Performance evaluation of phycocyanin probes for the monitoring of cyanobacteria
Christian
Bastien
*a,
Richard
Cardin
a,
Éloïse
Veilleux
a,
Christian
Deblois
a,
Annabelle
Warren
b and
Isabelle
Laurion
b
aCentre d'expertise en analyse environnementale, MDDEP, 2700 rue Einstein, Québec (Qc), G1P 3W8, Canada. E-mail: christian.bastien@mddep.gouv.qc.ca; Fax: +1 418 643-9023; Tel: +1 418 643-8225 ext. 227
bInstitut National de la Recherche Scientifique—Eau Terre Environnement, 490 de la Couronne, Québec (Qc), G1K 9A9, Canada
First published on 26th October 2010
Abstract
The performance of two field probes (YSI 6600 and TriOS), used for the measurement of in vivophycocyanin fluorescence, was compared and validated in the laboratory in 2008 and 2009 with cultures of Microcystis aeruginosa and field samples. The background noise of the two probes was low and the detection limits were estimated at 1500 cells mL−1 for the YSI and 0.69 µg PC L−1 for the TriOS. The linearity and repeatability of both probes have been excellent. Strong relationships were observed between the in vivo fluorescence and the total cyanobacterial biovolume (R2 = 0.82 YSI; 0.83 TriOS) or the abundance (R2 = 0.71 YSI; 0.75 TriOS) of cyanobacteria. However, the difference between cell densities determined by microscopy and measured by the YSI can be very large and has been associated to the variability of cell volume among cyanobacteria. This last observation makes the YSI a qualitative tool if a post-calibration is not done. The analysis of filtrated samples showed that dissolved phycocyanin (extracellular) may represent a significant fluorescence signal. No relationship could be established between the abundance, the total cyanobacterial biovolume or the in vivo fluorescence of phycocyanin and the concentrations of cyanotoxins (R2 ≤ 0.22).
Environmental impactField probes for the measurement of in vivo fluorescence of phycocyanin have been proposed as a complementary tool for monitoring cyanobacteria with the more classical laboratory analysis. Our paper discusses in detail the reliability of those probes and the limit to the interpretation of results. The impact of cell volume on results expressed in cells mL−1 is especially considered. In our knowledge it is the first report that discriminates the contribution of dissolved (extracellular) and intracellular phycocyanin to the fluorescence signal. We have also established a relationship between potentially harmful cyanobacteria and actual detection of cyanotoxins. We think that the paper brings original and useful information for those, like public organizations, who are questioning the practical application of these probes. |
Introduction
The proliferation of cyanobacteria in lakes of Québec (Canada) is of growing concern given the large number of blooms and degradation of many lakes reported in recent years, and the ability of cyanobacteria to produce toxins potentially at risk for human health.1 In order to protect public health, to improve knowledge related to cyanobacteria issues and to improve the water quality, the Government of Québec has established the Management Plan 2007–2017.2 From 2007 to 2009, an average of 148 lakes have been affected by blooms in Québec.3 In order to support this management plan, identification and enumeration of cyanobacteria are performed and analysed for cyanotoxins on a regular basis. These tests are essential to provide reliable data for assessing the state of lakes and take decisions on the basis of management thresholds expressed as cyanotoxin concentrations and abundance of cyanobacteria. However, these relatively time-consuming methods performed on grab samples do not allow monitoring of cyanobacteria with sufficient spatio-temporal resolution. In this context, field probes measuring the in vivo fluorescence of phycocyanin may represent an interesting approach for monitoring cyanobacteria in bringing a rapid tool to locate and follow the bloom forming cyanobacteria and for the selection of samples to be analyzed in the laboratory.Phycocyanin (PC) is a blue pigment which belongs to a group of light harvesting proteins named phycobiliproteins (PBPs). The major biological role of PBPs is photosynthetic light harvesting. PBPs are water-soluble and strongly fluorescent.4 PC is found as a major pigment (>10% of total pigments) in cyanophyceae and cryptophyceae, and as a trace pigment (<1% of total pigments) in rhodophyceae.5,6 It absorbs red and orange light at wavelengths between 610 and 630 nm (absorption peak at 620 nm) and emits fluorescence in the band of wavelengths from 600 to 700 nm (emission peak at 647 nm). In freshwaters, the PC is mainly associated with cyanobacteria and can be used as an indicator of their biomass.7 Previous research has demonstrated a very good relationship between the PC measured with TriOS probe and the cell density determined by microscopy on nearly 800 samples (R2 = 0.73).8 Field probes measuring PC fluorescence can then potentially be used to perform spatio-temporal monitoring of cyanobacteria blooms and to optimize sampling for laboratory analyses. Vertical profiles of PC could also be done to evaluate the proximity of cyanobacteria to drinking water intakes. In this situation, the use of such probes could provide early warning signals.9–12
The objectives of this study are to evaluate the reliability (background noise, detection limit, quantification limit, linearity, repeatability and accuracy) of two field probes, the YSI 6600 and the TriOS microFlu-blue, in controlled conditions in the laboratory, and to determine their potential for a use in the context of monitoring blooms of cyanobacteria.
