||
| Fig. 4 MS4 spectra of 1,3-dicaffeoyl-5-fumaroylquinic acid or 3, 5-dicaffeoyl-1-fumaroylquinic (9) and 1,5-dicaffeoyl-4-fumaroylquinic acid (10) (parent ion at m/z 613 in negative ion mode). | ||
Characterisation of putative dicaffeoyl-maloylquinic acids (Mr 632)
Targeted MSn experiments and selected ion monitoring located three dicaffeoyl-maloylquinic acids (11–13) at retention times between ∼36–42 min. These isomers have UV-spectra typical of chlorogenic acids. The first eluting isomer 11 produced the MS2 base peak at m/z 353 [(caffeoylquinic acid-H+)−] by the loss of a caffeoyl and a maloyl residue and the MS3 base peak at m/z 191 [(quinic acid-H+)−] by the loss of another caffeoyl residue (Fig. 5). The MS3 spectrum of this isomer was identical to the MS3 spectrum of 1,5-dicaffeoylquinic acid 32 which confirmed that the caffeoyl residues are found at the C1 and the C5 of the quinic acid moiety. Absence of the dehydrated secondary peaks in the MS2 spectrum confirmed that there is no vic acylation.2 Based on the above arguments, isomer 11 can be assigned as 1,5-dicaffeoyl-3-maloylquinic acid.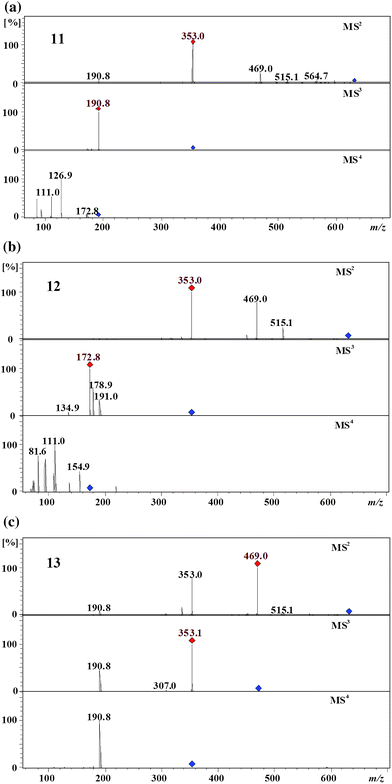 | ||
| Fig. 5 MS4 spectra of 1,5-dicaffeoyl-3-maloylquinic acid (11), 1,4-dicaffeoyl-3-maloylquinic acid (12), and 1,5-dicaffeoyl-4-maloylquinic acid (13) (parent ion at m/z 631 in negative ion mode). | ||
The second eluting isomer 12 produced the MS2 base peak at m/z 353 [(caffeoylquinic acid-H+)−] and secondary peaks at m/z 469 [(caffeoyl-maloylquinic acid-H+)−], 515 [(dicaffeoylquinic acid-H+)−], and 451 [(caffeoyl-maloylquinic acid-H2O–H+)−]. It produced the MS3 base peak at m/z 173 and secondary peaks at m/z 179 and 191 which was identical to the MS3 spectrum of 1,4-dicaffeoylquinic acid 2 (Fig. 5).2 Accordingly, this isomer can be assigned as 1,4-dicaffeoyl-maloylquinic acid. This isomer produced the MS2 secondary dehydrated peak at m/z 451 which suggested the vic diacylation of quinic acid,2 it also produced an intense peak at m/z 469 which suggested that the loss of the caffeoyl residue takes place easier than of the maloyl residue. Based on the above arguments, isomer 12 can be assigned as 1,4-dicaffeoyl-3-maloylquinic acid.
