Semiconductor/biomolecular composites for solar energy applications
Chuanhao
Li
,
Feng
Wang
and
Jimmy C.
Yu
*
Department of Chemistry, Environmental Science Programme and Centre of Novel Functional Materials, The Chinese University of Hong Kong, Shatin, New Territories, Hong Kong, China. E-mail: jimyu@cuhk.edu.hk; Fax: +852-26035057; Tel: +852-26096268
First published on 29th October 2010
Abstract
Solar energy is the world's most abundant renewable energy source. It has great potential to replace the depleting fossil fuels to power our society. Current photovoltaic and photocatalytic systems are almost entirely based on semiconductor materials. To enhance the efficiency of these systems for solar energy applications, various approaches have been developed. Considerable effort has been focused on the development of semiconductor/biomolecular composites for converting sunlight to electricity and fuels. This perspective is intended to discuss the recent research findings involving the bio-inspired photovoltaic devices, photoelectrochemical cells, and photocatalytic processes. The synthesis strategies for the hybrid materials and the working principles of the systems are presented.
 Chuanhao Li | Chuanhao Li obtained his BS (2005) and MS (2008) degrees from Anhui University, PR China. Now a PhD student under the supervision of Prof. Yu, Mr Li is investigating near-infrared-assisted photocatalysis. |
 Feng Wang | Feng Wang received a BS degree from Anhui University in 2005 and a MS degree from The University of Science and Technology of China in 2008. His PhD research project is about the preparation of photocatalysts for energy applications. |
 Jimmy C. Yu | Prof. Jimmy Yu received his PhD degree from the University of Idaho in 1985. His research interests are photocatalysis and nanomaterials. An author of many highly cited papers on photocatalytic nanomaterials, Dr Yu also holds six patents for their applications. |
Broader contextExploring alternatives to replace conventional fossil fuels is critical for sustainable development. Solar energy is a promising candidate because it is safe, clean and renewable. However, solar conversion devices are not in widespread use due to the relatively poor efficiency and high cost compared with traditional energy sources. Various approaches have been developed to solve these problems. One of the most promising solutions is the utilization of non-silicon based semiconductors. Recently, solar-responsive semiconductor/biomolecular composites have attracted a lot of attention. In this perspective, we describe the latest research progress in bio-inspired photovoltaic devices, photoelectrochemical cells, photocatalytic processes and artificial photosynthesis. |
1. Introduction
A rapid increase in human population and global economic development create a strong demand for energy, while the fossil fuel reserves that we rely heavily on are depleting. In addition to the large disparity between demand and supply, consumption of fossil fuels also causes consequential environmental problems, such as air pollution, global warming leading to climate changes, and destruction of biodiversity.1 In order to maintain the sustainable global development, exploring renewable energy with minimal adverse environmental effects is necessary. The promising candidates should be abundant, cheap, environmentally clean, and readily available on earth. Among the natural renewable energy including sunlight, wind, geothermal, ocean thermal, tidal and biomass energy, the solar energy is the most attractive option.2Solar light is the largest global renewable energy source. The sun delivers energy to the earth's surface at an average of 4.3 × 1020 J h−1, which is approximately equivalent to the world's total annual energy consumption in all forms.3 However, the solar energy has been utilized by humans with limited efficiency. In this regard, applications of solar energy in different areas were investigated with the view to enhancing utilization efficiency. The first one is the natural photosynthesis, in which plants convert solar energy to chemical energy to provide humans the essential needs.4 Based on the understanding of the photosynthesis mechanism, artificial photosynthesis has been achieved. The second one is photovoltaic devices.5 A solar cell is developed to generate electrical current from sunlight. The third is photoelectrochemical cells, which convert solar energy into chemical fuels.6 A typical example is the water splitting process to produce hydrogen, a clean energy. The fourth one is a photocatalysis process, which has application in environmental pollution.7 Solar energy is captured and then converted to induce a series of redox reactions to photodegrade pollutant molecules. Also some medical applications for solar energy have been demonstrated.8
So far, all the solar applications are based on the light absorption. Due to their unique electronic structure composed of a filled valence band (VB) and an empty conduction band (CB), semiconductors can absorb the light with energy equivalent to or higher than the band gap. Then electrons are excited to CBs, leaving positive holes in VBs.9 The photogenerated electrons or holes play very important roles in solar energy applications including solar photovoltaic, solar water photoelectrolysis and photocatalytic remediation. The unique tunable optical and electronic features make semiconductors the most attractive candidates for solar energy applications. The tunable optical property corresponds to a tunable energy band gap which facilitates the light absorption, while the tunable electronic property can help enhancing the lifetime of hole/electron pair.9 However, there are drawbacks with the single semiconductor system, such as narrow-spectrum response and photoerosion. To overcome these problems, semiconductor composites have been developed. A typical example is TiO2/CdS composite.10 TiO2 is a wide band gap semiconductor, which can be only activated by UV light. After combination with the narrow band gap semiconductor CdS, the photoabsorption is extended to visible light. And the photocorrosion of CdS is avoided. Some organic/inorganic hybrid systems have also been developed.11
Recently, biomolecules have been widely used to modify semiconductors to enhance the biocompatibility. Furthermore, some of the biomolecules such as chlorophylls have efficient absorption ability for solar energy. Biomolecule/semiconductor composites have been developed for different applications. Herein, we focus on the novel composite materials with great potential for solar energy applications. The mechanism and the advantages over traditional materials are discussed.
2. Photovoltaic technology
Since reported by Grätzel in 1991, dye-sensitized solar cells (DSSCs) have quickly attracted interest as an alternative to the expensive silicon-based photovoltaic systems.12–14 As shown in Fig. 1, dye molecules absorb sunlight and transfer/inject the excited electrons to a TiO2 anode to power an external load. Electrons are re-introduced into the cell through an iodide electrolyte.12,15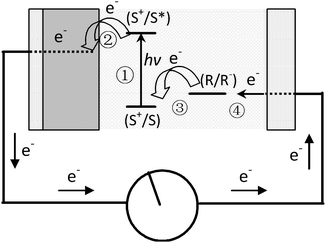 | ||
| Fig. 1 Schematic representation of the principle of the dye-sensitized photovoltaic cell. S, sensitizer; S*, electronically excited sensitizer; S+, oxidized sensitizer (adapted from ref. 12). | ||
The energy producing reactions in a dye-sensitized TiO2 solar cell are as follows:
| dye + light → dye* | (1) |
| dye* + TiO2 → e−1(TiO2) + oxidized dye | (2) |
| oxidized dye + 3/2I− → dye + 1/2I3− | (3) |
| 1/2I3 + e−1 → 3/2I− | (4) |
Semiconductor/biomolecular composites have a role to play in the future development of Grätzel cells. Natural dyes extracted from fruits, flowers, leaves and bacteria are easy to prepare, low in cost, non-toxic, environmentally friendly and fully biodegradable, compared with the synthetic ones. Recently, Bignozzi and co-authors gave a review about natural dye sensitized solar cell, involving, chlorophyll derivatives, anthocyanin dyes and betalain dyes.16 It has been reported that the natural dye sensitized solar cells have limitations being short lifetime and low conversion efficiency (5%) of the overall sunlight to electrical energy.15 An important parameter for its performance is the ratio of the forward (injection) to reverse (recombination) electron transfer rates. The speed of re-introducing electrons to the oxidized dye molecules from the redox mediator (I−/I3−) is also critical.17,18 Many ideas have been proposed to improve the overall efficiency by using suitable dye derivatives or proteins, co-sensitizers, near-infrared sensitizers, and utilising gold nanostructures as an electron relay.
Chlorophylls are key photosynthetic pigments in natural systems. Their functions include harvesting light and transferring energy and electrons.20 Chlorophyll a has a polycyclic porphyrinic structure with conjugated double bonds, which are responsible for absorption in the red and blue regions of the visible spectrum. There are two characteristic π–π* absorption regions, the weaker Q band at lower visible wavelengths (550–700 nm) and the intense Soret band in the near UV region (400–450 nm). Due to the strong light-harvesting capability of chlorophylls and their derivatives, they have been introduced into DSSCs as dye sensitizers. However, chlorophyll a (Fig. 2a) does not adsorb efficiently onto TiO2, due to lack of effective functional groups. The photocurrent efficiency, the incident photon to current conversion efficiency (IPCE), is 3.5% at 670 nm.
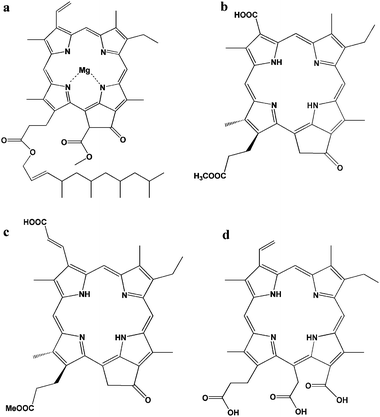 | ||
| Fig. 2 Structure of natural chlorophyll derivatives: (a) chlorophyll a,23 (b) pheophorbide a,19 (c) chlorin 2,21 and (d) chlorine-e6.23 | ||
Pheophorbide, one of the chlorophyll derivatives, with a modified carboxylic group allows a much stronger adsorption onto TiO2. Wang and co-authors reported pheophorbide (phe a) (Fig. 2b) modified TiO2 showed higher conversion efficiency than some other similar pheophorbide structures. Jsc, Voc, FF and η of phe a-sensitized solar cell using platinum-coated OTE as a counter electrode were 11.4 mA cm−2, 550 mV, 61.0% and 3.8%, respectively.19
Wang and co-authors synthesized a new and high-efficient methyl trans-32-carboxy-pyropheophorbide a (chlorin 2, Fig. 2c), which has an extended π-conjugation length along the Qy axis of the chlorin skeleton.21,22 The chlorin-2 had an efficient LUMO + 2 molecular orbital for electron injection, and an enhanced Qy band and a smaller Eox value for light-harvesting. Moreover, the HOMO and HOMO − 1 orbitals had their main distribution far from the TiO2 surface, which was suitable for electron transfer from the redox couple of I−/I3− in the electrolyte. The energy conversion efficiency (η value) for TiO2 and ZnO was reported to be 6.1% and 3.6% respectively.
Chlorine-e6 (Chl-e6), another kind of chlorophyll derivative, formed by the hydrolysis of chlorophyll has three carboxylate groups in the molecule (Fig. 2d). As Chl-e6 does not contain a heavy metal ion, Chl-e6 is a suitable photosensitizer from the point of view regarding environmental problems. The absorption spectrum of Chl-e6 is similar to that of chlorophyll. Amao and co-workers reported that the short-circuit photocurrent density (Isc), the open-circuit photovoltage (Voc), and the fill factor (FF) of solar cell using Chl-e6 adsorbed onto a nanocrystalline TiO2 film electrode were estimated to be 0.305 ± 0.012 mA cm−2, 426 ± 10 mV, and 45.0%, respectively. IPCE values could reach a maximum around the wavelength of absorption maximum 7.40% at 400 nm, 1.44% at 514 nm and 2.91% at 670 nm respectively.23,24
Anthocyanin dyes are natural compounds and are responsible for the red and purple colouration of many fruits, vegetables and leaves. The extracted anthocyanin sensitized semiconductors also have been used for the photocurrent production.26,27 As shown in Fig. 3, anthocyanin dyes appear red in acidic solution, due to an intense band centered at ca. 520 nm. The presence of three hydroxyl groups in the cyanine and nasunin (typical of the delphidine structure) structure favoured the chelating effect towards titanium (Ti4+). Hence, this leads to an efficient electron injection from the dye to TiO2.25
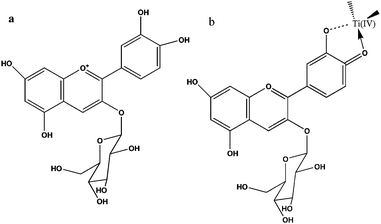 | ||
| Fig. 3 Chemical structures of cyanine (a) and schematic representation of cyanine (b) attachment by chelating effect to Ti(IV) sites (right) (adapted from ref. 25). | ||
Cherepy and co-authors demonstrated the ultrafast charge injection processes from anthocyanins to TiO2. However, the IPCE of anthocyanins was quite low (20%). It was due to dye aggregation, electron recombination with the oxidized sensitizer and electron recapture by I3−, which was dependent upon dye structure and surface coverage.28 Anthocyanins extracted from different plants performed differently. The quantum yield of TiO2 photosensitized by commercial anthocyanin dye was 52% with iodide/iodine in water (pH 1) or pure ethanol acting as the charge carriers.29 Blue-violet anthocyanins extracted from Jaboticaba (Myrtus cauliflora Mart) and Calafate (Berberis buxifolia Lam) were employed as TiO2 dye-sensitizers. Solar cells sensitized by Jaboticaba extracts could achieve up to Jsc = 9.0 mA cm−2, Voc = 0.59 V, and Pmax = 1.9 mW cm−2, while for Calafate sensitized cells the values determined were up to Jsc = 6.2 mA cm−2, Voc = 0.47 V, and Pmax = 1.1 mW cm−2.30 Calogero and co-workers used directly the juice of the Moro variety of red Sicilian orange as a natural sensitizer in DSSCs. They obtained solar energy conversion efficiency of 0.66% under AM 1.5 illumination. In the Moro juice, the high concentration of cyanine and the natural presence of citric acid or hydroxycinnamic acids acted as co-absorbers. These compounds, filling the free space between the dye molecules, partially blocked the physical contact between iodine solution and TiO2 semiconductor film surface, reduced the backward reaction of I3− and e− in TiO2 and inhibited dye aggregation.25
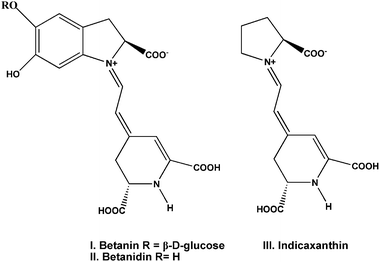
Betalain dyes also have been used as a sensitizer for the production of photocurrent. The betalain pigments (Fig. 4) comprise the red-purple betacyanins, betanin (I) and betanidin (II), with maximum absorptivity at λmax about 535 nm, and the yellow betaxanthins (III) with λmax near 480 nm. These pigments are present in the different parts of the plants including flowers, petals, fruits, leaves, stems and roots. The ground and excited state oxidation potentials of betalain pigments using DFT and TDDFT are calculated to be ca. 1.0 V and −1.3 V (vs. NHE) respectively, making the electron transfer from the dye molecule to the TiO2 conduction band thermodynamically favourable.32 McHale and co-workers reported the nanocrystalline TiO2 sensitized by natural betalain pigments, which was extracted from red beetroots. The fabricated materials gave a maximum photocurrent of 2.42 mA cm−2 and open-circuit photovoltage of 0.44 V in the presence of methoxypropionitrile containing I−/I3− redox mediator.31
Combining two dyes with complementary spectra would be possible to achieve higher efficiencies than with simple dye. In plants, the chloroplast containing many pigment molecules shows higher energy conversion efficiency than the extracted single one. To mimic the principles of natural photosynthesis including using multiple pigments for light-harvesting, several groups have already attempted to combine the appropriate synthetic dyes in “molecular cocktails” using ionic interactions, as well as supramolecular and materials chemistry.34–36 It was reported that a dye-sensitized solar cell was made by coating pigments of shisonin (λmax = 600 nm) and chlorophyll (λmax = 440 nm) on a nanocrystalline film of TiO2. An energy conversion efficiency of ∼1.3% was obtained after deposition of p-CuI. Both shisonin and chlorophyll contributed to light energy harvesting in fabricated cells.27 Besides the complementary adsorption spectra of used dyes, the energy levels in the complex were also important for high performance. Wang and co-authors reported that Phe a and Chl c2 co-sensitized TiO2 showed enhancement in conversion efficiency. The photogenerated electron transferred from the lowest singlet excited states (Qy) of Chl c2 to that of Phe a, followed by electron injection from Qy of Phe a to TiO2, as shown in Fig. 5. The power–conversion efficiency of 5.4% was obtained.37 In fact, to find a co-sensitizer with enhanced efficiency is not easy. For example, the mixed extracts from roselle and blue pea adsorbed onto TiO2 do not show synergistic light absorption and photosensitization compared to the individual extracts.38 Many factors would affect the efficiency of the co-sensitized system, for example, the overlap of the adsorption, the molecular structure, and the electron injection time between two dyes and semiconductors.
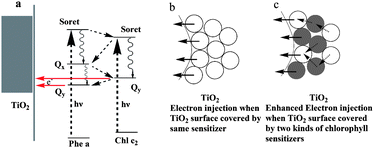 | ||
| Fig. 5 The mechanism of electron transportation upon co-sensitization. (a) Chl sensitizers form a monolayer on the TiO2 surface. (b) A schematic representation of how one kind of Chl sensitizer forms a multilayer on the TiO2 surface, and (c) how different kinds of Chl sensitizers form multilayers on the TiO2 surface.33 | ||
As the chlorophyll derivative molecules usually aggregate on the solid surface, the photogenerated electrons would recombine and decrease the sensitization activity. Interestingly, Amao and co-workers reported the formed aggregation from Zn chlorine-e6 (J-aggregates) resulted in a considerable red shift and showed an absorption maximum at 795 nm. A photoelectrochemical device based on the dye aggregates could absorb and convert near IR photons into electrons. The IPCE value was 7% at 800 nm for Zn chlorine-e6 aggregation.39
Spanning across the overall spectra of solar cell, the performance of aggregated dyes was less efficient than simple dyes. The aggregation of ZnChl-e6 molecules on the TiO2 was suppressed by cholic acid or myristic acid (Fig. 6).40 Without cholic acid, the IPCE values of ZnChl-e6/TiO2 at 400, 660 and 800 nm were 7.8, 6.9 and 7.5% respectively. With cholic acid and ZnChl-e6 co-adsorbed TiO2 electrode, the IPCE values at 400 and 600 nm increased to 12 and 8.0%, while the IPCE value at 800 nm decreased to 4.1%. The overall performance of ZnChl-e6/TiO2 was improved in the presence of cholic acid under a xenon lamp through suppressing aggregation.40
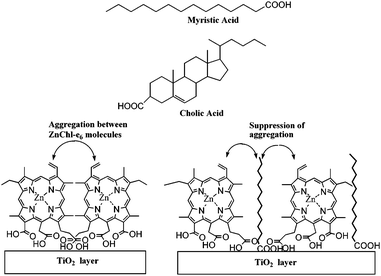 | ||
| Fig. 6 Chemical structures of myristic acid and cholic acid and schematic representation of ZnChl-e6 and fatty acid layers onto TiO2.40 | ||
Besides using extracted natural dyes as sensitizers, the use of proteins as sensitizers directly has been studied. Bacteriorhodopsin (bR), a natural light activated protein, which has been found in Halobacterium salinarum, shows application in solar energy conversion.42 In the work as reported in Fig.7 which also shows the structure of the triple mutant of bR i., four Glu side chains (Glu9, Glu74, Glu194 and Glu204) are located in the extracellular region of the protein. Two of these residues Glu194 and Glu204 together with water clusters were identified as major players in proton release mechanism in the study.43 The mutation of three of EC Glu residues (Glu9, Glu194 and Glu204) for Gln (E9Q/E194Q/E204Q) results in 3Glu protein with less negative charge density at the extracellular surface, which facilitates more favourable binding of bR to the TiO2 surface via electrostatic interactions. The lowest unoccupied molecular orbital (LUMO) and highest occupied molecular orbital (HOMO) of bR were determined to be −3.8 eV and −5.2 eV.44 The bR(3Glu) immobilized TiO2 film electrode solar cell produced a short-circuit photocurrent density (Jsc) of 0.089 mA cm−2 and the open-circuit photovoltage (Voc) of 0.35 V under an excitation intensity of 40 mW cm−2.41 Compared with chlorophylls, bR (3Glu) also has another advantage. Chlorophyll and its derivatives are considerably sensitive to extreme thermal conditions once extracted out of its source. bR has remarkable stability against thermal stability even up to ∼140 °C. Besides it is also stable under a high concentration of salt (up to 5 M NaCl) and functional under broad range of pH (5–11).