Material and methods
Measurement of the in vivophycocyanin fluorescence
The YSI 6600 probe, equipped with the optical sensor BGA-PC 6131, uses an orange light emitting diode for the excitation (peak at 610 nm) and a photodiode of high sensitivity for the detection of fluorescence emitted between 600 and 700 nm. The probe is designed to transform the fluorescence of the PC into an equivalent cell density of Microcystis aeruginosa. If the probe is used with the manufacturer's default calibration, its putative measuring range is between 0 and 280![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells mL−1. Cyanobacteria density here expresses the concentration of cells of a known cell volume, equivalent to dimensions of the species used for calibration (for M. aeruginosa: cell diameter ≈ 3.5 µm and cell volume ≈ 38 µm3). Three types of calibration can be used with that probe: the manufacturer's default calibration (which is done with a culture of M. aeruginosa), a calibration with rhodamine to estimate the temporal drift in the calibration and a two-point home calibration performed with a culture of cyanobacteria (e.g., 0 and 5000 cells mL−1). The two-point calibration with cyanobacteria is considered the optimal way to calibrate the probe.13 In 2008 and 2009, tests were made with manufacturer's calibration and two-point calibration with a culture of M. aeruginosa for the determination of background noise and linearity (see section below). For all others measurements, the YSI probe had a two-point calibration with a culture of M. aeruginosa at a cell density of 20000 cells mL−1.
000 cells mL−1. Cyanobacteria density here expresses the concentration of cells of a known cell volume, equivalent to dimensions of the species used for calibration (for M. aeruginosa: cell diameter ≈ 3.5 µm and cell volume ≈ 38 µm3). Three types of calibration can be used with that probe: the manufacturer's default calibration (which is done with a culture of M. aeruginosa), a calibration with rhodamine to estimate the temporal drift in the calibration and a two-point home calibration performed with a culture of cyanobacteria (e.g., 0 and 5000 cells mL−1). The two-point calibration with cyanobacteria is considered the optimal way to calibrate the probe.13 In 2008 and 2009, tests were made with manufacturer's calibration and two-point calibration with a culture of M. aeruginosa for the determination of background noise and linearity (see section below). For all others measurements, the YSI probe had a two-point calibration with a culture of M. aeruginosa at a cell density of 20000 cells mL−1.
The TriOS microFlu-blue probe has an excitation peak at 620 nm and reads the fluorescence emitted between 650 and 660 nm. This probe provides a result in terms of concentration of PC and its putative measuring range is between 0 and 200 µg L−1. The probe allows measurements to be made in two ranges of sensitivity, from 0 to 20 µg L−1 and from 0 to 200 µg L−1.14 The manufacturer's calibration was used for all tests and was validated with a standard solution of PC (see section below). The TriOS probe was tested only in 2009. Values of cyanobacteria biomass (µg PC L−1) measured with the TriOS must be converted to density equivalent (cells of ∼38 µm3 mL−1) or biomass (µg chl-a L−1) to evaluate risk associated to cyanobacteria on the basis of established guidelines.1,15
Culture of M. aeruginosa
A culture of M. aeruginosa (UTCC 299) was maintained in a BG-11 medium16 with continuous agitation under controlled light conditions (20 µmol m−2s−1), temperature (26 °C ± 1) and photoperiod (16 h light and 8 h dark). Under these conditions, the culture remained unicellular (without colony formation).The cell density and average cell volume of the culture were determined using a particle counter Coulter Multisizer 3. The results of counts were regularly validated using an inverted microscope (Nikon Eclipse TE 2000S) with the Utermöhl's method.17
Standard solution of phycocyanin
A standard solution of PC was made with the C-phycocyanin extracted from Spirulina (P2172 Sigma) in phosphate buffer at pH 7.6. The PC concentration was determined by spectrophotometry using the following equation:18where A is the absorbance at 615 or 652 nm.

Background noise
The average background noise of probes was determined with phosphate buffer at pH 7.4 and with raw water from St Charles River (near Quebec City) filtered through a nitrocellulose 0.22 µm filter (Millipore GSWP). The values of background noise were subsequently subtracted from all measures that followed.Method detection limit and method quantification limit
The method detection limit (MDL) and the method quantification limit (MQL) were considered, respectively, as the concentration equivalent to 3 times and 10 times the standard deviation of 10 measures done on 10 replicates of a sample at low density.19 For the YSI probe, the MDL and MQL were determined with the culture of M. aeruginosa maintained in the laboratory at concentrations of 1500 and 4000 cells mL−1. The MDL and MQL of the TriOS probe were determined using a standard solution of PC at a concentration of 3.1 µg L−1 and also with M. aeruginosa at 4000 cells mL−1.Linearity
The relationship between the in vivo fluorescence of PC measured with the YSI probe and the cell density of the culture of M. aeruginosa was determined with calibrations done with two cyanobacteria cell densities (5000 and 25000 cells mL−1) and for the manufacturer's calibration. A culture of M. aeruginosa at high cell density (1 × 106cells mL−1, ±15%) was quantified with a particle counter and validated by microscopy. The microscopy measured values were used as reference points and subsequently, a dilution series was performed to obtain aliquots between 0 and 100000 cells mL−1 (±15%). Some of the dilutions were then validated by microscopy.
With the TriOS probe, the relationship was established between the in vivo fluorescence and a standard solution of PC (from 0 to 150 µg L−1). The relationship was also determined with a culture of M. aeruginosa (from 0 to 100000 cells mL−1, ±15%).
Repeatability and accuracy
The repeatability and the accuracy of the YSI probe were determined by taking fluorescence measures of 10 replicates of a culture of M. aeruginosa at cell densities of 30000 and 65000 cells mL−1. The repeatability of the TriOS probe was determined by measuring 10 replicates of a culture of M. aeruginosa at densities of 30000 and 75000 cells mL−1 and its accuracy was determined by compiling measures of 10 replicates of two concentrations (20 and 75 µg L−1) of the PC standard solution.
The repeatability was calculated as follows:19
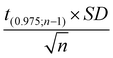
The accuracy was calculated as the difference between the “true” value and the measured value. In the case of the YSI, the “true” value is the cell density determined by microscopy, while for the TriOS probe, it is the PC concentration determined by spectrophotometry. The accuracy was calculated as follows:

| accuracy (%) = 100 − relative error (%) |
Validation of probes with field samples
The 91 samples used for this part of the study were taken from 20 lakes at the surface (portion 0 to 1 metre depth) or in surface scum between May and November in 2008 and 2009. Samples were preserved on ice during transport and at 4 ± 1 °C in the laboratory for a maximum storage time of 5 days before fluorometric measurement. A parallel experiment, conducted to ensure that the storage time of samples was appropriate, indicated no significant loss of PC (α < 0.05) (data not shown). All in vivo fluorescence measurements were performed in the laboratory. It should also be noted that the samples analyzed in this study were obtained from several lakes visited upon alerts given by citizens and were not following a time sequence monitoring.In 2008, only the fluorescence of total PC (intra and extracellular) was measured with the YSI probe. In 2009, the in vivo fluorescence was measured with both probes before and after the filtration of samples through a 0.22 µm filter (Millipore GSWP). This was done in order to discriminate the signal contribution by the intracellular PC and the dissolved or extracellular PC as well as the effect of other possible sources of interference. The identifications, enumerations and estimations of total cyanobacterial biovolume were made from sub-samples preserved with 1% Lugol solution and analyzed using inverted microscopy. All identifications were done on the basis of reference books.20–23 The cyanotoxins (microcystin LR, RR, YR, LA, LY, LW, LF and anatoxin-a) were analyzed using liquid chromatography coupled to mass spectrometers in tandem (HPLC-MS/MS).
Statistical analysis
All results were transformed into logarithms to meet the requirements of normality and homoscedasticity for the statistical treatment. The statistical analysis was performed using the software Sigma Plot 2001 (version 7101) and coefficients of determination (R2) are presented.Results and discussion
Background noise
The background noise of both probes was low. The average background noise of the YSI probe was less than 310 cells mL−1, no matter the type of calibration used, and it was estimated at 0.56 µg PC L−1 for the TriOS probe (Table 1). Negative numbers were only recorded when the YSI probe was calibrated with M. aeruginosa.| YSI probe | N | Mean | SD | Min | Max |
|---|---|---|---|---|---|
| Cells mL−1 | |||||
| Manufacturer's calibration | 10 | 307 | 58 | 213 | 387 |
| Calibration with 5000 cells mL−1 of M. aeruginosa | 10 | −219 | 350 | −1034 | 364 |
| TriOS probe | N | Mean | SD | Min | Max |
|---|---|---|---|---|---|
| µg PC L−1 | |||||
| Manufacturer's calibration | 9 | 0.56 | 0.32 | 0.24 | 1.0 |
Method detection limit and method quantification limit
The method detection limit (MDL) and the method quantification limit (MQL) of the YSI probe were determined at two cell densities (Table 2). The average MDL obtained with these two tests was 1500 cells mL−1, which is higher than the 220 cells mL−1 reported by the manufacturer.13 The MDL of the TriOS probe was estimated at 0.93 µg PC L−1 with the standard solution of PC and at 0.69 µg PC L−1 with a suspension of M. aeruginosa. These MDL expressed in PC concentrations represent respectively around 1170 and 870 cells mL−1 of M. aeruginosa. The variability in the response of the TriOS probe was lower in the culture of cyanobacteria which has contributed to a lower MDL. The YSI showed an average MQL of 5000 cells mL−1 for the two dilutions of M. aeruginosa tested and the TriOS showed a MQL of 2.3 µg PC L−1 (or 2900 cells mL−1) with the suspension of M. aeruginosa.| YSI probe | N | Mean | SD | MDL (3 × SD) | MQL (10 × SD) |
|---|---|---|---|---|---|
| Cells mL−1 | |||||
| 1500 cells mL−1 of M. aeruginosa | 10 | 1688 | 407 | 1221 | 4070 |
| 4000 cells mL−1 of M. aeruginosa | 10 | 4374 | 594 | 1782 | 5940 |
| TriOS probe | N | Mean | SD | MDL (3 × SD) | MQL (10 × SD) |
|---|---|---|---|---|---|
| µg PC L−1 | |||||
| Standard solution of PC at 3.1 µg L−1 | 10 | 3.2 | 0.31 | 0.93 | 3.1 |
| 4000 cells mL−1 of M. aeruginosa | 10 | 3.8 | 0.23 | 0.69 | 2.3 |
A MQL between 3000 and 6000 cells mL−1 is acceptable as it allows quantitative detection of cyanobacteria with a pure culture of M. aeruginosa. This limit is also low enough for an application in the context of monitoring raw and treated water for human consumption. However, the MDL and MQL may be higher where the colonial or filamentous forms of cyanobacteria are dominating, as in the natural environment.24
MDL of 102 and 103cells mL−1 is reported in the literature for M. aeruginosa measured in the laboratory using a spectrophotometer.25,26 MDL ranging from 103 to 107cells mL−1 for other species of cyanobacteria was also reported.25 An MDL of 102cells mL−1 was found in a study with the fluoroprobe designed by BBE Moldaenke.7 In another study done on a reservoir affected mainly by M. aeruginosa, a MDL of 1113 cells mL−1 was obtained using the fluorometer Turner Designs 10-AU-005.10
In order to release the PC, the use of a fluorometer coupled to an ultrasonic device to break the colonies and filaments and even cause cell lysis has been proposed in which a sonication of 20 minutes would increase by ten the fluorescence response at low cell density.25 The sonication of samples prior to the reading of fluorescence could thus substantially decrease the MDL. Such an approach has the potential to increase sensitivity and reduce variability related to the morphological diversity of cyanobacteria. However, sonication is not easily applicable in the field.
Linearity
Overall, the linearity of both probes was excellent with M. aeruginosa cultures in controlled conditions in the laboratory. With the manufacturer's calibration, the linear regression between cell density measured by microscopy in the range 0 to 100000 cells mL−1 and the density determined with the YSI probe was highly significant (R2 = 0.996, P < 0.0001; Fig. 1a). The linearity was also very good even if only the range 0 to 10000 cells mL−1 was considered (R2 = 0.951, P = 0.0001; data not shown). However, this calibration of the YSI probe leads to results that are largely underestimated (by 70 to 93%) in comparison to values determined by microscopy.
 | ||
| Fig. 1 Relationships between cell density of M. aeruginosa measured by microscopy and the YSI probe calibrated (a) by the manufacturer or (b) with cyanobacteria suspensions (filled circle with 5000 cells mL−1 and empty circles with 25000 cells mL−1), and relationships between (c) a standard PC solution and the TriOS probe or (d) relationship between cell density measured by microscopy and the TriOS probe (the dotted lines indicate 1:1 relationships). | ||
When the YSI probe was calibrated with cyanobacteria at densities of 5000 and 25000 cells mL−1, the linear regressions were again highly significant in the range 0 to 100000 cells mL−1 (R2 > 0.993, P < 0.0001; Fig. 1b). When considering the relationship in the range 0 to 10000 cells mL−1 only, R2 was similar as for the entire range for the calibration done at 5000 cells mL−1. However, it was slightly lower for the calibration done at 25000 cells mL−1 (R2 = 0.815, P = 0.0009; data not shown). According to our results, the probe seems more efficient when it is calibrated at 5000 cells mL−1 in comparison with a calibration at 25000 cells mL−1. Moreover, the calibration with cyanobacteria significantly improved the accuracy beyond 5000 cells mL−1 by comparison to the manufacturer's calibration. The stability of the YSI calibration was followed during the summer 2009 by repeated measurements using a solution of 100 µg L−1 of rhodamine. This monitoring has shown an excellent stability of the calibration over the entire period of the evaluation (N = 40, mean = 289000 cells mL−1, and SD = 10789 cells mL−1).
The linearity of the TriOS probe determined with a standard solution of PC was highly significant (R2 = 0.998, P < 0.0001; Fig. 1c). Moreover, the results are near the nominal concentrations expected. The differences between the probe and the 1:1 relationship were of 30% or less in the concentration range 5 to 150 µg L−1. When the exercise was done with a culture of M. aeruginosa, the linearity was also highly significant (R2 = 0.999, P < 0.0001) for intracellular PC concentrations of about 1 ng per cell (Fig. 1d).
Repeatability and accuracy
The repeatability of measurements was excellent for both probes (Table 3). The coefficients of variation (CV) of the YSI probe calibrated by the manufacturer are of 2.0 and 1.1% depending on the cell density measured. However, the results show relatively large differences between the probe and the nominal values determined by microscopy (the probe overestimated cell density by 38 and 59%, respectively at 30000 and 60000 cell mL−1). The CV of the TriOS probe varied between 1.2 and 4.8% depending on the type of sample used. Although the TriOS probe response was more variable than that of the YSI probe, it proved to be more accurate (with differences with “true” values lower or equal to 15%) in comparison to the YSI probe even if this last one was calibrated with M. aeruginosa. Considering this aspect, the TriOS probe appears more efficient than the YSI probe, although it is also possible that the use of pure PC extracts artificially improved its accuracy.
| YSI probe | N | Mean | SD | CV | Repeatability | Accuracy |
|---|---|---|---|---|---|---|
| Cells mL−1 | % | Cells mL−1 | % | |||
| 30000 cells mL−1 of M. aeruginosa |
10 | 41401 |
832 | 2.0 | 44401 ± 540 |
+38 |
| 65000 cells mL−1 of M. aeruginosa |
10 | 103451 |
122 | 1.1 | 103451 ± 802 |
+59 |
| TriOS probe | N | Mean | SD | CV | Repeatability | Accuracy |
|---|---|---|---|---|---|---|
| µg L−1 of PC | % | µg L−1 of PC | % | |||
| 30000 cells mL−1 of M. aeruginosa |
10 | 30 | 0.49 | 1.6 | 30 ± 0.32 | na |
| 75000 cells mL−1 of M. aeruginosa |
10 | 59 | 0.71 | 1.2 | 59 ± 0.51 | na |
| Standard solution of PC at 20 µg L−1 | 10 | 23 | 1.1 | 4.8 | 23 ± 0.71 | +15 |
| Standard solution of PC at 75 µg L−1 | 10 | 69 | 2.4 | 3.5 | 69 ± 1.6 | −8.4 |
Validation of probes with field samples
In 2008 and 2009, the in vivo fluorescence of PC measured by both probes was strongly related to the cell density and to the total cyanobacterial biovolume measured by microscopy (R2 > 0.705, P < 0.0001; Fig. 2). However, the differences between YSI probe and microscopy results varied from −97 to 1239% in 2008 and from −93 to 890% in 2009. For the TriOS probe, the relationships had slightly higher R2 by comparison to the YSI probe. TriOS accuracy with field samples cannot be assessed because the PC concentrations were not determined by a reference method (e.g., with pigment extraction and spectroscopy). | ||
| Fig. 2 Relationships between the YSI probe in 2008–2009 for field samples and (a) the cell density measured by microscopy; (b) the biovolume measured by microscopy; and relationships between the TriOS probe in 2009 and (c) the cell density measured by microscopy; and (d) the biovolume measured by microscopy (the dotted line indicates 1:1 relationship). | ||
The samples for which density was underestimated by the YSI probe contained in most cases less than 30000 cells mL−1 while the overestimated densities were observed in samples having more than 30000 cells mL−1 (Fig. 2a). The underestimated samples generally exhibited diverse communities of cyanobacteria, including picocyanobacteria that are often dominant in terms of abundance. Inversely, the overestimated samples often contained bloom-forming species dominated by one or two genera and where picocyanobacteria were rarely identified. This dominance of picocyanobacteria in samples of low density is also reflected in the relationship between the in vivo fluorescence measured with the YSI probe and the total cyanobacterial biovolume in 2008–2009 (Fig. 2b) where nearly 15% of samples containing a total cyanobacterial biovolume that was very small. It is possible that picocyanobacteria have been underestimated in samples with the presence of bloom-forming species. Moreover, no false-positives of PC have been observed in this study but some authors have reported false-positives in the presence of high turbidity.27
In order to better understand the effect of picocyanobacteria on the relationships observed, linear regressions have also been applied after the picocyanobacteria genera quantified by microscopic analysis (Aphanothece, Aphanocapsa, Cyanodictyon, Merismopedia, Planktolyngbya, Limnothrix, Phormidium and Synechococcus) have been removed from the data series. It was found that the withdrawal of these genera did not affect R2 values between the total cyanobacterial biovolume and both probe estimations, suggesting their small contribution to the total cyanobacterial biovolume. However, for the relationships with cell density, the withdrawal of the picocyanobacteria genera diminished the R2 values for both probes (R2 = 0.655 YSI, 0.610 TriOS) (data not shown). This diminution is clearly due to samples dominated by picocyanobacteria. In this regard, it may be wise to use the YSI raw data (in relative fluorescence units) instead of the result transformed in cells per mL since the raw data are directly related to the quantity of PC and then to the total cyanobacterial biovolume, the same way as for the TriOS probe. In addition to the variability in cell volume of different genera of cyanobacteria and the possible effect of turbidity, there are several other factors that may influence the in vivo fluorescence of PC. They comprise the growth stage of cyanobacteria,28 the level of light saturation,29 the aggregation of cells into colonies more or less voluminous,25,30 and the historical conditions of light and nutrients.31,32
Because in vivo PC fluorescence measurements are qualitative in the absence of data correction following post-calibration, it can be difficult to evaluate whether or not a decisional threshold is reached. However, some authors suggested management thresholds based on PC levels for the determination of the appropriate sampling frequency.8 Some also suggested using direct PC thresholds (measured in the laboratory with a spectrophotometer) but they warn of the need to give special attention to samples having an in vivo fluorescence near the management threshold given the variability between probes in terms of reliability and calibration.33 In the context of optimization of field sampling, the PC in vivo fluorescence could be used as criteria to decide whether or not a sample should be taken for identification and enumeration of cyanobacteria by microscopic analysis in the laboratory. Given the qualitative aspect of the measures of PC, a threshold corresponding to a density of 15000 cells mL−1 of cyanobacteria or about 15 µg PC L−1 seems appropriate as decision criteria, as one of the management thresholds is 20000 cells mL−1. However, a more precise validation is needed near the threshold value of 20000 cells mL−1, especially for the YSI probe. Also, the combined use of PC in vivo fluorescence and cyanotoxins screening tests (microcystins strip test34) seems optimal by avoiding false negative (i.e., detectable presence of cyanotoxins with low density of cyanobacteria or conversely high density of cyanobacteria and no cyanotoxins detection). The latter immunochromatographic strip test is being already tested in the Government of Québec Management Plan.
The simultaneous variations of both probes with the cell densities and total cyanobacterial biovolume measured by microscopy are illustrated in Fig. 3. The curves show a close agreement between probe estimations and the total cyanobacterial biovolume. However, differences sometimes exceeding an order of magnitude are seen between cell densities estimated with the YSI probe and those measured by microscopy. The samples for which the YSI probe presented a good accuracy were dominated by Microcystis or a mixture of Anabaena and Aphanothece. The latter potentially results in an average cell volume similar to the one of M. aeruginosa used for the calibration of the instrument in the laboratory.
 | ||
| Fig. 3 Comparison between the cell density of field samples and the biovolume measured by microscopy: (a) the YSI probe for 2008–2009 and (b) the TriOS probe for 2009. | ||
In 2009, measurements obtained on filtrated samples indicated the presence of extracellular PC in many samples. There are significant relationships between the fluorescence signal in the filtrate measured by the YSI probe and the cell density (R2 = 0.600, P < 0.0001; data not shown) or total cyanobacterial biovolume (R2 = 0.733, P < 0.0001; Fig. 4a), as well as for the TriOS probe signal with cell density (R2 = 0.570, P < 0.0001; data not shown) or total cyanobacterial biovolume (R2 = 0.725, P < 0.0001; Fig. 4b). On average, 21 to 25% of the fluorescence signal was related to the filtrate, and therefore to extracellular PC. The variations of the filtrate fluorescence signal were important between samples and the ratio of extracellular PC to intracellular PC varied from 1 to 100%. In some cases, the whole PC signal was therefore extracellular. Interferences caused by varying concentrations of chromophoric dissolved organic matter may have contributed to the fluorescence of filtrates. However, the relationship between the filtrate PC signal and total cyanobacterial biovolume suggests that the fluorescence was indeed related to the extracellular PC released by lysed cells of cyanobacteria. This observation raises questions on the expression of the YSI results in terms of cells mL−1 because the extracellular PC is included in the computed abundance. Measurement of the in vivo fluorescence must be interpreted in terms of total PC and the subtraction of a field blank obtained from filtered samples will allow an estimate of intracellular PC. The presence of extracellular PC has already been raised as a hypothesis to explain some data showing no apparent relationship with cell density.8 No relationship could be established between the presence of extracellular PC and the cyanobacteria genera observed or the time of the year. However, in some cases where the extracellular PC was elevated, high densities of the genus Anabaena were observed.
 | ||
| Fig. 4 Relationships between the fluorescence of the filtrate of field samples and the biovolume measured by microscopy for (a) the YSI probe in 2008–2009 and (b) the TriOS probe in 2009. | ||
During the two years of the study, cyanotoxins have been identified in 36 of the 91 samples. When all samples were considered, no relationship was established between the concentration of cyanotoxins and the cell density (R2 = 0.235) or the total cyanobacterial biovolume (R2 = 0.198) or with the probe estimations (R2 = 0.172 and 0.161, respectively for the YSI and the TriOS probes). This result confirms that there is no direct link between the presence of cyanobacteria and the cyanotoxin concentrations measured. It is known that the gene expression for microcystin or other toxin production may vary from one strain to another.35 The activation of these genes is also strongly influenced by environmental conditions, such as the concentrations of nitrogen and phosphorus.36–38 Moreover, there is a wide range of cyanotoxin types39 and many are not currently quantified. This could also contribute to the weakness of the relationship between in vivo PC fluorescence and the concentrations of cyanotoxins. A genomic approach has been proposed to distinguish toxic and non-toxic genotypes of Microcystis.40
It is important to note that the 36 samples with detectable concentrations of cyanotoxins contained in most cases high densities of cyanobacteria. The presence of cyanobacteria remains therefore an indicator of risk even if a quantitative relationship cannot be established.
Conclusion
In vivo PC fluorescence measurements using field probes present an interest for spatio-temporal monitoring of algal blooms in natural environments, for the optimization of sampling strategies and for the early detection of cyanobacteria in drinking water intakes. The relationships observed with the total cyanobacterial biovolume and cell density are significant and the quantification limits of fluorescence probes are low enough for the purposes of such monitoring. However, the results for the YSI probe are qualitative if no correction is applied following a post-calibration, as suggested by the manufacturer.The TriOS probe appears interesting for monitoring cyanobacterial blooms because of its ease of use, its reliability and its expression of the results in terms of PC concentration which is not biased by the variability of cell volume. However, thresholds generally used in management plans are in cells per mL which disadvantages the TriOS probe as this provides results in terms of concentration of PC. The advantage of using a multi-probe system such as proposed by YSI, where for example data on water temperature, dissolved oxygen, total chlorophyll-a and PC are collected simultaneously, is certainly considerable and should also be taken into account in monitoring studies.41 Improvement of converting equations provided by manufacturers is needed. The lack of quantitative relationship between cyanobacteria and cyanotoxin concentrations shows that these two parameters must be used in a complementary way to manage risk appropriately.
Acknowledgements
The authors would like to thank Gaëlle Triffault-Bouchet for her support to the revision of the document. They also want to thank all the people from the different regional divisions of the ministry for their contribution to the field sampling.References
-
I. Chorus and J. Bartram, Toxic Cyanobacteria in Water. A Guide to their Public Health Consequences, Monitoring and Management, E & FN Spon on behalf of the World Health Organization, 1999, pp. 1–416 Search PubMed
.
- Ministère du Développement Durable, de l'Environnement et des Parcs, Plan de gestion des épisodes de fleurs d'eau, 2010http://www.mddep.gouv.qc.ca/eau/algues-bv/plan_intervention_2007-2017.pdf.
- Ministère du Développement Durable, de l'Environnement et des Parcs, Bilan final des plans d'eau touchés par une fleur d'eau d'algues bleu-vert, 2010http://www.mddep.gouv.qc.ca/eau/flrivlac/algues.htm.
- D. A. Bryant, G. Guglielmi, N. Tandeau de Marsac, A.-M. Castets and G. Cohen-Bazire, Arch. Microbiol., 1979, 123, 113–127 CrossRef CAS
-
S. W. Jeffrey and R. F. C. Mantoura, in Phytoplankton Pigments in Oceanography: Guidelines to Modern Methods, ed. S. W. Jeffrey, R. F. C. Mantoura and S. W. Wright, UNESCO, Paris, 1997, pp. 19–36 Search PubMed
-
R. G. Wetzel, Limnology–Lake and River Ecosystems, Academic Press, 3rd edn, 2001, pp. 331–332 Search PubMed
- J. Gregor, B. Marsalek and H. Sipkova, Water Res., 2007, 41, 228–234 CrossRef CAS
- L. Brient, M. Lengronne, E. Bertrand, D. Rolland, A. Sipel, D. Steinmann, I. Baudin, M. Legeas, B. Le Rouzic and M. Bormans, J. Environ. Monit., 2008, 10, 248–255 RSC
- J. Gregor, R. Geris, B. Marsalek, J. Hetesa and P. Marvan, Hydrobiologia, 2005, 548, 141–151 CrossRef
- K. Izydorczyk, M. Tarczynska, T. Jurczak, J. Mrowczynski and M. Zalewski, Environ. Toxicol., 2005, 20, 425–430 CrossRef CAS
- K. Izydorczyk, C. Carpentier, J. Mrowczynski, A. Wagenvoort, T. Jurczak and M. Tarczynska, Water Res., 2009, 43, 989–996 CrossRef CAS
- C. Leboulanger, U. Dorigo, S. Jacquet, B. Le Berre, G. Paolini and J. F. Humbert, Aquat. Microb. Ecol., 2002, 30, 83–89 Search PubMed
-
YSI incorporated, 6-series Multiparameter Water Quality Sondes, User Manual, October 2006 Search PubMed
-
Trios Optical Sensor, MicroFlu Manual, ver. 2.0, December 2009 Search PubMed
- W. W. Carmichael, Hum. Ecol. Risk Assess., 2001, 7, 1393–1407 CrossRef
- R. J. Rippka, J. Deruelles, J. Waterbury, M. Herdman and R. Stanier, J. Gen. Microbiol., 1979, 111, 1–61
- H. Utermöhl, Mitt.–Int. Ver. Theor. Angew. Limnol., 1958, 9, 1–38 Search PubMed
- A. Bennett and L. Bogorad, J. Cell Biol., 1973, 58, 419–435 CAS
-
Centre d'Expertise en Analyse Environnementale du Québec, Protocole pour la validation d'une méthode d'analyse en chimie DR-12-VMC, Québec, 2009, 29 p. Search PubMed
- J. Komarek and K. Anagnostidis, Arch. Hydrobiol., Suppl., 1989, 82, 249–345 Search PubMed
-
J. Komarek and K. Anagnostidis, Cyanoprokaryota. 1. Teil: Chroococcales. Süßwasserflora von Mitteleuropa, Spectum Akademischer Verlag, 1998, vol. 19/1, ISBN: 3-8274-0890-3 Search PubMed
-
J. Komarek and K. Anagnostidis, Cyanoprokaryota. Oscillatoriales. Süßwasserflora von Mitteleuropa, Spektrum Akademischer Verlag, 2005, vol. 19/2, ISBN: 3-8274-0919-5 Search PubMed
-
D. J. Wehr and R. G. Sheath, Freshwater Algae or North America, Ecology and Classification, in Aquatic Ecology Series, Academic Press, 2003, pp. 59–196 Search PubMed
- A. Warren, unpublished work.
- R. Asai, S. McNiven, K. Ikebukuro and I. Karube, Field Anal. Chem. Technol., 2000, 4, 53–61 CrossRef CAS
- T. Lee, M. Tsuzuki, T. Takeuchi, K. Yokoyama and I. Karube, J. Appl. Phycol., 1994, 6, 489–495 Search PubMed
- L. Bowling, personal communication.
-
R. L. Oliver and G. G. Ganf, in The Ecology of Cyanobacteria, ed. B. A. Whitton and Potts, Kluwer Academic Publishers, The Netherlands, 2000, pp. 2149–2194 Search PubMed
- M. Beutler, K. H. Wittshire, B. Meyer, C. Moldaenke, C. Luring, M. Meyerhofer, U. P. Hansen and H. Dau, Photosynth. Res., 2002, 72, 39–53 CrossRef CAS
- Z.-X. Wu and L.-R. Song, Phycologia, 2008, 47, 98–104 CrossRef CAS
- A. R. Grossman, D. Bhaya and Q. He, J. Biol. Chem., 2001, 276, 11 449–11 452 CrossRef CAS
- C. W. Mullineaux, Mol. Microbiol., 2001, 41, 965–971 CrossRef CAS
- C. Y. Anh, S. H. Joug, S. K. Yoon and H. M. Oh, J. Microbiol., 2007, 45, 98–104 Search PubMed
- Abraxis, http://www.abraxiskits.com/, 2010.
- N. L. Saker, J. Fastner, E. Dittmann, G. Christiansen and V. M. Vasconcelos, J. Appl. Microbiol., 2005, 99, 749–757 CrossRef
- T. G. Downing, C. Meyer, M. M. Gehringer and M. Van de Venter, Environ. Toxicol., 2005, 20, 257–262 CrossRef CAS
- A. Giani, D. F. Bird, Y. T. Prairie and J. F. Lawrence, Can. J. Fish. Aquat. Sci., 2005, 62, 2100–2109 CrossRef CAS
- A. Rolland, D. F. Bird and A. Giani, J. Plankton Res., 2005, 27, 683–694 CrossRef CAS
- G. A. Codd, L. E. Morrisson and J. S. Metcalf, Toxicol. Appl. Pharmacol., 2005, 203, 264–272 CrossRef CAS
- I. Janse, W. Edwin, A. Kardinaal, M. Mejma, J. Fastner, P. M. Visser and G. Zwart, Appl. Environ. Microbiol., 2004, 70, 3979–3987 CrossRef CAS
- B. Karrasch and M. Herzog, Acta Hydrochim. Hydrobiol., 2005, 33, 165–169 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2011 |