The most hydrophobic isomer 13 produced the MS2 base peak at m/z 469 [(caffeoyl-maloylquinic acid-H+)−] and secondary peaks at m/z 353 [(caffeoylquinic acid-H+)−] and 335 [(caffeoylquinic acid-H2O–H+)−] (Fig. 5). It produced the MS3 base peak at m/z 353 [(caffeoylquinic acid-H+)−] by the loss of a maloyl residue and the secondary peak at m/z 191 by the loss of a caffeoyl and a maloyl residues. Its MS4 spectrum was identical to the MS3 spectrum of 1,5-dicaffeoylquinic acid2 which identified the compound as being 1,5-dicaffeoyl-maloylquinic acid. There are two possibilities for the maloyl residue: it can be at either the C3 or the C4 position of the quinic acid moiety. The MS2 base peak at m/z 469 and the secondary dehydrated peak at m/z 335 suggested 4-acylation.2 Accordingly, this isomer was tentatively assigned as 1,5-dicaffeoyl-4-maloylquinic acid.
Characterisation of putative tricaffeoyl-succinoylquinic acid (Mr 778)
Isomer 14 produced the parent ion at m/z 777 (∼48 min) and was tentatively assigned as tricaffeoyl-succinoylquinic acid. This isomer produced the MS2 base peak at m/z 353 ([3-caffeoylquinic acid-H+]−) by the loss of a succinoyl and two caffeoyl residues with secondary peaks at m/z 453 ([caffeoyl-succinoylquinic acid-H+]−), 497 ([dicaffeoylquinic acid-H2O–H+]−), 515 ([dicaffeoylquinic acid-H+]−), and 597 ([tricaffeoyl-succinoylquinic acid-H2O–H+]−) (Fig. 6). It produced the MS3 base peak at m/z 191 accompanied by m/z 179 and 135 which suggested that two caffeoyl residues are at the C1 and C5 positions, with the succinoyl at the C4 position of the quinic acid moiety (Fig. 6).2 Thus, isomer 14 was tentatively assigned as 1,3,5-tricaffeoyl-4-succinoylquinic acid. It is worth noting that this derivative in burdock as well as the compounds described below are to our knowledge the first tetraacyl chlorogenic acids described in dietary plants. | ||
| Fig. 6 MS4 spectra of 1,3,5-tricaffeoyl-4-succinoylquinic acid (14) (parent ion at m/z 777 in negative ion mode). | ||
Characterisation of putative dicaffeoyl-disuccinoylquinic acid (Mr 716)
Isomer 15 produced the parent ion at m/z 715 (∼50 min) and was tentatively assigned as dicaffeoyl-disuccinoylquinic acid. This isomer produced the MS2 base peak at m/z 353 ([caffeoylquinic acid-H+]−) by the loss of a caffeoyl and two succinoyl residues (Fig. 7). It showed the MS3 spectrum at m/z 191 ([quinic acid- H+]−) by the loss of a caffeoyl residue and this was identical to the MS3 spectrum of 1,5-dicaffeoylquinic acid2 which suggested that caffeoyl residues were present at the C1 and C5 positions of the quinic acid moiety. Accordingly, both succinoyl residues were identified at the C3 and C4 positions of the quinic acid moiety. Based on the above arguments, isomer 15 was tentatively assigned as 1,5-dicaffeoyl-3,4-disuccinoylquinic acid. | ||
| Fig. 7 MS4 spectra of 1,5-dicaffeoyl-3,4-disuccinoylquinic acid (15) (parent ion at m/z 715 in negative ion mode). | ||
Characterisation of putative dicaffeoyl-succinoyl-fumaroylquinic acids (Mr 714)
Two peaks were detected at m/z 713 eluting at 41 min and 44 min. Both isomers 16 and 17 produced an MS2 base peak at m/z 615 ([dicaffeoyl-succinoylquinic acid-H+]−) by the loss of a fumaroyl residue (Fig. 8). Isomers 16 and 17 have the MS3 and the MS4 spectra identical to the MS2 and the MS3 spectra of 1,5-dicaffeoylquinic acid 3 and 3,5-dicaffeoylquinic acid 4,2 respectively (Fig. 8). Accordingly, isomer 16 is 1,5-dicaffeoyl-succinoyl-fumaroylquinic acid and 17 is 3,5-dicaffeoyl-fumaroylquinic acid. The second eluting isomer 17 produced a dehydrated MS2 secondary peak at m/z 435 (6% of the base peak) by the loss of a caffeoyl residue, a fumaroyl residue, and a H2O molecule which is characteristic to the vic 3,4- or 4,5-diacyl chlorogenic acids,2 this peak is absent in the first eluting isomer 16. From this it is clear that isomers 16 and 17 have each a succinoyl residue at the C3 and the C4, respectively. Based on the above arguments, isomers 16 and 17 were tentatively assigned as 1,5-dicaffeoyl-3-fumaroyl-4-succinoylquinic acid and 1-fumaroyl-3,5-dicaffeoyl-4-succinoylquinic acid, respectively.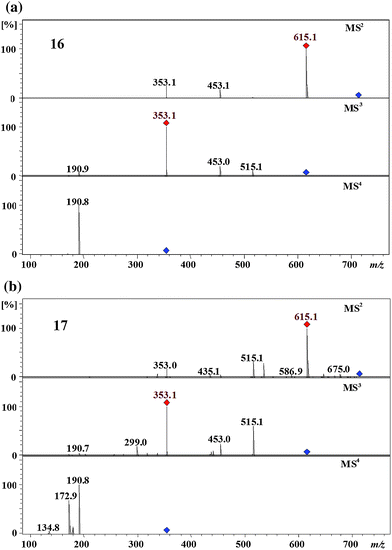 | ||
| Fig. 8 MS4 spectra of 1,5-dicaffeoyl-3-fumaroyl-4-succinoylquinic acid (16) and 1-fumaroyl-3,5-dicaffeoyl-4-succinoylquinic acid (17) (parent ion at m/z 713 in negative ion mode). | ||
Characterisation of putative dicaffeoyl-dimaloylquinic acid (Mr 748)
Isomer 18 produced the parent ion at m/z 747 (∼46 min) and was tentatively assigned as dicaffeoyl-dimaloylquinic acid. This isomer produced the MS2 base peak at m/z 631 ([dicaffeoyl-maloylquinic acid-H+]−) by the loss of a maloyl residue which suggested that one of the two maloyl residues is at the C1 of the quinic acid moiety (Fig. 9). It showed the MS3 spectrum at m/z 353 ([caffeoylquinic acid- H+]−) by the loss of a caffeoyl residue and a maloyl residue which suggested that one of the caffeoyl residues was at either the C5 or the C3. In the MS4 spectrum it produced a base peak at m/z 191 accompanied by m/z 179 which suggested that the caffeoyl residues were found at the C1 and the C3 of the quinic acid moiety.2 Based on the above arguments, isomer 18 was tentatively assigned as 1,3-dicaffeoyl-4,5-dimaloylquinic acid or 3, 5-dicaffeoyl-1, 4,-dimaloylquinic acid. | ||
| Fig. 9 MS4 spectra of 1,3-dicaffeoyl-4,5-maloylquinic acid or 3, 5-dicaffeoyl-1, 4,-dimaloylquinic acid (18) (parent ion at m/z 747 in negative ion mode). | ||
Characterisation of putative dicaffeoyl-maloyl-succinoylquinic acid (Mr 732)
Isomer 19 produced the parent ion at m/z 731 (∼46 min) and was tentatively assigned as dicaffeoyl-maloyl-succinoylquinic acid. This isomer produced the MS2 base peak at m/z 469 ([caffeoyl-maloylquinic acid-H+]−) by the loss of a caffeoyl residue and a succinoyl residue, accompanied by m/z 569 ([caffeoyl-maloyl-succinoylquinic acid-H+]−) and 631 ([dicaffeoyl-maloylquinic acid-H+]−) (Fig. 10). The secondary peak at m/z 569 ([caffeoyl-maloyl-succinoylquinic acid-H+]−) in the MS2 spectrum was more intense than the peak at m/z 631 ([dicaffeoyl-maloylquinic acid-H+]−) which suggested that the loss of caffeoyl residue takes place easier than that of the succinoyl residue (Fig. 10). Additionally, it showed the MS3 spectrum at m/z 353 ([caffeoylquinic acid- H+]−) by the loss of a maloyl residue. In the MS4 spectrum it produced a base peak at m/z 191 which suggested that the caffeoyl residues were at the C1 and the C5 of the quinic acid moiety.2 Based on the above arguments, isomer 19 was tentatively assigned as 1,5-dicaffeoyl-3-succinoyl-4-maloylquinic acid. | ||
| Fig. 10 MS4 spectra of 1,5-dicaffeoyl-3-succinoyl-4-maloylquinic acid (19) (parent ion at m/z 731 in negative ion mode). | ||
Intermediates in biosynthesis of maloylquinic acid derivatives
In this study we observed a full series of compounds (1,5-dicaffeoyl-4-acylquinic acids) which are biochemical intermediates. These biochemical intermediates allowed us to propose a biosynthetic route for the synthesis of maloylquinic acids. This proposed route is based on the structures of three intermediates observed in this study and shown in Fig. 11. We propose to start from 1,5-dicaffeoyl-4-succinoylquinic acid 7; succinic dehydrogenase enzyme removes one dihydrogen molecule from succinoyl residue and produces a fumaroyl residue, intermediate 1,5-dicaffeoyl-4-fumaroylquinic acid 10. In the final step addition of a H2O molecule (hydroxyl functionality) by the enzyme fumarase to the si-face of the fumaroyl double bond, would yield 1,5-dicaffeoyl-4-maloylquinic acid 13. For the further evidences, this proposed biosynthetic pathway needs to be tested in a genetically altered plant with and without the presence of enzymes involved in the pathway. | ||
| Fig. 11 Proposed pathway for the biosynthesis of maloylquinic acid derivatives. | ||
Conclusions
In this study we observed a series of novel chlorogenic acid derivatives in burdock containing next to two or three caffeoyl moieties fumaric, succinic, and malic acid side chains. We have shown the contrary to cinnamoyl and galloyl quinic acids, where the loss of an ester residue follows the order 1 ≈ 5 > 3 > 4, fumaroyl, succinoyl, and maloyl residues are fragmented more facile independent of their regiochemistry. For regiochemistry assignment of these novel CGA derivatives we have therefore provided a significant extension of our previous hierarchical key. Here we employ targeted MSn experiments to define caffeoyl regiochemistry followed by further targeted MSn experiments to define the aliphatic side chain regiochemistry. Additionally the assignment of malic acid regioisomers by tandem MS could be established.Experimental
Chemicals and materials
All the chemicals (analytical grade) were purchased from Sigma-Aldrich (Bremen, Germany). Burdock roots were collected from a Botanical Garden in Bremen (Germany).Methanolic extract
Burdock roots (10 g) were minced with aqueous methanol (90%, 100 mL) at room temperature for 30 min and ultra-sonicated for 5 min. The methanol and the water were removed in vacuo and the residue was stored at −20 °C until required, thawed at room temperature, dissolved in methanol (40 mg in 10 mL), filtered through a membrane filter and then used for LC-MS.Hydrolysis
Burdock root extract (20 mg) was stirred with 1M NaOH solution (5 mL) at room temperature for 1 h. The mixture was neutralized using 1 M HCl and used directly for HPLC-MS analysis in the negative ion mode using the HPLC method with peaks identified corresponding to fumaric, succinic and malic acid.LC-MSn
The LC equipment (Agilent 1100 series, Karlsruhe, Germany) comprised a binary pump, an auto sampler with a 100 μL loop, and a DAD detector with a light-pipe flow cell (recording at 320 and 254 nm and scanning from 200 to 600 nm). This was interfaced with an ion-trap mass spectrometer fitted with an ESI source (Bruker Daltonics HCT Ultra, Bremen, Germany) operating in full scan, Auto MSn mode to obtain fragment ion m/z. As necessary, MS2, MS3, and MS4 fragment-targeted experiments were performed to focus only on compounds producing a parent ion at m/z 353, 367, 515, 613, 615, 631, 677, 715, 731, 747, and 777. Tandem mass spectra were acquired in an Auto-MSn mode (smart fragmentation) using a ramping of the collision energy. Maximum fragmentation amplitude was set to 1 V, starting at 30% and ending at 200%. MS operating conditions (negative mode) had been optimized using 5-caffeoylquinic acid 1 with a capillary temperature of 365 °C, a dry gas flow rate of 10 L min−1, and a nebulizer pressure of 10 psi. High resolution LC-MS was carried out using the same HPLC equipped with a MicrOTOF Focus mass spectrometer (Bruker Daltonics, Bremen, Germany) fitted with an ESI source and internal calibration was achieved with 10 mL of a 0.1 M sodium formate solution injected through a six port valve prior to each chromatographic run. Calibration was carried out using the enhanced quadratic mode.HPLC
Separation was achieved on a 150 × 3 mm i.d. column containing diphenyl 5 μm, with a 5 mm × 3 mm i.d. guard column (Varian, Darmstadt, Germany). Solvent A was water/formic acid (1000![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.005 v/v) and solvent B was methanol. Solvents were delivered at a total flow rate of 500 μL min−1. The gradient profile was from 10% B to 70% B linearly in 60 min followed by 10 min isocratic, and a return to 10% B at 90 min and 10 min isocratic to re-equilibrate.
:0.005 v/v) and solvent B was methanol. Solvents were delivered at a total flow rate of 500 μL min−1. The gradient profile was from 10% B to 70% B linearly in 60 min followed by 10 min isocratic, and a return to 10% B at 90 min and 10 min isocratic to re-equilibrate.
Notes and references
- Biochem. J., 1976, 153, 23–31.
- M. N. Clifford, J. Sci. Food Agric., 1999, 79, 362–372 CrossRef CAS; M. N. Clifford, J. Sci. Food Agric., 2000, 80, 1033–1043 CrossRef CAS; M. N. Clifford, K. L. Johnston, S. Knight and N. Kuhnert, J. Agric. Food Chem., 2003, 51, 2900–2911 CrossRef CAS; M. N. Clifford and S. Knight, Food Chem., 2004, 87, 457–463 CrossRef CAS; M. N. Clifford, S. Knight and N. Kuhnert, J. Agric. Food Chem., 2005, 53, 3821–3832 CrossRef CAS; M. N. Clifford, S. Knight, B. Surucu and N. Kuhnert, J. Agric. Food Chem., 2006, 54, 1957–1969 CrossRef CAS; M. N. Clifford, S. Marks, S. Knight and N. Kuhnert, J. Agric. Food Chem., 2006, 54, 4095–4101 CrossRef CAS; M. N. Clifford, S. Stoupi and N. Kuhnert, J. Agric. Food Chem., 2007, 55, 2797–2807 CrossRef CAS; M. N. Clifford, W. G. Wu, J. Kirkpatrick and N. Kuhnert, J. Agric. Food Chem., 2007, 55, 929–936 CrossRef CAS; M. N. Clifford, W. G. Wu and N. Kuhnert, Food Chem., 2006, 95, 574–578 CrossRef CAS; M. N. Clifford, W. Zheng and N. Kuhnert, Phytochem. Anal., 2006, 17, 384–393 CrossRef CAS.
- R. Jaiswal and N. Kuhnert, Rapid Commun. Mass Spectrom., 2010, 24, 2283–2294 CrossRef CAS; R. Jaiswal, M. A. Patras, P. J. Eravuchira and N. Kuhnert, J. Agric. Food Chem., 2010, 58, 8722–8737 CrossRef CAS; R. Jaiswal, T. Sovdat, F. Vivan and N. Kuhnert, J. Agric. Food Chem., 2010, 58, 5471–5484 CrossRef CAS.
- E. L. Eliel and M. B. Ramirez, Tetrahedron: Asymmetry, 1997, 8, 3551–3554 CrossRef CAS.
- Y. Kono, K. Kobayashi, S. Tagawa, K. Adachi, A. Ueda, Y. Sawa and H. Shibata, Biochim. Biophys. Acta, Gen. Subj., 1997, 1335, 335–342 CrossRef CAS.
- M. R. Luzia, K. C. C. DaPaixao, R. Marcilio, L. C. Trugo, L. M. C. Quinteiro and C. A. B. DeMaria, Int. J. Food Sci. Technol., 1997, 32, 15–19 CrossRef CAS.
- M. D. del Castillo, J. M. Ames and M. H. Gordon, J. Agric. Food Chem., 2002, 50, 3698–3703 CrossRef CAS.
- A. W. Herling, H. J. Burger, D. Schwab, H. Hemmerle, P. Below and G. Schubert, Am. J. Physiol., 1998, 274, G1087–G1093 CAS.
- J. Shearer, A. Farah, T. de Paulis, D. P. Bracy, R. R. Pencek, T. E. Graham and D. H. Wasserman, J. Nutr., 2003, 133, 3529–3532 CAS.
- H. Hemmerle, H. J. Burger, P. Below, G. Schubert, R. Rippel, P. W. Schindler, E. Paulus and A. W. Herling, J. Med. Chem., 1997, 40, 137–145 CrossRef CAS.
- W. J. Arion, W. K. Canfield, F. C. Ramos, P. W. Schindler, H. J. Burger, H. Hemmerle, G. Schubert, P. Below and A. W. Herling, Arch. Biochem. Biophys., 1997, 339, 315–322 CrossRef CAS.
- K. L. Johnston, M. N. Clifford and L. M. Morgan, Am. J. Clin. Nutr., 2003, 78, 728–733 CAS.
- W. E. Robinson, M. Cordeiro, S. AbdelMalek, Q. Jia, S. A. Chow, M. G. Reinecke and W. M. Mitchell, Mol. Pharmacol., 1996, 50, 846–855 CAS; W. E. Robinson, M. G. Reinecke, S. AbdelMalek, Q. Jia and S. A. Chow, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 6326–6331 CrossRef CAS.
- B. McDougall, P. J. King, B. W. Wu, Z. Hostomsky, M. G. Reinecke and W. E. Robinson, Antimicrob. Agents Chemother., 1998, 42, 140–146 CAS.
- A. Trute, J. Gross, E. Mutschler and A. Nahrstedt, Planta Med., 1997, 63, 125–129 CrossRef CAS.
- H. F. Stich, M. P. Rosin and L. Bryson, Mutat. Res., Fundam. Mol. Mech. Mutagen., 1982, 95, 119–128 CrossRef CAS.
- K. Morita, T. Kada and M. Namiki, Mutat. Res., Fundam. Mol. Mech. Mutagen., 1984, 129, 25–31 CrossRef CAS.
- Z. X. Lou, H. X. Wang, J. Li, S. W. Chen, S. Zhu, C. Y. Ma and Z. P. Wang, J. Food Sci., 2010, 75, C413–C419 CAS; Z. X. Lou, H. X. Wang, S. Zhu, M. Zhang, Y. Gao, C. Y. Ma and Z. P. Wang, J. Chromatogr., A, 2010, 1217, 2441–2446 CrossRef CAS.
- Y. Maruta, J. Kawabata and R. Niki, J. Agric. Food Chem., 1995, 43, 2592–2595 CrossRef CAS.
- B. S. Wang, G. C. Yen, L. W. Chang, W. J. Yen and P. D. Duh, Food Chem., 2007, 101, 729–738 CrossRef CAS.
- C. C. Lin, J. M. Lin, J. J. Yang, S. C. Chuang and T. Ujiie, Am. J. Chin. Med., 1996, 24, 127–137 Search PubMed.
- S. C. Lin, T. C. Chung, C. C. Lin, T. H. Ueng, Y. H. Lin, S. Y. Lin and L. Y. Wang, Am. J. Chin. Med., 2000, 28, 163–173 Search PubMed; S. C. Lin, C. H. Lin, C. C. Lin, Y. H. Lin, C. F. Chen, I. C. Chen and L. Y. Wang, J. Biomed. Sci., 2002, 9, 401–409.
- R. Wang, H. Ayano, T. Furumoto, A. Kondo and H. Fukui, Journal of the Japanese Society for Food Science and Technology-Nippon Shokuhin Kagaku Kogaku Kaishi, 2001, 48, 857–862 Search PubMed.
- L. Z. Lin and J. M. Harnly, J. Agric. Food Chem., 2008, 56, 10105–10114 CrossRef CAS.
Footnotes |
| † This paper is dedicated to Prof. Dr Joachim Treusch on the occasion of his 70th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c0fo00125b |
| This journal is © The Royal Society of Chemistry 2011 |