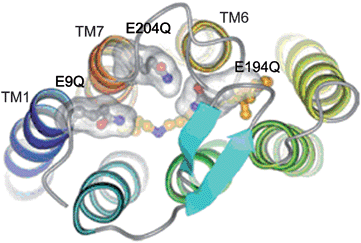 | ||
| Fig. 7 Extracellular region of 3Glu mutant of bR. The three Glu residues mutated to Gln are shown in separated transparent surfaces. Retinal and Lys216 carbon atoms are colored in orange.41 | ||
The efficiency of dye sensitized solar cells is influenced by the charge recombination. Hotchandani and co-workers found the gold nanoparticles enhanced the charge separation in a Chl a-sensitized TiO2 solar cell. Besides accepting electrons, gold nanoparticles could also act as an electric relay due to its electron storing properties.46,47 For an OTE/TiO2/Au/Chl a electrode, upon excitation with visible light, which was predominantly absorbed by Chl a, excited Chl a (Chl a*), being at a higher energy level (−1.1 V vs. NHE), injected electrons into gold nanoparticles (EF = 0.5 V vs. NHE). The electron accumulation in gold nanoparticles raised their Fermi level (EF) to more negative potentials until it reached the Fermi level of TiO2 (approx. −0.5 V vs. NHE) whereupon a quick shuttling of electrons from Au to TiO2 took place (Fig. 8). The electrons transferred to TiO2 particles were collected at the back OTE contact to generate an anodic photocurrent. A nearly 300% increase in IPCE at 440 nm was noticed in the OTE/TiO2/Au/Chl a system in comparison with OTE/TiO2/Chl a. The enhanced photoresponse of OTE/TiO2/Au/Chl a system compared to that for OTE/TiO2/Chl a was mainly due to the ability of Au nanoparticles in accepting and quickly shuttling the electrons from excited Chl a to TiO2, thus preventing charge recombination.45 The fluorescence investigation indicated a large value of association constant (104 M−1) provided there were favourable conditions for the heterogeneous charge-transfer process at the Chl a–Au nanoparticles interface. The electrochemical modulation of the emission intensity of Chl a supported the electron-transfer process between excited Chl a and Au. Nanosecond laser flash experiments of Chl a demonstrated that Au nanoparticles, besides accepting electrons, could also mediate electrons to another acceptor thus aiding in charge separation.48
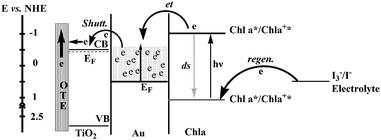 | ||
| Fig. 8 Energy level diagram and mechanism of photocurrent generation in the photoelectrochemical cell using OTE/TiO2/Au/Chl a as photoanode. CB and VB are, respectively, the conduction and valence bands of TiO2; other terms have been defined.45 | ||
For the natural dye sensitized solar cells, the performance of thin films can be favourably altered by translation from a flat, two-dimensional surface to a three-dimensional architecture with nanoscale features, which can both increase the available surface area and add infrastructure to the system. Agostiano and co-workers prepared a pigment modified quantum-sized ZnO nanocrystals electrode by adsorbing either chlorophyll a, carotenoids or their mixture onto a film of ZnO nanoparticles. The photoconversion process was found to be greatly enhanced at the nanocrystalline electrodes compared with that of the bulk one. Terasaki and co-authors reported that a PSI modified Au nanoparticles electrode showed a high photocurrent generation.49
3. Photoelectrochemical cells
Hydrogen is a clean and affordable energy that can minimize our dependence on oil and reduce environmental pollution. Hydrogen production from water under solar light irradiation has been regarded as an ideal long-term solution to energy-related environmental problems. In 1971, Fujishima and Honda reported the generation of H2 using TiO2 under UV light irradiation.50 Since then, hydrogen generation based on various semiconductors has been extensively studied. A number of visible-light-driven photocatalysts have been prepared by dye-sensitization including synthetic dyes and natural dyes extracted from plants. Another interesting way to produce hydrogen is to use hydrogenases directly as the plants do in photosynthesis. It was also found that coupling hydrogenases with noble metals could harvest the photochemically produced hydrogen in vitro. In this review, we focus on the combination of bio-molecular and metal or metal oxide photocatalysts for H2 production using hydrogenases as active centers in the hydrogenase modified semiconductor photocatalysts.51 Readers interested in other methods can refer to a series of review articles about semiconductors,52–54 dye-sensitized semiconductors and hydrogenases system.55,56Hydrogenases represent an alternative approach for the production of hydrogen (H2). The two main classes of hydrogenases are known as [NiFe]- or [FeFe]-hydrogenases depending on the metal ion content of their catalytically active sites. Coordination of iron or iron and nickel by thiolates, CO and CN ligands at the active site forms an extremely efficient catalyst.56,57 Combination of hydrogenases with semiconductors is a promising alternative photocatalyst for H2 production. For Pt as active center for Pt doped semiconductor catalysts, there are several drawbacks: (i) lack of selectivity (at low potentials it catalyses O2 reduction at diffusion controlled rates), (ii) poisoning by trace inhibitors (e.g., CO) which is difficult to reverse, and (iii) high cost/limited resources, in particular considering large-scale applications.58 There are obstacles to be overcome for using hydrogenases including the coupling of the light-induced charge separation and the multi-electron catalytic processes leading to H2 production, the cost of the catalyst, its instability and the lifetime of the reaction.51
Since the early 1980s, different hydrogenases have been coupled with semiconductors for catalyzing the reaction 2H+ + 2e− = H2. It was reported that the hydrogenases could efficiently promote hydrogen evolution in these types of systems. The photobiocatalytic hydrogen processes are shown in Fig. 9.59 Methyl viologen (MV2+) is used as an electron mediator. The photogenerated electrons from semiconductors are transferred to the active sites of hydrogenase through MV2+ for H2 evolution.60,61
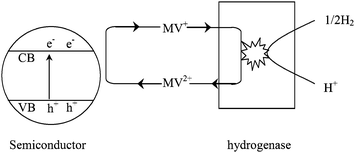 | ||
| Fig. 9 Photobiocatalytic hydrogen production using semiconductors combined with bacterial hydrogenases.59 | ||
In the early studies, various hydrogenases were added to an aqueous suspension of semiconductor powders for photochemical hydrogen formation.62–64 Hydrogenase was adsorbed by TiO2 by hydrogen bonding or electrostatic interactions. The rate of hydrogen formation for TiO2 and hydrogenase isolated from the phototrophic bacteria Thiocapsa roseopersicina generated H2 at a rate of <0.05 µL H2 min−1 in the absence of MV2+, indicating a direct electron transfer from the semiconductor to the active site of the enzyme.65 The addition of 0.5 mM of MV2+ increased the rate of H2 production to 3.1 µL H2 min−1. It was further increased to 5.0 µL H2 min−1 by adding 1.6 mM of Ca2+ indicating that the Ca2+ increased hydrogenase adsorption on TiO2.66
Different bacteria (Rhodopseudomonas capsulate, Rhodospirillum rubrum and Escherichia coli) were investigated for photobiocatalytic production of hydrogen in the presence of Bi2O3 semiconductor, MV2+ as an electron mediator. It was found that the presence of carbohydrates (fructose, dextrose and starch) and organic acids (oxalic acid, EDTA and ascorbic acid) as electron donors with the above system could enhance hydrogen production. Addition of divalent metal ions to the semiconductor/MV2+/donor/bacterial system also increased the hydrogen formation efficiency. Maximum efficiency was observed for Mn2+. The reason was that proteins had a tendency to bind with metal ions and Mn2+ acted as a potent activator of enzyme.59
Pedroni extracted tetrameric hydrogenase from the hyperthermophilic archaeon Pyrococcus furiosus and adsorbed it onto TiO2. The rate of hydrogen production reached 11 µM min−1 mg−1 with an electron donor of sodium dithionite even in the absence of MV2+. The reaction lifetime of using hyperthermophilic archaeon at 60 °C was prolonged to 8 h, and it would be beneficial for large amounts of H2 generation.67
The adsorption and correct orientation of the hydrogenases on the semiconductor surface, their structure features, as well as their proximity to the surface of the powder, are important considerations in H2 evolution processes. In the absence of MV2+, Clostridium pasteurianum was found to be more efficient than P. furiosus and Acetomicrobium flavidum. This indicated effective electron transfer from TiO2 to C. pasteurianum. With the addition of MV2+ as an electron carrier, P. furiosus hydrogenase catalyses gave the highest amount of hydrogen production. This suggested the availability of highly active sites in the P. furiosus hydrogenase.68
Gurunathan fabricated visible-light-driven bio-metal oxide photocatalysts. TiO2 was coupled with intact bacterial cells of R. capsulate species. Sensitization of TiO2 was induced in three ways: (1) using organic dyes, (2) using Cu(II) ion doping, and (3) loading with a low-band gap semiconductor (CdS). Among the catalyst systems investigated, Cu(II)/TiO2 with Mn2+ and MV2+ produced the highest amount of hydrogen (1.8 mL h−1), followed by Rhodamine B sensitized TiO2 (1.6 mL h−1).69
One of the major limitations for the development of solar powered bio-H2 production is the O2-sensitivity of hydrogenases. Recently, Armstrong and co-workers fabricated a new kind of [NiFeSe]-hydrogenase. One of the terminal cysteine ligands to the nickel in the [NiFe] active site was replaced by selenocysteine, resulting in the subclass known as [NiFeSe]-hydrogenases (Fig. 10). Conventional [NiFe] and [FeFe] hydrogenases, in addition to catalyzing H2/H+ cycling, also react with O2, resulting in inactivation due to oxidation and introduction of oxygen-derived species at the active sites.71 The [NiFeSe]-hydrogenases reacted with O2, reversibly, causing inactivation. However, the rates of reactivation for inactive [NiFeSe] increased to about 102 fold, corresponding to inactive [NiFe]. It was also found that the presence of an H2 atmosphere was not a requirement to complete the reactivation of the [NiFeSe]-hydrogenases at very negative potentials. The excellent performance of [NiFeSe]-hydrogenases was mostly related to the weaker chemical bonds formed by selenium compared with sulfur. Firstly, a selenol group is more acidic than a thiol (the pKa of SeCys is 5.2, whereas that of the cysteine thiol is 8.0). This implies that a SeCys can more readily donate a proton to an acceptor in its vicinity, and thus favour H2 production at the hydrogenase active site. Secondly, in regard to the reaction with O2, a Se–O bond should be weaker than a S–O bond, so any oxygen-bound inactive state of the [NiFe] active site should be more easily restored to its normal form in a selenium-containing hydrogenase and this might then provide a protective mechanism and some degree of O2 tolerance.70
![Comparison of the structures of [NiFe]- and [NiFeSe]-hydrogenase active sites. The Desulfovibrio gigas structure corresponds to an as-prepared oxidized form of a [NiFe]-hydrogenase with a putative bridging (hydro)peroxo ligand with 70% occupancy. This is the unready or Ni–A form of enzyme. The [NiFeSe]-hydrogenase structure corresponds to a H2-reduced form of the enzyme and it is in an active state.70](/image/article/2011/EE/c0ee00162g/c0ee00162g-f10.gif) | ||
| Fig. 10 Comparison of the structures of [NiFe]- and [NiFeSe]-hydrogenase active sites. The Desulfovibrio gigas structure corresponds to an as-prepared oxidized form of a [NiFe]-hydrogenase with a putative bridging (hydro)peroxo ligand with 70% occupancy. This is the unready or Ni–A form of enzyme. The [NiFeSe]-hydrogenase structure corresponds to a H2-reduced form of the enzyme and it is in an active state.70 | ||
As shown in Fig. 11 [NiFeSe]-hydrogenases were able to produce H2 under visible light irradiation from a hybrid enzyme–TiO2 nanoparticle system with a co-attached synthetic Ru photosensitizer. The turnover numbers were 1.9 × 105 and 100 for [NiFeSe]-hydrogenases and RuP, respectively, after 8 h irradiation with RuP–TiO2 (5 mg, 0.1 µmol RuP) and [NiFeSe]-hydrogenases (10 µL, 5 µM solution, pH 7.0) suspended in EDTA/Tris buffer (4.5 mL).58 As compared to several hydrogenases, Chlamydomonas reinhardtii [FeFe]-H, E. coli [NiFe]-Hyd-1, Ralstonia eutropha [NiFe], Clostridium acetobutylicum [FeFe]-H, Allochromatium vinosum Miyazaki F (Av) [NiFe]-MBH, and synthesized Desulfomicrobium baculatum, the [NiFeSe]-hdrogenase showed a higher activity.72
![Schematic representation of visible-light driven H2 evolution with RuP–TiO2–H2ase, i.e., [NiFeSe]-H2ase attached to RuP dye sensitized TiO2 nanoparticles. Upon excitation by visible light in the presence of a sacrificial electron donor D, RuP injects an electron into the conduction band of TiO2. The electrons are transferred directly to the adsorbed [NiFeSe]-H2ase, thereby reducing H+ from the buffered aqueous solution. The three [4Fe4S]-clusters (responsible for electron transfer to and from the protein active site) and the active site of [NiFeSe]-H2ase are shown in the protein structure at the top left, respectively, and the structure of the sensitizer RuP is shown at the top right.58](/image/article/2011/EE/c0ee00162g/c0ee00162g-f11.gif) | ||
| Fig. 11 Schematic representation of visible-light driven H2 evolution with RuP–TiO2–H2ase, i.e., [NiFeSe]-H2ase attached to RuP dye sensitized TiO2 nanoparticles. Upon excitation by visible light in the presence of a sacrificial electron donor D, RuP injects an electron into the conduction band of TiO2. The electrons are transferred directly to the adsorbed [NiFeSe]-H2ase, thereby reducing H+ from the buffered aqueous solution. The three [4Fe4S]-clusters (responsible for electron transfer to and from the protein active site) and the active site of [NiFeSe]-H2ase are shown in the protein structure at the top left, respectively, and the structure of the sensitizer RuP is shown at the top right.58 | ||
4. Photocatalysis
Solar-driven semiconductor-mediated photocatalysis is an attractive environmental remediation technology because it makes use of the clean, safe, and renewable sunlight to solve environmental problems.9Fig. 12 shows a typical photocatalysis process involving TiO2 as the photocatalyst.9 Excitation of semiconductors with suitable light causes the formation of negative electrons and positive holes. Then the formed electron–hole pairs migrate to the semiconductor surface. The holes are thought to induce various redox processes at the semiconductor/solution interface.9 However, the lifetime of the electron–hole pairs is very short. The photogenerated electrons and holes recombine and dissipate the input energy as heat very quickly, which is the main problem that limits the photocatalysis efficiency. A promising solution is to derivatize the surface of the semiconductor with some trapping materials to inhibit the recombination. Various organic molecules having functional groups including hydroxyl, carboxyl, silanyl, and phosphonate groups have been demonstrated as efficient trapping molecules.73 Most biomolecules contain such functional groups, including amino acids, sugars and biopolymers. Thus surface modification by biomolecules has been widely used to enhance photocatalytic efficiency.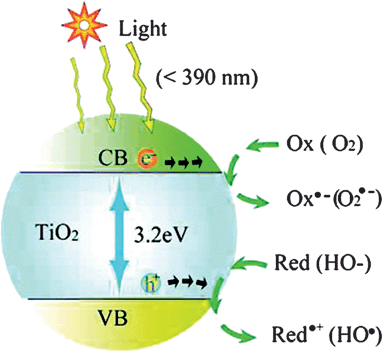 | ||
| Fig. 12 Photoexcitation of a semiconductor (e.g., TiO2) and the subsequent generation of an electron and hole, which are trapped by an oxidant (Ox) and a reductant (Red), respectively. For TiO2 photocatalysis, the “Ox” is a surface-adsorbed oxygen molecule and the “Red” is a surface-bound hydroxyl group.9 | ||
4.1. Amino acids
Amino acid modified semiconductor photocatalysts have attracted extensive interest. Enhanced photoreduction of lead ions was observed by using cysteine modified TiO2 nanoparticles in the study of Rajh et al.74 Based on the findings from electron paramagnetic resonance and infrared spectra, they proposed a mechanism as shown in Fig. 13. For unmodified TiO2, upon excitation, photogenerated electrons were trapped at metal centers as Ti(III), while the holes were trapped on the surface OH groups as a (TiO2)nTiIVO˙. Recombination of electrons and holes occurred easily as the separation distance was short. However, in the case of cysteine-modified TiO2, the holes were trapped on cysteine to form carboxyl radicals, while the electrons were trapped on the TiO2 particle. The resulting increased separation distance inhibited the electron/hole recombination. Cysteine-capped ZnS, CdS nanocrystals also showed high photocatalytic activity.75,76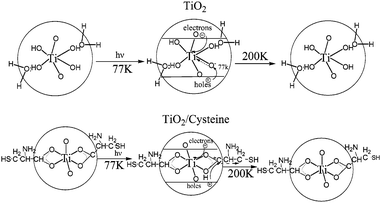 | ||
| Fig. 13 Schematic representation of structure and electron transfer reactions in (a) TiO2 and (b) TiO2/cysteine.74 | ||
Makarova et al. reported arginine-modified TiO2 nanoparticles with enhanced adsorption and photodecomposition of nitrobenzene (NB) as shown in Fig. 14a.77 They found that the electron-withdrawing nature of the nitro group of NB provided an electron deficient aromatic ring to attract the electron-rich surface modifier arginine. Such interactions improved the coupling between nitrobenzene and TiO2, and facilitated the transfer of photogenerated electrons from the TiO2 conduction band to the adsorbed NB. The quantum yield for NB photodegradation of arginine-modified TiO2 nanoparticles was 0.31, as compared to the value of 0.15 for unmodified TiO2 and 0.18 for Degussa P25. As an efficient hole trapper, methanol was added into the system to further enhance photoactivity. The arginine-modified TiO2 nanoparticles also exhibited selective photocatalytic activity for the decomposition of nitrobenzene.78 Since the amine groups of arginine had different charges according to the solution pH, the effect of pH values on the photocatalytic efficiency had been investigated by degrading 4-nitrophenol. Results showed that the optimal pH was 9, where the positively charged amine groups of arginine effectively coupled with negatively charged 4-NP.79
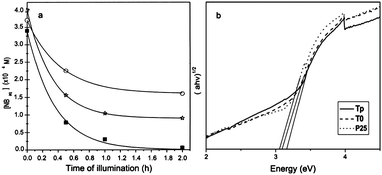 | ||
| Fig. 14 (a) Photocatalytic degradation of NB vs. time of illumination over bare (circles) and arginine-modified (square) nanocrystalline TiO2 and Degussa P25 TiO2 (stars).77 (b) UV-vis diffuse reflectance spectra of the sample P25, pure TiO2 (T0), and functionalized TiO2 nanorods (Tp).80 | ||
In another case, a nonaqueous sol–gel route was developed to prepare L-hydroxyproline functionalized anatase TiO2 nanorods.80 The L-hydroxyproline functionalization results in enhanced surface area and larger energy band gap (Fig. 14b). Accordingly, the photocatalytic activity was higher than uncoated counterparts. They summarized that the enhancement was caused by three reasons: the larger surface area to absorb more hydroxyl radicals, the presence of hydroxyproline as the inhibitor for electron–hole recombination, and larger band gap with more powerful redox ability.
Histidine and alanine were also used to modify TiO2 nanoparticles.81 The Fermi level of modified TiO2 was found to shift towards negative potentials at least 400 mV to achieve efficient photoreduction of cadmium ions.
4.2. Biopolymers
Cellulose (C6H10O5)n is the most abundant natural biopolymer on earth. It is a polysaccharide consisting of a linear chain of several hundred to over ten thousand of glucose units.82 Its typical morphology is nanofiber with around several hundred nanometres in length and cross-sectional dimensions of 10 nm, resulting in the large specific surface area.82 Anchoring a photocatalyst on supporting materials of large surface area, on which pollutants would be accumulated, can obviously enhance photodecomposition efficiency. Another advantage is the ease of recovering a large composite material compared to the fine photocatalyst particles.Fig. 15 shows the immobilization of P25 TiO2 nanoparticles onto the paper by the bioconjugation of a bacterial cellulose.37 Bacterial cellulose nanofibers were biosynthesized by Acetobacter xylinum NUST5.2. A layer of water molecules was covered on the surface of cellulose nanofibers by hydrogen bonds. The hydrolysis of Ti(OBu)4 at the ordered water layer of the BC nanofibers caused the formation of uniform hybrid nanofiber structures. The BC/TiO2 hybrid nanofibers were used to photodegrade methyl orange under UV irradiation, and they showed higher efficiency than that of the P25.
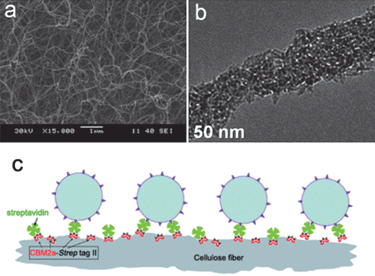 | ||
| Fig. 15 (a and b) SEM and TEM images of bacterial cellulose/TiO2 hybrid nanofibers.37 (c) Immobilization of P25 TiO2 nanoparticles onto paper by the bioconjugation of fiber–CBM2a–Strep-tag II–streptavidin–biotinylated TiO2 nanoparticles.86 | ||
An interesting Au/TiO2/cotton cellulose nanocomposite textile was prepared at low temperature by the sol–gel process.83 The Au/TiO2 nanoparticles formed a homogeneous thin film on the fibre surface. The resulting materials displayed a purple colour and were photocatalytically active under solar light.
Paper is made of cellulose fibers. The immobilization of photoactive TiO2 nanoparticles onto cellulose fibers provides the paper self-cleaning property.84 A possible approach to prepare such paper is to make the TiO2 nanoparticles surface positively charged by adjusting the pH of precursor solution.85 Then the electrostatic attraction force between TiO2 and negatively charged fibers produces the TiO2-containing paper. Also some additives, such as cationic polymers,84 have been used as bridges to combine negatively charged nanoparticles and fibers together. Recently TiO2 nanoparticles have been immobilized on the paper by a bioconjugated method. As indicated in Fig. 15c, cellulose fibers are modified by CBM2a–Strep-tag II protein and then streptavidin.86 Finally the attachment of biotinylated TiO2 nanoparticles to streptavidin through the biotin–streptavidin interaction causes the formation of the bioconjugated system. It has been found that TiO2 nanoparticles are uniformly immobilized onto paper surface and the bioconjugated paper is more resistant to UV irradiation damage. This paper exhibits efficient photodegradation ability for anionic reactive black 5 dyes.
As the second most abundant natural biopolymers next to cellulose, chitosan, β-(1,4)-2-amino-2-deoxy-D-glucosamine, is a kind of polysaccharide derived from the alkaline deacetylation of chitin.87 The biopolymer contains a large amount of amino (NH2) and hydroxyl (OH) functional groups, which can serve as coordination and reaction sites. Numerous studies have demonstrated the effectiveness of chitosan and derived products for the absorption of metal cations and organic pollutants.87 The high absorption favours the high photocatalytic efficiency. Also the mechanical property of chitosan is very good, which facilitates the recovery of composite photocatalyst.
Fig. 16a and b show a highly ordered 3D macroporous TiO2/chitosan scaffold prepared by ice segregation induced self-assembly method.88 The morphology and microstructure of the composite are controllable by varying solute concentrations and freezing rate. The enhanced photocatalytic activity is demonstrated by degrading methylene blue and Orange II. It has been found that the composite is of excellent mechanical property, which makes it a reusable photocatalyst. Zainal et al. used chitosan as binder to anchor the reactive TiO2 onto the smooth surface of glass plates.89 They proposed a photodegradation–adsorption process to explain the enhanced photocatalytic activity. Based on the findings of FTIR study, the reactive –NH2, –OH, and metal oxide contents in the prepared photocatalyst were responsible for the photodegradation–adsorption effect. Torres et al. prepared Nb2O5/chitosan composite with different semiconductor loadings.90 Nb2O5 was homogeneously dispersed on the surface of the composites. Although the thermal stabilities of the composites were slightly weaker than that of the pure biopolymer, the materials exhibited very high catalytic efficiencies after 15 cycles of reuse.
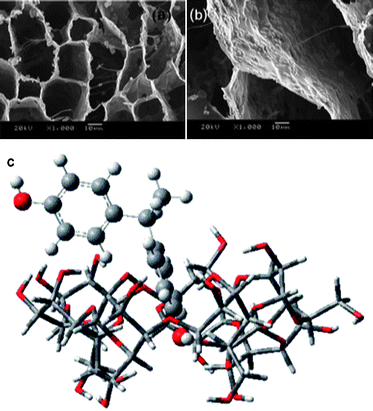 | ||
| Fig. 16 (a and b) High magnification SEM images of ordered 3D macroporous TiO2/chitosan sponges.88 (c) Proposed structure of the bisphenol E/CD inclusion complex.99 | ||
Chitosan has also been used to assist the visible light photocatalysis. Zhu et al. presented the crosslinked chitosan/CdS nanocomposite catalyst prepared by simulating biomineralization process.91 The effects of the photocatalyst amount, initial dye concentration, the solution pH and the co-existing anions on the photocatalytic activity were investigated. And they found that the photocatalytic efficiency in acidic media was higher than alkaline media. The presence of NO3− accelerated the photoreaction. In the same system, Jiang et al. examined the recycle and reuse of the catalyst.92 And results showed that photocatalytic efficiency could retain 80% after 60 min of reaction time when the catalyst was used for 5 times. In another case, needle shaped Cu2O/chitosan nanocomposites showed enhanced photocatalytic activity under visible light.93
Cyclodextrins (CDs), produced from starch by means of enzymatic conversion, are macrocyclic oligosaccharides commonly composed of 6, 7, or 8 glucosidic units bearing the names α-, β-, and γ-CD, respectively. They form a structure with the hydrophobic internal cavity and hydrophilic external surface.94 Based on the unique structure, various semiconductor/cyclodextrin hybrid materials with different morphologies have been developed. Feng et al. reported a self-assembly approach to fabricate photoactive TiO2–cyclodextrin wires by using CDs as bifunctional linkers.95 Ma et al. described three-dimensionally ordered macroporous titanium phosphonate frameworks functionalized by β-cyclodextrin.94 Li et al. synthesized the anatase TiO2 nanoparticles with β-cyclodextrin as a supramolecular shell.96
The special molecular structure allows cyclodextrins to form host/guest inclusion complexes with various guest molecules including both organic and inorganic complexes, which is helpful for photocatalysis. β-CD has been found to be beneficial to improve the kinetics for charge transfer from the photoexcited semiconductor to electron acceptors absorbed in the cavity.97 By using low-temperature EPR and cyclic voltammetry, Dimitrijevic and co-workers revealed the localization of valence band holes at carboxyl groups of surface-conjugated cyclodextrin and conduction band electrons at lattice Ti atoms.98
Wang et al. studied the inclusion of bisphenol E molecules into the CD cavity as shown in Fig. 16c.99 They proposed that the moderate inclusion-depth allowed sufficient proximity of bisphenol E to catalytically activate the secondary hydroxyl groups of the CD. Thus they observed the enhanced photodegradation of bisphenol E under UV light. And the photodegradation rate depended on CD concentration, pH and BPE initial concentration. Lu et al. investigated the photocatalytic oxidation of dyes and reduction of Cr(VI) in aqueous solutions by CD modified TiO2.100 For azo dyes, the photodegradation efficiency was increased by three to seven times. And the photocatalytic reduction rate for Cr(VI) was enhanced by 12–17 times. The β-CD/TiO2 hybrid material was also synthesized through a photo-induced self-assembly method.101 The catalytic degradation of paracetamol was significantly enhanced under visible light irradiation. A sonochemical method was explored to synthesize porous Cu2O nanospheres by using ascorbic acid as the reducing agent and β-CD as the capping agent.102
4.3. Other biomolecules
The modification of photocatalyst with some other small biomolecules has also been investigated. Rajh et al. carried out a preliminary investigation on the optical and charge separation properties of nanocrystalline TiO2 by surface modification with ascorbic acid.103 As shown in Fig. 17a, the binding of modifiers on the surface of TiO2 nanocrystals induced a 1.6 eV of red shift in the onset of absorption, compared to unmodified nanocrystallites. They explained that electron–hole pairs were divided into two phases: the holes on the donating organic modifier and the electrons in the conduction band of TiO2. A similar red shift was also reported by Ou and co-workers.104 In their study, the photocatalytic activity was measured by degrading methyl orange. The enhanced photocatalytic efficiency was observed upon UV and solar light irradiation.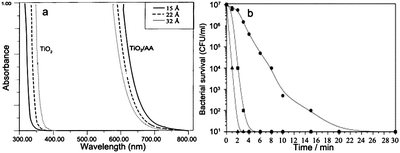 | ||
| Fig. 17 (a) Absorption spectra of 0.05 M TiO2 nanoparticles with different sizes before and after surface modification with ascorbic acid.103 (b) Inactivation of E. coli under UV illumination over catalyst systems. (■) Ag–TiO2, (●) 1 wt% Ag–HAP, and (▲) 5 wt% Ag–TiO2/HAP.107 | ||
Citric acid (2-hydroxy-propane-1,2,3-tricarboxylic acid) is a strong chelating agent for many heavy metal ions. It is also employed as a good donor scavenger to deplete the photogenerated holes from TiO2-based catalysts.105,106 As a result, the photoreduction of Cr(VI) is enhanced.
Some inorganic biomolecule/semiconductor composites have also been reported. Hydroxyapatite (HAP, Ca10(PO4)6(OH)2) is the major inorganic component in natural bones and teeth.107 Due to the excellent absorption towards organic molecules, TiO2 coupled with HAP has been developed to improve the photocatalytic decomposition activity against bacteria, virus, biohazardous substances and chemicals. Yang and Zhang employed the quartz crystal microbalance technique to investigate the adsorption and degradation behavior of bilirubin on HAP-modified titania coatings.108 They found that the optimal content for HAP in the coating was 0.7 wt%. The high bacterial adsorption of different bacteria onto HAP made the HAP-supported photocatalyst a great disinfection property. As shown in Fig. 17b, Reddy et al. reported that the photocatalytic properties of hydroxyapatite-supported Ag–TiO2 composite resulted in 100% E. coli bacteria reduction within 2 min.107 Park demonstrated the HAP-precipitated potassium titanate whiskers as promising candidate photocatalysts for environmental purification.109
4.4. Chlorophyll-based photocatalyst
To increase the photocatalytic activity, a primary requirement is to enhance the photoabsorption in the visible light region. In the natural world, chlorophylls are the most efficient molecules to absorb visible light. Furthermore, in photosynthesis chlorophylls are excited by visible light, and then transfer the absorbed energy to the photoreaction center and trigger a series of redox reactions, which is very similar to the role of photocatalyst in photocatalysis. So researchers have demonstrated chlorophyll-based photocatalysts. To increase the photostability, chlorophylls are immobilized in mesoporous nanomaterials functionalized by monoethanolamine (MEA).110 Four different mesoporous matrices are tried, including silica gel, meso-alumina, meso-titania and MCM-41. And the MCM-41/MEA matrix shows the highest chlorophyll immobilization. As indicated in Fig. 18a, the obtained photocatalyst shows significant absorption in visible light. The novel photocatalyst displays a significant photocatalytic activity towards methyl orange under visible light. The possible mechanism is shown in Fig. 18b. Chlorophylls are excited to produce complex (MCM-41/MEA/Chl*) by sunlight. Then the excited chlorophylls release the electron and generate cationic form (MCM-41/MEA/Chl+). The released electron is responsible for the reduction of MO. Ethanol present in the solution supplies electron to the cationic form to reproduce MCM-41/MEA/Chl.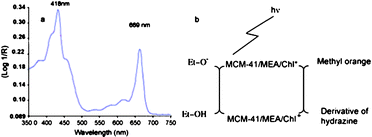 | ||
| Fig. 18 (a) Diffuse reflectance spectra of chlorophyll adsorbed onto MCM-41/MEA. (b) Schematic illustration for the possible mechanism for the photoreduction using MCM-41/MEA/Chl.110 | ||
5. Artificial photosynthesis
Photosynthesis is a process that converts light to chemical energy by plants. It is vital for life on earth because the solar energy that is stored in the form of coal, oil and natural gas, and that is required by the biological world is generated from the photosynthesis process. Photosynthesis captures approximately 100 terawatts every year, which is about six times larger than the energy consumption of human civilization.111 Thus it is considered as the most efficient biological machine in nature.112Fig. 19 shows the typical photosynthetic process.113 Light is captured in photosystem II (PSII) by the antenna complex composed of chlorophylls, accessory pigments and proteins. Then the excitation energy is transferred to the photosynthetic reaction center, photosystem I (PSI), to trigger a series of redox reactions to produce organic compounds. During the process, the solar energy is transferred to chemical energy and the crucial oxygen for human is released. The quantum yield for absorbing a photon within the whole process has been determined to be close to 100%.112 With the deep investigation on the mechanism, this process is no longer nature's secret. Researchers, including biologists, chemists, and physicists, have put much effort to mimicking the process.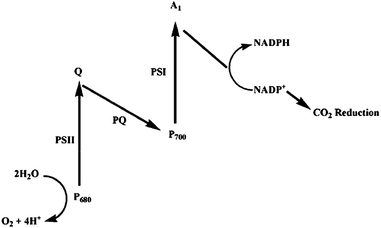 | ||
| Fig. 19 Z-Scheme of the photosynthetic processes at chloroplasts. The reaction centres PSI and PSII feature light harvesting units denoted as P680 and P700. The primary electron acceptor and shuttle between the reaction centres involve quinone species (Q, PQ).113 | ||
According to the natural model, the primary requirement for an artificial photosynthetic system is to construct PSII to harvest sunlight. In the natural system, chlorophyll molecules bind to proteins to form stable chlorophyll–protein conjugates. However, chlorophyll extracted from living organisms has rarely been used in artificial photosynthetic systems because of its instability.114 It was found that chlorophylls could be stabilized by hybridization with inorganic hosts such as clays or zeolites. In addition, there have been some reports concerning the immobilization of chlorophylls by incorporation into mesoporous silica materials. Nanoscale space in pores provides hosting sites to include chlorophyll molecules.
Folded silica mesoporous material (FSM) has a honeycomb (hexagonal) structure with ordered cylindrical channels of 2–10 nm in diameter. The chlorophyll–FSM conjugate has been prepared based on the interaction between chlorophyll molecules inside the mesoporous spaces of FSM.114,115 The amount of the adsorbed chlorophylls is dependent on the pore size. The larger pore diameter is corresponding to higher chlorophyll content, longer absorption maximum wavelength and enhanced photostability. As shown in Fig. 20 larger pore size provides enough space for not only the interaction between chlorophyll and the supporting silica materials, but also the interaction between neighboring chlorophyll molecules, which is very important to enhance performance. The energy and electron transfer in the conjugate are confirmed.116 The chlorophyll–FSM conjugate catalyzes the photoreduction and hydrogen gas production under visible light. By using the same approach, functional membrane protein complex, light-harvesting protein LH2, has been introduced into FSM.117
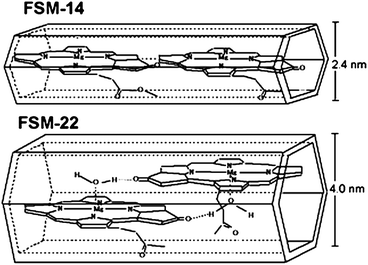 | ||
| Fig. 20 Tentative arrangement in chlorophyll–FSM.114 | ||
Besides these fundamental studies, some operational artificial photosystems have been fabricated recently. For example, thylakoids, photosynthetic sub-cellular plant structures, have been entrapped into porous silica network by using a sol–gel approach.118 To examine the photosynthesis efficiency, oxygen generation has been investigated as shown in Fig. 21. The bioactivity of the entrapped thylakoids can be extended to several weeks, while the free thylakoid suspension loses activity within several days. The bioactivity is influenced by the pore volume of the silica matrix, corresponding to the different silica precursor concentrations.
![Comparison of enzymatic stability (10 °C) of entrapped thylakoids within different ratios of silica gels to free thylakoids. The gels are synthesized with different silica precursor concentrations: HG1: [Si]sol = 1.0 M; HG2: [Si]sol = 1.2 M; HG3: [Si]sol = 1.6 M. Time zero corresponds to the moment at which thylakoids have been isolated.118](/image/article/2011/EE/c0ee00162g/c0ee00162g-f21.gif) | ||
| Fig. 21 Comparison of enzymatic stability (10 °C) of entrapped thylakoids within different ratios of silica gels to free thylakoids. The gels are synthesized with different silica precursor concentrations: HG1: [Si]sol = 1.0 M; HG2: [Si]sol = 1.2 M; HG3: [Si]sol = 1.6 M. Time zero corresponds to the moment at which thylakoids have been isolated.118 | ||
Photosynthesis begins with solar energy absorption by chlorophylls. As such, enhancing the photoabsorption ability of chlorophylls can increase the photosynthesis efficiency. By coupling with inorganic nanoparticles, especially metallic and semiconductor nanoparticles, the fluorescence of organic dyes has been enhanced.119 Plasmon excitations in a nanoparticle lead to the amplification of electromagnetic fields in the vicinity of the dye molecules, resulting in the enhanced emission and absorption. As well, chlorophylls have been involved in the hybrid system. In the PSI/CdTe nanoconjugate, an enhancement factor as high as 100 has been observed.120 The great enhancement results from the combined effect of the building blocks. The semiconductor nanoparticles strongly absorb light, while the PSI efficiently separates electrons and holes. Compared with the natural photosynthetic systems, the hybrid can harvest light energy in a much broader spectral range. Besides, some metallic nanoparticles have also been used to enhance the absorption of chlorophylls.121
Some practical examples focusing on the effect of semiconductor nanomaterials on the plant growth have been reported. In a preliminary work, the effects of TiO2 nanoparticles on the growth of spinach seeds were investigated.122 It was reported that the germination rate of the seeds treated with TiO2 was higher than the non-treated samples. And the chlorophyll content, the ribulosebisphosphate carboxylase/oxygenase activity, and the photosynthetic rate were increased. To further study the effect of TiO2 on the growth, spinach was treated with TiO2 suspensions.123 As shown in Fig. 22, the spinach with TiO2 nanoparticles treatment grew faster because the leaves were larger and greener than the nontreated and bulk TiO2 treated spinach. During the growth process, the fresh weight, dry weight, contents of total nitrogen, chlorophyll, and protein of spinach by TiO2 treatment were found to be highly enhanced compared with control samples. However, under N-deficient conditions, the improvement was not very obvious. Thus the improvement was considered to be related to N2 photoreduction caused by TiO2. Upon light irradiation, N2 was absorbed by TiO2 and then photoreduced to NH3, and then transformed to organic nitrogen to improve spinach growth. To further study the mechanism, PSII was extracted from spinach leaves and then treated with TiO2.124 Then the effect of TiO2 treatment on the energy transfer in PSII was investigated by spectroscopy and on oxygen evolution. They found the PSII microenvironment was changed along with the enhanced visible light absorption. Also the energy transfer in PSII was improved. The improvement resulted in the higher photosynthesis rate and enhanced growth.
 | ||
| Fig. 22 Effect of nano-anatase TiO2 on growth of spinach. (a) Cultured by Hoagland solution. (b) Cultured by N-deficient Hoagland solution.123 | ||
6. Other applications
Photodynamic therapy (PDT) is a technique that uses drugs along with light to kill cancer cells. The known mechanism is that upon light irradiation of the drugs, which are photosensitizers, they produces a large amount of highly reactive singlet oxygen (RSO), which causes the destruction of cancer cells.125 Due to the noninvasive property, PDT is considered as a promising treatment for many kinds of diseases. The conventional PDT drugs are organic molecules. Also, inorganic semiconductor nanocrystals have been widely used in PDT because of excellent capability for producing RSO.125 However, photoactivation of the PDT by using pure CdSe quantum dots provides a very low efficiency for RSO production.125 The reason is the recombination of electron/hole pairs, which is similar to the phenomenon as discussed in photocatalysis part. The toxicity of pure inorganic nanocrystals is another problem. To overcome the said issues, inorganic nanocrystals coupled with various kinds of biomolecules have been developed including peptides,126 avidin,127etc. Compared with the conventional organic drug systems, bioconjugated inorganic semiconductors show better photostability and broader absorption cross-section. Besides the combination of inorganic semiconductors provides PDT some unique properties. For example, magnetic Fe3O4 nanoparticles/photosensitizer/chitosan, a good drug delivery system, could be used for PDT, magnetic resonance imaging (MRI).128Global warming, the most serious environmental problem of our time, is mainly caused by the increasing amount of carbon dioxide (CO2) released from the burning of fossil fuels. An attractive solution to global warming is to convert CO2 to valuable hydrocarbon fuels.129 In recent years, the photoreduction of CO2 by using semiconductor photocatalysts has been widely studied. The products include CO, HCOOH, HCHO, CH3OH, and CH4.129,130 Very recently, an interesting enzyme–nanoparticle hybrid system involving TiO2 modified by CO2-reducing enzyme CODH I and a photosensitizer has been reported.131 As shown in Fig. 23, upon visible light irradiation, the photosensitizer was reported to produce an electron, which was injected into the conduction band of TiO2, and then to CODH I. Then CO2 was reduced to CO. A two-electron pathway was proposed to explain the high activity of producing CO.
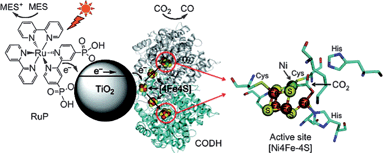 | ||
| Fig. 23 A schematic representation of the CO2 photoreduction system using Ch CODH I attached to RuP-modified TiO2 NPs. A catalytic intermediate of the active site of the closely related enzyme CODH II with bound substrate (CO2, indicated with an arrow) is also shown. The oxidized photosensitizer is recovered by the sacrificial electron donor MES. The enzyme structure used in the cartoon is CODH II, created using PyMOL.131 | ||
7. Conclusions
In response to the increasing demand for solar energy applications, various novel semiconductor nanomaterials have been developed along with the conventional silicon-based solar conversion devices. Many interesting ideas have been proposed to enhance the efficient use of sunlight. In this perspective, we have reviewed the promising bioconjugated semiconductor hybrid systems. Environmental friendliness and low toxicity are the apparent advantages of the conjugated systems, and these are very important considerations for future commercial applications.Acknowledgements
This work was supported by the Group Research Scheme 2009-10 and the Strategic Investments Scheme administrated by The Chinese University of Hong Kong.References
- J. Nowotny, Energy Environ. Sci., 2008, 1, 565–572 RSC.
- J. Baxter, Z. X. Bian, G. Chen, D. Danielson, M. S. Dresselhaus, A. G. Fedorov, T. S. Fisher, C. W. Jones, E. Maginn, U. Kortshagen, A. Manthiram, A. Nozik, D. R. Rolison, T. Sands, L. Shi, D. Sholl and Y. Y. Wu, Energy Environ. Sci., 2009, 2, 559–588 RSC.
- X. G. Zhu, S. P. Long and D. R. Ort, Curr. Opin. Biotechnol., 2008, 19, 153–159 CrossRef CAS.
- G. Calzaferri, Top. Catal., 2010, 53, 130–140 CrossRef CAS.
- L. M. Goncalves, V. D. Bermudez, H. A. Ribeiro and A. M. Mendes, Energy Environ. Sci., 2008, 1, 655–667 RSC.
- Y. Inoue, Energy Environ. Sci., 2009, 2, 364–386 RSC.
- M. D. Hernandez-Alonso, F. Fresno, S. Suarez and J. M. Coronado, Energy Environ. Sci., 2009, 2, 1231–1257 RSC.
- A. M. Smith, H. W. Duan, A. M. Mohs and S. M. Nie, Adv. Drug Delivery Rev., 2008, 60, 1226–1240 CrossRef CAS.
- X. L. Hu, G. S. Li and J. C. Yu, Langmuir, 2010, 26, 3031–3039 CrossRef CAS.
- G. S. Li, D. Q. Zhang and J. C. Yu, Environ. Sci. Technol., 2009, 43, 7079–7085 CrossRef CAS.
- T. Kida, R. Oshima, K. Nonaka, K. Shimanoe and M. Nagano, J. Phys. Chem. C, 2009, 113, 19986–19993 CrossRef CAS.
- B. Oregan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef CAS.
- A. S. Polo, M. K. Itokazu and N. Y. M. Iha, Coord. Chem. Rev., 2004, 248, 1343–1361 CrossRef CAS.
- R. Argazzi, N. Y. M. Iha, H. Zabri, F. Odobel and C. A. Bignozzi, Coord. Chem. Rev., 2004, 248, 1299–1316 CrossRef CAS.
- G. P. Smestad, Sol. Energy Mater. Sol. Cells, 1998, 55, 157–178 CrossRef CAS.
- G. Calogero, G. Di Marco, S. Caramori, S. Cazzanti, R. Argazzi and C. A. Bignozzi, Energy Environ. Sci., 2009, 2, 1162–1172 RSC.
- J. R. Durrant, S. A. Haque and E. Palomares, Coord. Chem. Rev., 2004, 248, 1247–1257 CrossRef.
- S. Y. Huang, G. Schlichthorl, A. J. Nozik, M. Gratzel and A. J. Frank, J. Phys. Chem. B, 1997, 101, 2576–2582 CrossRef CAS.
- X. F. Wang, Y. Koyama, H. Nagae, Y. Wada, S. I. Sasaki and H. Tamiaki, J. Phys. Chem. C, 2008, 112, 4418–4426 CrossRef CAS.
- H. Tamiaki, R. Shibata and T. Mizoguchi, Photochem. Photobiol., 2007, 83, 152–162 CAS.
- X. F. Wang, O. Kitao, E. Hosono, H. S. Zhou, S. Sasaki and H. Tamiaki, J. Photochem. Photobiol., A, 2010, 210, 145–152 CrossRef CAS.
- X. F. Wang, O. Kitao, H. S. Zhou, H. Tamiaki and S. Sasaki, Chem. Commun., 2009, 1523–1525 RSC.
- Y. Amao, Y. Yamada and K. Aoki, J. Photochem. Photobiol., A, 2004, 164, 47–51 CrossRef CAS.
- Y. Amao and T. Komori, Biosens. Bioelectron., 2004, 19, 843–847 CrossRef CAS.
- G. Calogero and G. Di Marco, Sol. Energy Mater. Sol. Cells, 2008, 92, 1341–1346 CrossRef CAS.
- C. G. Garcia, A. S. Polo and N. Y. M. Iha, J. Photochem. Photobiol., A, 2003, 160, 87–91 CrossRef CAS.
- G. R. A. Kumara, S. Kaneko, M. Okuya, B. Onwona-Agyeman, A. Konno and K. Tennakone, Sol. Energy Mater. Sol. Cells, 2006, 90, 1220–1226 CrossRef CAS.
- N. J. Cherepy, G. P. Smestad, M. Gratzel and J. Z. Zhang, J. Phys. Chem. B, 1997, 101, 9342–9351 CrossRef CAS.
- Q. Dai and J. Rabani, J. Photochem. Photobiol., A, 2002, 148, 17–24 CrossRef CAS.
- A. S. Polo and N. Y. M. Iha, Sol. Energy Mater. Sol. Cells, 2006, 90, 1936–1944 CrossRef.
- D. Zhang, S. M. Lanier, J. A. Downing, J. L. Avent, J. Lum and J. L. McHale, J. Photochem. Photobiol., A, 2008, 195, 72–80 CrossRef CAS.
- C. Y. Qin and A. E. Clark, Chem. Phys. Lett., 2007, 438, 26–30 CrossRef.
- X. F. Wang, Y. Koyama, O. Kitao, Y. Wada, S. Sasaki, H. Tamiaki and H. S. Zhou, Biosens. Bioelectron., 2010, 25, 1970–1976 CrossRef CAS.
- J. J. Cid, J. H. Yum, S. R. Jang, M. K. Nazeeruddin, E. M. Ferrero, E. Palomares, J. Ko, M. Gratzel and T. Torres, Angew. Chem., Int. Ed., 2007, 46, 8358–8362 CrossRef CAS.
- A. Ehret, L. Stuhl and M. T. Spitler, J. Phys. Chem. B, 2001, 105, 9960–9965 CrossRef CAS.
- J. N. Clifford, E. Palomares, K. Nazeeruddin, R. Thampi, M. Gratzel and J. R. Durrant, J. Am. Chem. Soc., 2004, 126, 5670–5671 CrossRef.
- D. P. Sun, J. Z. Yang and X. Wang, Nanoscale, 2010, 2, 287–292 RSC.
- K. Wongcharee, V. Meeyoo and S. Chavadej, Sol. Energy Mater. Sol. Cells, 2007, 91, 566–571 CrossRef CAS.
- Y. Amao and Y. Yamada, Langmuir, 2005, 21, 3008–3012 CrossRef CAS.
- Y. Amao and Y. Yamada, Biosens. Bioelectron., 2007, 22, 1561–1565 CrossRef CAS.
- V. Thavasi, T. Lazarova, S. Filipek, M. Kolinski, E. Querol, A. Kumar, S. Ramakrishna, E. Padros and V. Renugopalakrishnan, J. Nanosci. Nanotechnol., 2009, 9, 1679–1687 CrossRef CAS.
- P. Bertoncello, D. Nicolini, C. Paternolli, V. Bavastrello and C. Nicolini, IEEE Trans. NanoBiosci., 2003, 2, 124–132 CrossRef.
- F. Garczarek, L. S. Brown, J. K. Lanyi and K. Gerwert, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 3633–3638 CrossRef CAS.
- L. S. Li, T. Xu, Y. J. Zhang, J. Jin, T. J. Li, B. S. Zou and J. P. Wang, J. Vac. Sci. Technol., A, 2001, 19, 1037–1041 CrossRef CAS.
- S. Barazzouk and S. Hotchandani, J. Appl. Phys., 2004, 96, 7744–7746 CrossRef CAS.
- N. Fishelson, I. Shkrob, O. Lev, J. Gun and A. D. Modestov, Langmuir, 2001, 17, 403–412 CrossRef CAS.
- T. Baum, D. Bethell, M. Brust and D. J. Schiffrin, Langmuir, 1999, 15, 866–871 CrossRef CAS.
- S. Barazzouk, P. V. Kamat and S. Hotchandani, J. Phys. Chem. B, 2005, 109, 716–723 CrossRef CAS.
- N. Terasaki, N. Yamamoto, T. Hiraga, Y. Yamanoi, T. Yonezawa, H. Nishihara, T. Ohmori, M. Sakai, M. Fujii, A. Tohri, M. Iwai, Y. Inoue, S. Yoneyama, M. Minakata and I. Enami, Angew. Chem., Int. Ed., 2009, 48, 1585–1587 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CAS.
- J. H. Alstrum-Acevedo, M. K. Brennaman and T. J. Meyer, Inorg. Chem., 2005, 44, 6802–6827 CrossRef CAS.
- R. M. N. Yerga, M. C. A. Galvan, F. del Valle, J. A. V. de la Mano and J. L. G. Fierro, ChemSusChem, 2009, 2, 471–485 CrossRef CAS.
- F. E. Osterloh, Chem. Mater., 2008, 20, 35–54 CrossRef CAS.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- W. J. Youngblood, S. H. A. Lee, K. Maeda and T. E. Mallouk, Acc. Chem. Res., 2009, 42, 1966–1973 CrossRef CAS.
- J. C. Fontecilla-Camps, A. Volbeda, C. Cavazza and Y. Nicolet, Chem. Rev., 2007, 107, 4273–4303 CrossRef CAS.
- A. A. Karyakin, S. V. Morozov, O. G. Voronin, N. A. Zorin, E. E. Karyakina, V. N. Fateyev and S. Cosnier, Angew. Chem., Int. Ed., 2007, 46, 7244–7246 CrossRef CAS.
- E. Reisner, J. C. Fontecilla-Camps and F. A. Armstrong, Chem. Commun., 2009, 550–552 RSC.
- P. Maruthamuthu, S. Muthu, K. Gurunathan, M. Ashokkumar and M. V. C. Sastri, Int. J. Hydrogen Energy, 1992, 17, 863–866 CrossRef CAS.
- P. Cuendet, M. Gratzel and M. L. Pelaprat, J. Electroanal. Chem., 1984, 181, 173–185 CrossRef CAS.
- I. Okura, Coord. Chem. Rev., 1985, 68, 53–99 CrossRef CAS.
- A. A. Krasnovskii, G. P. Brin and V. V. Nikandrov, Dokl. Akad. Nauk SSSR, 1976, 229, 990–993 CAS.
- V. V. Nikandrov, G. P. Brin and A. A. Krasnovskii, Dokl. Akad. Nauk SSSR, 1981, 256, 1249–1253 CAS.
- P. Cuendet, K. K. Rao, M. Gratzel and D. O. Hall, Biochimie, 1986, 68, 217–221 CrossRef CAS.
- P. Cuendet, M. Gratzel, K. K. Rao and D. O. Hall, Photobiochem. Photobiophys., 1984, 7, 331–340 Search PubMed.
- V. V. Nikandrov, M. A. Shlyk, N. A. Zorin, I. N. Gogotov and A. A. Krasnovsky, FEBS Lett., 1988, 234, 111–114 CrossRef CAS.
- P. Pedroni, G. M. Mura, G. Galli, C. Pratesi, L. Serbolisca and G. Grandi, Int. J. Hydrogen Energy, 1996, 21, 853–858 CrossRef CAS.
- G. M. Mura, M. L. Ganadu, P. Lombardi, G. Lubinu, M. Branca and V. Maida, J. Photochem. Photobiol., A, 2002, 148, 199–204 CrossRef CAS.
- K. Gurunathan, J. Mol. Catal. A: Chem., 2000, 156, 59–67 CrossRef CAS.
- A. Parkin, G. Goldet, C. Cavazza, J. C. Fontecilla-Camps and F. A. Armstrong, J. Am. Chem. Soc., 2008, 130, 13410–13416 CrossRef CAS.
- K. A. Vincent, A. Parkin, O. Lenz, S. P. J. Albracht, J. C. Fontecilla-Camps, R. Cammack, B. Friedrich and F. A. Armstrong, J. Am. Chem. Soc., 2005, 127, 18179–18189 CrossRef CAS.
- E. Reisner, D. J. Powell, C. Cavazza, J. C. Fontecilla-Camps and F. A. Armstrong, J. Am. Chem. Soc., 2009, 131, 18457–18466 CrossRef CAS.
- S. H. Toma, J. A. Bonacin, K. Araki and H. E. Toma, Surf. Sci., 2006, 600, 4591–4597 CrossRef CAS.
- T. Rajh, A. E. Ostafin, O. I. Micic, D. M. Tiede and M. C. Thurnauer, J. Phys. Chem., 1996, 100, 4538–4545 CrossRef CAS.
- W. Bae and R. K. Mehra, J. Inorg. Biochem., 1998, 70, 125–135 CrossRef CAS.
- V. Rajendran, M. Lehnig and C. M. Niemeyer, J. Mater. Chem., 2009, 19, 6348–6353 RSC.
- O. V. Makarova, T. Rajh, M. C. Thurnauer, A. Martin, P. A. Kemme and D. Cropek, Environ. Sci. Technol., 2000, 34, 4797–4803 CrossRef CAS.
- D. Cropek, P. A. Kemme, O. V. Makarova, L. X. Chen and T. Rajh, J. Phys. Chem. C, 2008, 112, 8311–8318 CrossRef CAS.
- W. Y. Ahn, S. A. Sheeley, T. Rajh and D. M. Cropek, Appl. Catal., B, 2007, 74, 103–110 CrossRef CAS.
- H. M. Jia, W. J. Xiao, L. Z. Zhang, Z. Zheng, H. L. Zhang and F. Deng, J. Phys. Chem. C, 2008, 112, 11379–11384 CrossRef CAS.
- I. A. Ruvarac-Bugarcic, Z. V. Saponjic, S. Zec, T. Rajh and J. M. Nedeljkovic, Chem. Phys. Lett., 2005, 407, 110–113 CrossRef CAS.
- G. Morandi, L. Heath and W. Thielemans, Langmuir, 2009, 25, 8280–8286 CrossRef CAS.
- M. J. Uddin, F. Cesano, D. Scarano, F. Bonino, G. Agostini, G. Spoto, S. Bordiga and A. Zecchina, J. Photochem. Photobiol., A, 2008, 199, 64–72 CrossRef CAS.
- Y. Iguchi, H. Ichiura, T. Kitaoka and H. Tanaka, Chemosphere, 2003, 53, 1193–1199 CrossRef CAS.
- R. Pelton, X. L. Geng and M. Brook, Adv. Colloid Interface Sci., 2006, 127, 43–53 CrossRef CAS.
- L. Ye, C. D. M. Filipe, M. Kavoosi, C. A. Haynes, R. Pelton and M. A. Brook, J. Mater. Chem., 2009, 19, 2189–2198 RSC.
- G. Y. Zhang, R. J. Qu, C. M. Sun, C. N. Ji, H. Chen, C. H. Wang and Y. Z. Niu, J. Appl. Polym. Sci., 2008, 110, 2321–2327 CrossRef CAS.
- C. Suwanchawalit, A. J. Patil, R. K. Kumar, S. Wongnawa and S. Mann, J. Mater. Chem., 2009, 19, 8478–8483 RSC.
- Z. Zainal, L. K. Hui, M. Z. Hussein, A. H. Abdullah and I. R. Hamadneh, J. Hazard. Mater., 2009, 164, 138–145 CrossRef CAS.
- J. D. Torres, E. A. Faria, J. R. SouzaDe and A. G. S. Prado, J. Photochem. Photobiol., A, 2006, 182, 202–206 CrossRef CAS.
- H. Y. Zhu, R. Jiang, L. Xiao, Y. H. Chang, Y. J. Guan, X. D. Li and G. M. Zeng, J. Hazard. Mater., 2009, 169, 933–940 CrossRef CAS.
- R. Jiang, H. Y. Zhu, X. D. Li and L. Xiao, Chem. Eng. J., 2009, 152, 537–542 CrossRef CAS.
- J. Y. Chen, P. J. Zhou, J. L. Li and Y. Wang, Carbohydr. Polym., 2008, 72, 128–132 CrossRef CAS.
- T. Y. Ma, X. J. Zhang, G. S. Shao, J. L. Cao and Z. Y. Yuan, J. Phys. Chem. C, 2008, 112, 3090–3096 CrossRef CAS.
- J. Feng, A. Miedaner, P. Ahrenlkiel, M. E. Himmel, C. Curtis and D. Ginley, J. Am. Chem. Soc., 2005, 127, 14968–14969 CrossRef CAS.
- L. D. Li, X. H. Sun, Y. L. Yang, N. J. Guan and F. X. Zhang, Chem.–Asian J., 2006, 1, 664–668 CrossRef CAS.
- I. Willner and Y. Eichen, J. Am. Chem. Soc., 1987, 109, 6862–6863 CrossRef CAS.
- N. M. Dimitrijevic, T. Rajh, Z. V. Saponjic, L. de la Garza and D. M. Tiede, J. Phys. Chem. B, 2004, 108, 9105–9110 CrossRef CAS.
- G. Wang, F. Wu, X. Zhang, M. Luo and N. Deng, J. Hazard. Mater., 2006, 133, 85–91 CrossRef CAS.
- P. Lu, F. Wu and N. S. Deng, Appl. Catal., B, 2004, 53, 87–93 CrossRef CAS.
- X. Zhang, F. Wu and N. S. Deng, Catal. Commun., 2010, 11, 422–425 CrossRef CAS.
- L. Xu, L. P. Jiang and J. J. Zhu, Nanotechnology, 2009, 20, 045605 CrossRef.
- T. Rajh, J. M. Nedeljkovic, L. X. Chen, O. Poluektov and M. C. Thurnauer, J. Phys. Chem. B, 1999, 103, 3515–3519 CrossRef CAS.
- Y. Ou, J. D. Lin, H. M. Zou and D. W. Liao, J. Mol. Catal. A: Chem., 2005, 241, 59–64 CrossRef CAS.
- J. M. Meichtry, M. Brusa, G. Mailhot, M. A. Grela and M. I. Litter, Appl. Catal., B, 2007, 71, 101–107 CrossRef CAS.
- L. X. Yang, Y. Xiao, S. H. Liu, Y. Li, Q. Y. Cai, S. L. Luo and G. M. Zeng, Appl. Catal., B, 2010, 94, 142–149 CrossRef CAS.
- M. P. Reddy, A. Venugopal and M. Subrahmanyam, Water Res., 2007, 41, 379–386 CrossRef CAS.
- Z. P. Yang and C. J. Zhang, Catal. Commun., 2008, 10, 351–354 CrossRef CAS.
- J. Park, J. Alloys Compd., 2010, 492, L57–L60 CrossRef CAS.
- M. Joshi, S. R. Kamble, N. K. Labhsetwar, D. V. Parwate and S. S. Rayalu, J. Photochem. Photobiol., A, 2009, 204, 83–89 CrossRef CAS.
- K. H. Nealson and P. G. Conrad, Philos. Trans. R. Soc. London, Ser. B, 1999, 354, 1923–1939 CrossRef CAS.
- H. Imahori, Y. Mori and Y. Matano, J. Photochem. Photobiol., C, 2003, 4, 51–83 CrossRef CAS.
- R. Lahtinen, D. J. Fermin, K. Kontturi and H. H. Girault, J. Electroanal. Chem., 2000, 483, 81–87 CrossRef CAS.
- T. Itoh, K. Yano, Y. Inada and Y. Fukushima, J. Mater. Chem., 2002, 12, 3275–3277 RSC.
- T. Itoh, K. Yano, Y. Inada and Y. Fukushima, J. Am. Chem. Soc., 2002, 124, 13437–13441 CrossRef CAS.
- T. Itoh, K. Yano, T. Kajino, S. Itoh, Y. Shibata, H. Mino, R. Miyamoto, Y. Inada, S. Iwai and Y. Fukushima, J. Phys. Chem. B, 2004, 108, 13683–13687 CrossRef CAS.
- I. Oda, K. Hirata, S. Watanabe, Y. Shibata, T. Kajino, Y. Fukushima, S. Iwai and S. Itoh, J. Phys. Chem. B, 2006, 110, 1114–1120 CrossRef CAS.
- C. F. Meunier, P. Van Cutsem, Y. U. Kwon and B. L. Su, J. Mater. Chem., 2009, 19, 1535–1542 RSC.
- H. Szmacinski and J. R. Lakowicz, Anal. Chem., 2008, 80, 6260–6266 CrossRef CAS.
- A. O. Govorov and I. Carmeli, Nano Lett., 2007, 7, 620–625 CrossRef CAS.
- S. Mackowski, S. Wormke, A. J. Maier, T. H. P. