Inhibitory effects of 1,3-bis-(2-substituted-phenyl)-propane-1,3-dione, β-diketone structural analogues of curcumin, on chemical-induced tumor promotion and inflammation in mouse skin
Chuan-Chuan
Lin
*a,
Yue
Liu
b,
Chi-Tang
Ho
c and
Mou-Tuan
Huang
b
aDepartment of Food Science, China University of Science and Technology, 245 Yen-Chiu-Yuan Rd., Sec. 3, Nankang, Taipei, 115, Taiwan. E-mail: cclin@cc.cust.edu.tw; Fax: +886 2 2786 4291; Tel: +886 2 2782 1862
bLaboratory for Cancer Research, School of Pharmacy, Rutgers, The State University of New Jersey, Piscataway, NJ 08854-8020, USA
cDepartment of Food Science, Rutgers, The State University of New Jersey, New Brunswick, NJ 08901-8520, USA
First published on 10th November 2010
Abstract
Dibenzoylmethane (DBM), a β-diketone structural analogue of curcumin, has been reported to exhibit anti-tumorigenic and chemopreventive activities. Due to the structural resemblance of DBM to the anti-inflammatory curcumin and an aspirin-like skeleton of DBM derivatives, we tested the anti-inflammatory effects of DBM and its derivatives, 1,3-bis-(2-substituted-phenyl)-propane-1,3-dione, on 12-O-tetradecanoylphorbol-13-acetate (TPA)-induced tumor promotion as well as TPA- and arachidonic acid-induced mouse ear edema in skin of CD-1 mice. Topical application of 10 μmol DBM together with TPA on the back of mice previously treated with 7,12-dimethylbenz[α]anthracene (DMBA) inhibited TPA-induced skin tumor promotion significantly. In addition, 1,3-bis-(2-acetoxy phenyl)-propane-1,3-dione was a superior anti-inflammatory agent to aspirin (80% of inhibition), on TPA-induced mouse ear edema and reduced the production of prostaglandin E2 (PGE2), comparable to aspirin. Taken together, 1,3-bis-(2-acetoxyphenyl-propane-1,3-dione merits a valuable anti-inflammatory agent substituting aspirin in therapeutic treatment as well prevention of cancer.
1 Introduction
Dibenzoylmethane (DBM), a β-diketone structural analogue of curcumin, has been reported to exhibit anti-tumorigenic and chemopreventive activities.1–7 Several isoprenoid-substituted dibenzoylmethanes were isolated from different licorice species, a sweet-tasting Glycyrrhiza (Leguminosae) root being used as a flavoring and anti-ulcer, anti-inflammatory agent in Eastern and Western countries.8,9 In addition, DBM and its derivatives have been used as sun-screening agents.10 It modulates the Phase I/Phase II metabolic systems, induces apoptosis in various cancer cells, and, as a metal-chelator, exerts beneficial effects for the ischemic diseases.1,3,5,11 Talalay's group indicated that DBM and its structural derivatives are potent inducers of Phase 2 detoxification enzymes.5Dibenzoylmethane has been reported to exhibit chemopreventive activities in several animal models, including mammary, colon and prostate tumorigenesis during the past few years.2,12–16Tumor promotion is a process that favors the clonal outgrowth of single mutated (initiated) cells to premalignant lesions and chronic inflammation is a recognized risk factor for epithelial carcinogenesis. Inflammation, induced either by irritation and wounding or by tumor promoting agents, increases the synthesis of PGs, at least in part, due to upregulation of COX-2.17 Tumor-promoting agent, 12-O-tetradecanoylphorbol-13-acetate (TPA), induce COX-2 gene expression by activating the PKC pathway, including the downstreamAP-1 transcription factor complex.18 AP-1 heterodimers are important for activating COX-2 promoter activity. Therefore, metabolic processes regulated by such pathways and essential for tumor development are potential targets of cancer chemoprevention. An example of chemopreventive agents in this category, curcumin, has been shown to inhibit the PKC pathway and AP-1mediated gene expression.19 In addition, curcumin inhibits phorbol ester-mediated induction of COX-2.20
Aspirin, the most widely used non-steroid anti-inflammatory drug, has been used to reduce pain and inflammation and cardio-vascular disease. The report indicated that long-term regular use of four to six aspirin per week reduced the risk of colorectal cancer by almost 50%.21 The anti-inflammatory action of aspirin has been attributed to the direct acetylation of cyclooxygenase, thereby inhibiting the production of inflammatory prostaglandins22 Recent studies have suggested that aspirin's therapeutic effects come from the inactivation as well as suppression of gene expression of COX-2, inducible isoform by several inflammatory mediators; while the anti-thrombotic and ulcerogenic side effects arise from the inactivation of COX-1, although the statement still remains controversial.23
Due to the structural resemblance of DBM to the anti-inflammatory curcumin and an aspirin-like skeleton of DBM derivatives (Fig. 1), we tested the anti-inflammatory effects of DBM and its 2,2′-substituted derivatives (1,3-bis-(2-substituted-phenyl)-propane-1,3-dione) on TPA-induced tumor promotion as well as TPA- and arachidonic acid (AA)-induced mouse ear edema in skin of CD-1 mice. The production of biomarker PGE2 in mouse ears was measured by ELISA method. These results were compared with those of aspirin to determine whether these modified DBM derivatives could be a superior anti-inflammatory agent to aspirin.
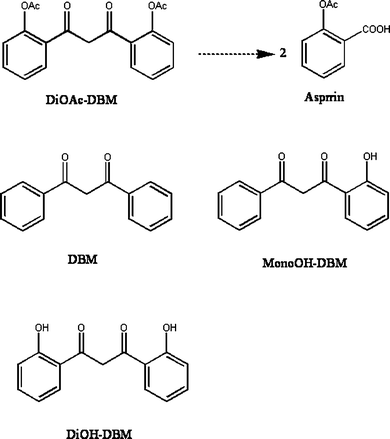 | ||
| Fig. 1 The aspirin-like structures of mono, di-OH and mono, di-OAc derivatives of DBM. | ||
2 Materials and methods
2.1 Chemicals
The reagents for synthesis, potassium t-butoxide, sodium hydride, pyridine, acetic anhydride, 2- and 3-methoxybenzoic acid, acetophenone, 2′-hydroxyacetophenone, methyl salicylate were purchased from Sigma-Aldrich (St. Louis, MO). 12-O-Tetradecanoylphorbol-13-acetate, sodium phosphate, methyl formate, dibenzoylmethane, aspirin were purchase from Sigma Chemical Co. (St. Louis, MO). Arachidonic acid was purchased from Cayman Chemical Company (Ann Arbor, MI). 1-(2-Hydroxyphenyl)-3-phenyl-1,3-propanedione was purchased from Aldrich. All solvents (HPLC grade) were purchased from Fisher Scientific Co. (Springfield, NJ).2.2 TPA-induced tumor promotion in skin of CD-1 mice previously initiated with DMBA
The procedure for tumor formation in mice was according to the reference by Huang et al.24 Female CD-1 mice (30 per group; 40 for the positive control group) were initiated with 200 nmol of 7,12-dimethylbenz[α]anthracene (DMBA) in 200 μl acetone. One week later, the mice were promoted with TPA (5 nmol) or TPA (5 nmol) with DBM (3 μmol or 10 μmol) in 200 μl acetone twice weekly for 20 weeks. Skin tumors with diameter greater than 1 mm were counted and recorded once every 2 weeks. All experiments were conducted in the Animal facility at Rutgers University (Busch Campus, Piscataway, NJ, USA) according to the NIH guideline.2.3 Synthesis of 1,3-bis-(2-hydroxylphenyl)-propane-1,3-dione (2,2′-diOH-DBM)
The procedure of synthesis of 2,2′-diOH-DBM was according to the previous published literature.25 A solution of 2-hydroxyacetophenone (13.6 g, 0.1 mol) and sodium hydride (19.2 g, 0.8 mol) in anhydrous xylene (200 ml) was refluxed for 10 min. Methyl ester of salicylic acid (15.2 g, 0.1 mol) was then added dropwise to the mixture and the solution was stirred under reflux for 7 h. The precipitate formed in the mixture was filtered, washed with ethyl ether and refiltered after 300 ml of ice water/acetic acid solution (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was added. The crude product was chromatographed on silica gel column eluted with n-hexane/ethyl acetate (5:1) to afford a yellowish powder: mp. 118–120 °C; MS(DIP-DCI-MS), m/z 257 (M + 1); keto-enolic forms with the ratio of 1:4 were observed: 1H-NMR (500 MHz, CD3COCD3): keto form: 11.85 (2H, s, OH), 7.98 (2H, d, J = 7.8 Hz, aromatic protons), 7.56–7.59 (2H, m, aromatic protons), 7.01–6.97 (4H, m, aromatic protons), 4.96 (2H, s, H-2); enolic form: 11.78 (2H, s, OH), 7.87 (2H, d, J = 7.0 Hz, aromatic protons), 7.23–7.27 (2H, m, aromatic protons), 6.74–6.77 (5H, m, aromatic protons, H-2).
:1) was added. The crude product was chromatographed on silica gel column eluted with n-hexane/ethyl acetate (5:1) to afford a yellowish powder: mp. 118–120 °C; MS(DIP-DCI-MS), m/z 257 (M + 1); keto-enolic forms with the ratio of 1:4 were observed: 1H-NMR (500 MHz, CD3COCD3): keto form: 11.85 (2H, s, OH), 7.98 (2H, d, J = 7.8 Hz, aromatic protons), 7.56–7.59 (2H, m, aromatic protons), 7.01–6.97 (4H, m, aromatic protons), 4.96 (2H, s, H-2); enolic form: 11.78 (2H, s, OH), 7.87 (2H, d, J = 7.0 Hz, aromatic protons), 7.23–7.27 (2H, m, aromatic protons), 6.74–6.77 (5H, m, aromatic protons, H-2).
2.4 Synthesis of 1,3-bis-(2-acetoxyphenyl)-propane-1,3-dione (2,2′-diOAC-DBM)
A solution of 2,2′-diOH-DBM (50mg, 0.195 mmole) in 25 ml of dichloromethane was treated with 4 eq. of pyridine (about 200 μl) and 6 eq. of acetic anhydride (about 200 μl) at room temperature. The solution turned from cloudy to clear overnight and was monitored by TLC with n-hexane and ethyl acetate (3:2) as solvent. After 20 h, the reaction was quenched by adding 1 N of NaHCO3(aq) and adjusted to neutrality. The mixture was extracted with EtOAc, dried over MgSO4 and concentrated to give a yellowish oily liquid. The crude residue was chromatographed on silica gel with petroleum ether : ethyl acetate (60% : 40%) to separate the products. The products were collected and identified by comparing with standard compounds that were obtained from reverse-phase HPLC separation with acetonitrile as eluent. 1,3-bis-(2-acetoxyphenyl)-propane-1,3-dione (2,2′-diOAC-DBM): yield 15%; mp 138–140 °C; MS (DIP-DCI-MS), m/z 341 (M + 1); 1H-NMR (500 MHz, CD3OD): 2.08 (3H, s, OCOCH3), 2.41 (3H, s, OCOCH3), 7.10 (1H, d, J = 8.1 Hz, aromatic protons), 7.30 (1H, t, J = 7.6 Hz, aromatic protons), 7.40 (1H, t, J = 7.8 Hz, aromatic protons), 7.44 (1H, d, J = 8.5 Hz, aromatic protons), 7.46 (1H, s, H-2), 7.55 (1H, t, J = 7.0 Hz, aromatic protons), 7.68 (1H, t, J = 7.3 Hz, aromatic protons), 7.78 (1H, d, J = 7.8 Hz, aromatic protons), 8.15 (1H, d, J = 7.9 Hz, aromatic protons). 1-(2-Acetoxyphenyl)-propane-1,3-dione (2-OAc-DBM) was obtained followed the same procedure using 1-(2-hydroxyphenyl)-3-phenyl-1,3-propanedione as starting material: yield 48%; mp. 86–88 °C; MS (DIP-DCI-MS), m/z 283 (M + 1); 1H-NMR (500 MHz, CD3OD): 2.30 (3H, s, OCOCH3), 6.68 (1H, s, H-2), 7.14 (1H, d, J = 8.1 Hz, aromatic protons), 7.34 (1H, t, J = 7.6 Hz, aromatic protons), 7.45–7.55 (4H, m, aromatic protons), 7.79 (1H, d, J = 7.8 Hz, aromatic protons), 7.92–7.94 (2H, m, aromatic protons).
2.5 TPA- and arachidonic acid-induced ear edema
Female CD-1 mice (4–5 mice/group) were treated topically on each ear with a inflammatory agent, TPA (1.25 nmol) or arachidonic acid (0.75 mg), or test compound (1 μmol) together with TPA or AA in 20 μl acetone. The mice were sacrificed 5 h after TPA or 1 h after arachidonic acid, and ear punches (6 or 8 mm in diameter) were weighed. The increased weight of ear punch is used as an indicator of ear edema (inflammation).2.6 Determination of PGE2 in TPA- and arachidonic acid-induced ear edema by enzyme-linked immunosorbent assay (ELISA)
The method for extraction and determination of PGE2 concentration from tissue sample is described in Prostaglandin E2 kit (Oxford Biomedical Research, Inc., MI). Briefly, Female CD-1 mice (4–5 mice/group), from treatment with TPA or AA as described previously, were killed after 20 min for AA-treated group or 1 h for TPA-treated group. Ear punches were frozen to −80 °C once removed (6 or 8 mm diameter) and used to determine the PGE2 level. Ear punches were then weighed and suspended in 15% methanol in 0.1 M sodium phosphate buffer, pH 7.5 (about 100 mg in 1 ml methanol buffer). The tissues were homogenized and centrifuged for 5 min at 10000 g. The supernatant was loaded on a preconditioning C18 Sep-Pak column (Waters corporation). After the washing procedure, PGE2 was eluted by 2 ml of methyl formate.
An enzyme-linked immunosorbent assay (ELISA) was used to measure the concentration of PGE2. The concentrated residue was dissolved in extraction buffer provided by commercial kit. The samples and enzyme conjugate (PGE2-horseradish peroxidase) were added to microplate wells precoated with the corresponding antibody and the mixture was incubated at room temperature for one hour. During this time, competition for the limited binding sites was take place. After washing 3 times with wash buffer to remove unbound material, the quantity of bound enzyme conjugate was determined by adding substrate (3,3′,5,5′-tetramethylbenzidine and hydrogen peroxide). The color was developed after 30 min of incubation and the absorbance reading of each well at 650 nm was measured by a microplate reader. The calculation of hormone concentration in samples was performed according to the manual by using a standard curve.
3 Results and discussion
3.1 Inhibitory effect of DBM on TPA-induced tumor promotion in skin of CD-1 mice previously initiated with DMBA
Dibenzoylmethane (DBM), a β-diketone structural analogue of curcumin, has been reported to exhibit anti-tumorigenic and chemopreventive activities.1–7 Both DBM and its derivatives have been used as sun-screening agents for UVA/UVB photoprotection.10 In our study, the antitumor activity of DBM on TPA-induced tumor promotion was performed in a two-stage skin tumorigenesis model (Table 1). Topical application of 5 nmol TPA twice weekly for 20 weeks to the backs of mice previously treated with DMBA resulted in the formation of 19.5 skin tumors per mouse, and 93% of the mice had tumors. In a parallel group of mice, in which the animals were treated with 3 or 10 μmol DBM together with TPA, the number of tumors per mouse was decreased by 65 or 98% respectively. The percentage of tumor-bearing mice was decreased by 29 or 50% after 20 weeks of treatment. In addition, such tumor promotion inhibition appeared to be dose dependent, as observed using different dosage of DBM application. The inhibitory ability was comparable with curcumin.27 As for TPA-induced short-term biomarker changes, curcumin inhibits expression and activity of ornithine decarboxylase (ODC), epidermal DNA synthesis and skin inflammation, formation of DNA base damages (8-hydroxydeoxyguanosine) in epidermis, expression of c-Fos and c-Jun proteins and arachidonic acid metabolism (see review in ref. 28). Several studies have shown that dietary curcumin inhibits carcinogenesis and tumorigenesis in several animal models, including chemically-induced skin, forestomach, duodenal, and colon tumorigenesis.26–32| Treatment | ||||||
|---|---|---|---|---|---|---|
| Week Of TPA promotion | TPA | TPA + DBM (3 μmol) | TPA + DBM (10 μmol) | |||
| Tumors per mouse | % Tumor incidence | Tumors per mouse | % Tumor incidence | Tumors per mouse | % Tumor incidence | |
| a Female CD-1 mice (30 per group; 40 for the positive control group) were initiated with 200 nmol of DMBA. One week later, the mice were promoted with TPA (5 nmol) or TPA (5 nmol) with DBM (3μmol or 10 μmol) in 200 μl acetone twice weekly for 20 weeks. Skin tumors with diameter greater than 1 mm were counted and recorded once every 2 weeks. Data in parentheses are percent of inhibition. | ||||||
| 8 | 5.1 ± 1.1 | 83 | 1.3 ± 0.8 (75%) | 24 (71%) | 0.0 ± 0.0 (100%) | 0 (100%) |
| 10 | 10.6 ± 1.9 | 85 | 3.3 ± 1.4 (69%) | 59 (31%) | 0.2 ± 0.1 (98%) | 7 (92%) |
| 12 | 15.5 ± 2.0 | 93 | 4.6 ± 1.5 (70%) | 52 (44%) | 0.5 ± 0.3 (97%) | 24 (74%) |
| 14 | 20.1 ± 2.3 | 93 | 6.2 ± 1.8 (69%) | 62 (33%) | 1.3 ± 0.5 (94%) | 41 (56%) |
| 16 | 19.5 ± 2.3 | 93 | 6.8 ± 1.9 (65%) | 66 (29%) | 1.3 ± 0.4 (93%) | 47 (50%) |
| 18 | 19.2 ± 2.0 | 93 | 8.9 ± 2.2 (54%) | 75 (19%) | 1.3 ± 0.9 (95%) | 57 (39%) |
| 20 | 19.5 ± 2.3 | 93 | 6.8 ± 1.9 (65%) | 68 (29%) | 1.3 ± 0.3 (98%) | 47 (50%) |
3.2 Effect of DBM and its derivatives on TPA- and arachidonic acid-induced mouse ear edema
Due to the exceptional antitumor activity of DBM on TPA-induced tumor promotion, several DBM derivative, 2,2′-substituted -DBM were prepared and the test of their anti-inflammatory activity was performed. The original idea came from the following reasons: (1) DBM and its derivatives belong to the C6-C3-C6 structural class (flavonoid, chalcone are in this class) that are normally found in a variety of plant extracts for the treatment of inflammation; (2) 2,2′-diacetoxy-DBM can be considered as a molecule constructed by two symmetrical aspirin molecules condensed with a methylene (CH2) group; (3) finding a better aspirin with selective inactivation of COX-2 (responsible for inducible inflammatory responses) over COX-1 (responsible for cytoprotective effect in gastric mucosa).The anti-inflammatory effects of DBM and its 2,2′-substituted derivatives were tested by determining the extents of TPA- and arachidonic acid-induced CD-1 mouse ear edema. Topically application of 1.25 nmol of TPA to the ear of a mouse caused the increase of ear punch weigh (8 mm in diameter) from 14.0 mg (control) to 28.0 mg after 5 h. Application of 1.25 nmol TPA together with DBM, mono-OH, mono-OAc, di-OH, di-OAc-DBM inhibited the TPA-induced mouse ear edema by 56, 49, 47, 59, 98%, respectively (Table 2). Due to the similarity in structural feature and anti-inflammatory activity between aspirin and DBM derivatives, the inhibitory effect of aspirin on TPA-induced edema were compared with those of DBM derivatives. The results indicated that di-OAc-DBM (aspirin-like molecule) was a superior anti-inflammatory agent to aspirin (80% of inhibition), while the other DBM derivatives showed weaker activities than aspirin. In the other study, 0.75 mg of AA and 1 μmol of test compounds were topically applied to the mouse ears. DBM, mono-OH, mono-OAc, di-OH, di-OAc-DBM and aspirin inhibited the AA-induced mouse ear edema by 7, 0, 6, 62, 50, and 24%, respectively (Table 3). The results showed a less effective anti-inflammatory activity of DBM derivatives and aspirin on AA-induced ear edema, but the order of inhibitory activity was similar to that on TPA-induced ear edema.
| Treatment | Weight of ear punch/mg | % of inhibition |
|---|---|---|
| a Female CD-1 mice (25 days old, 5 mice per group) were treated topically 20 μl acetone, TPA (1.25 nmol) or test compound (1 μmol) together with TPA in 20 μl acetone on each ears. Five hours later, the mice were sacrificed and ear punches (8 mm in diameter) were weighed. Data are expressed as the mean ± S.E. from 4–5 mice. b Statistically difference from TPA-treated group (P < 0.05) as determined by the Student's t-test. | ||
| Acetone | 14.0 ± 0.6 | — |
| TPA | 28.0 ± 1.5 | — |
| TPA + DBM | 20.1 ± 0.6 | 56b |
| TPA + 2-OH-DBM | 21.2 ± 1.3 | 49b |
| TPA + 2-OAc-DBM | 21.5 ± 1.7 | 47b |
| TPA + 2,2′-diOH-DBM | 19.7 ± 1.0 | 59b |
| TPA + 2,2′-diOAc-DBM | 14.3 ± 0.5 | 98b |
| Aspirin | 16.6 ± 1.3 | 80b |
| Treatment | Weight of ear punch/mg | % of inhibition |
|---|---|---|
| a Female CD-1 mice (26 days old) were treated topically 20 μl acetone, arachidonic acid AA (0.75 mg) or test compound (1 μmol) together with AA in 20 μl acetone on each ears. One hours later, the mice were sacrificed and ear punches (6 mm in diameter) were weighed. Data are expressed as the mean ± S.E. from 4–5 mice. b Statistically difference from AA-treated group (P < 0.05) as determined by the Student's t-test. | ||
| Acetone | 7.1 ± 0.3 | — |
| AA | 13.5 ± 0.4 | — |
| AA + DBM | 13.1 ± 0.6 | 7 |
| AA + 2-OH-DBM | 14.3 ± 0.3 | 0 |
| AA + 2-Oac-DBM | 13.1 ± 0.9 | 6 |
| AA + 2,2′-diOH-DBM | 9.5 ± 0.4 | 62b |
| AA + 2,2′-diOAc-DBM | 10.3 ± 0.6 | 50b |
| Aspirin | 11.9 ± 0.5 | 24b |
3.3 Effect of DBM and its derivatives on the production of PGE2 on TPA- and arachidonic acid-induced mouse ear edema
A enzyme-immunoassay (ELISA) was used to measure the effect of DBM derivatives and aspirin on the metabolism of arachidonic acid to PGE2 by cyclooxygenase system in vivo. Topically application of 1.25 nmol of TPA to the ear of a mouse caused the increase of PGE2 in ear punch (8 mm in diameter) from 9.76 (control) to 12.77 ng per punch 3 h later. Application of 1.25 nmol TPA together with DBM, di-OH, di-OAc-DBM and aspirin reduced the production of PGE2 to 8.10, 8.71, 5.24, 3.90 ng per punch, respectively (Fig. 2), which were all below the basal level. Besides, it was noticeable that even though aspirin exhibited the strongest inhibitory effect on the PGE2 production among the test compounds, the anti-inflammatory effect measured by ear punch weight was not correlated with the decrease of PGE2 concentration. In the other experiment, application of 0.75 mg of AA and 1 μmol of test compounds to the mouse ears increased the concentration of PGE2 to 47.8 ng per punch (6 mm in diameter) in AA-induced mouse ear. DBM, mono-OH, mono-OAc, di-OH, di-OAc-DBM and aspirin inhibited the production of PGE2 on the metabolism of AA to 39.3, 46.1, 41.5, 24.7, 29.7 and 5.2 ng per punch, respectively (Fig. 3). A weaker inhibitory activity on AA-induced PGE2 production was observed in DBM series, but not aspirin, which showed markedly inhibition of PGE2 concentration.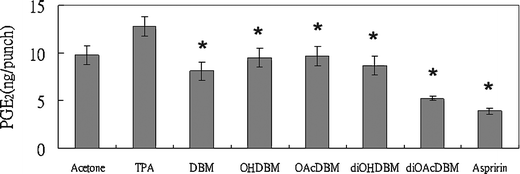 | ||
| Fig. 2 Effect of DBM and its derivatives on PGE2 production in TPA-induced ear edema in CD-1 mice. Female CD-1 mice (30 days old) were treated topically on each ear with 1.25 nmol TPA in 20 ml acetone or test compound (1 μmol) together with TPA in 20 ml acetone. Three hours later, the mice were killed and the PGE2 amount of ear punches (8 mm in diameter) were determined by enzymeimmunoassay (ELISA). Each value represents an average of 4 ear punch determinants. | ||
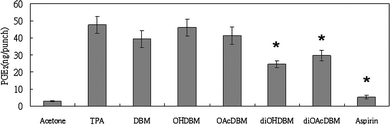 | ||
| Fig. 3 Effect of DBM and its derivatives on PGE2 production in arachidonic acid-induced ear edema in CD-1 mice. Female CD-1 mice (32 days old) were treated topically on each ear with 0.75 mg arachidonic acid in 20 ml acetone or test compound (1 μmol) together with AA in 20 ml acetone. The mice were killed after 20 min and the PGE2 amount of ear punches (6 mm in diameter) were determined by enzyme immunoassay (ELISA). Each value represents an average of 5 ear punch determinants. | ||
The mechanisms by which an increase in COX-2 activity promotes tumor formation could be explained with a study done by Tsujii and DuBois in 1995.33 They found that elevated COX-2 levels in the early stages of epithelial transformation could possibly prevent apoptosis in epithelial cellsin vivo. Therefore, the effects of aspirin and other NSAIDs would be to maintain the normal apoptotic potential of colonic epithelial cells in the initial stages of the transformation process. The other possibility is that the products of COX-2, prostaglandins, would cause tumor promotion through paracrine or autocrine signaling through prostaglandin receptors, members of the G protein-coupled family of receptor.34
In this study, the anti-inflammatory activities of DBM and its derivatives were tested. The most important finding is that 2,2′-diOAc-DBM is a superior anti-inflammatory agent to aspirin, proven by the studies of inhibitory effects on both TPA and AA-induced mouse ear edema. However, aspirin exhibited a dominant inhibitory potency against the production of PGE2 on both TPA- and AA-induced ear edema. The discrepancy between the inhibition of prostaglandin synthesis and inhibition of inflammation implied additional mechanisms, beside COX-2 inhibition, may be involved and play important roles in the exceptional anti-inflammatory action of 2,2′-diOAc-DBM. Furthermore, by comparing the results from TPA- and AA-induced cases, weaker inhibitory activities on AA-induced ear edema and PGE2 production were observed in DBM series. It can be concluded that the less blocking action might be resulted from high dosage of applied AA or the specific inhibitory effect of DBM series on TPA-induced inflammatory-responsible enzyme (COX-2) that is not normally found in AA-induced condition.
The questions concerning the structure–activity relationship in DBM series, why mono-substituted-DBMs were less active than di-substituted ones, why diOH-DBM (salicylate-like molecule) exerted an inconsistent inhibitory effect toward TPA- and AA-induced ear edema, still remain unclear and further studies are underway.
4 Conclusion
In summary, 2,2′-diOAc-DBM possesses a structure feature for dual actions: it is an aspirin-like molecule for acetylation inhibition of cyclooxygenase and its skeleton is similar to the structure of common nonsteroidal anti-inflammatory drugs. In our in vivo studies, it has been shown to exhibit extraordinary anti-inflammatory activity, especially on TPA-induced mouse ear edema, suggesting a potential selective inhibitor of COX-2. Taken together, 2,2′-diOAc-DBM merits a valuable anti-inflammatory agent substituting aspirin in therapeutic treatment as well as prevention of cancer.Acknowledgements
We thank Dr L.Y. Hsu at the Department of Biological Science, China University of Science and Technology, for assistance with the spectra analysis.References
- K. M. Jackson, M. DeLeon and C. R. Verret, et al. , Cancer Lett., 2002, 178, 161–165 CrossRef CAS.
- C. C. Lin, Y. P. Lu and Y. R. Lou, et al. , Cancer Lett., 2001, 168, 125–132 CrossRef CAS.
- C. J. MacDonald, H. P. Ciolino and G. C. Yeh, Cancer Res., 2001, 61, 3919–3924 CAS.
- K. Singletary and C. MacDonald, Cancer Lett., 2000, 155, 47–54 CrossRef CAS.
- A. T. Dinkova-Kostova and P. Talalay, Carcinogenesis, 1999, 20, 911–914 CrossRef CAS.
- M. T. Huang, Y. R. Lou and J. G. Xie, et al. , Carcinogenesis, 1998, 19, 1697–1700 CrossRef CAS.
- K. Singletary, C. MacDonald and M. Iovinelli, et al. , Carcinogenesis, 1998, 19, 1039–1043 CrossRef CAS.
- S. Demizu, K. Kajiyama and Y. Hiraga, et al. , Chem. Pharm. Bull., 1992, 40, 392–395 CAS.
- S. Shibata, in Food phytochemicals for cancer prevention II, ed. C. T. Ho, T. Osawa and M. T. Huang, et al., American Chemical Society, Washington, DC, 1992, pp. 308–321 Search PubMed.
- M. A. Nogueira, E. G. Magalhaes and A. F. Magalhaes, et al. , Farmaco, 2003, 58, 1163–1169 CrossRef CAS.
- N. J. Mabjeesh, M. T. Willard and W. B. Harris, et al. , Biochem. Biophys. Res. Commun., 2003, 303, 279–286 CrossRef CAS.
- C. C. Lin, C. T. Ho and M. T. Huang, Proc. Natl. Sci. Counc. Repub. China B., 2001, 25, 158–165 Search PubMed.
- C. C. Lin, Y. L. Tsai and M. T. Huang, et al. , Carcinogenesis, 2005, 27, 131–136 CrossRef.
- K. L. Cheung, T. O. Khor and M. T. Huang, et al. , Carcinogenesis, 2009, 31, 880–885.
- T. O. Khor, S. Yu and A. Barve, et al. , Cancer Res., 2009, 69, 7096–7102 CrossRef.
- G. Shen, T. O. Khor and R. Hu, et al. , Cancer Res., 2007, 67, 9937–9944 CrossRef CAS.
- D. A. Kujubu, B. S. Fletcher and B. C. Varnum, et al. , J. Biol. Chem., 1991, 266, 12866–12872 CAS.
- M. Karin, J. Biol. Chem., 1995, 270, 16483–16486 CAS.
- T. S. Huang, S. C. Lee and J. K. Lin, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 5292–5296 CrossRef CAS.
- D. J. Kelly, P. G. Sacks and J. T. Ramonetti, et al. , Proc. Am. Assoc. Cancer Res., 1996, 37, 909.
- E. Biovannucci, K. M. Egan and D. J. Hunter, et al. , N. Engl. J. Med., 1995, 333, 610–614.
- J. R. Vane, Nature, 1971, 231, 232–235 CAS.
- D. DeWitt and W. L. Smith, Cell, 1995, 83, 345–348 CrossRef CAS.
- M. T. Huang, C. T. Ho and T. Ferraro, et al. , Carcinogenesis, 1992, 13, 947–954 CrossRef CAS.
- F. Eiden and H. D. Schweiger, Synthesis, 1974, 511–513 CrossRef CAS.
- M. T. Huang, R. C. Smart, C. Q. Wong and A. H. Conney, et al. , Cancer Res., 1988, 48, 5941–5946 CAS.
- T. Huang, Z. Y. Wang and C. A. Georgiadis, et al. , Carcinogenesis, 1992, 13, 2183–2186 CrossRef.
- M. T. Huang, F. M. Robertson and T. Lysz, et al., in Phenolic Compounds in Food and Their Effects on Food and Health II, ed. M. T. Huang, C. T. Ho, C. L. Lee, American Chemical Society, Washington, DC, 1992, pp. 338–349 Search PubMed.
- M. T. Huang, Y. R. Lou and W. Ma, et al. , Cancer Res., 1994, 54, 5841–5847 CAS.
- M. T. Huang, W. Ma and Y. P. Lu, et al. , Carcinogenesis, 1995, 16, 2493–2497 CrossRef CAS.
- M. T. Huang, H. L. Newmark and K. Frenkel, J. Cell. Biochem., 1997, 67, 26–34 CrossRef.
- M. T. Huang, T. Lysz and T. Ferraro, et al. , Cancer Res., 1991, 51, 813–819 CAS.
- M. Tsujii and R. N. DuBois, Cell, 1995, 83, 493–501 CrossRef CAS.
- S. M. Prescott and R. L. White, Cell, 1996, 87, 783–786 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |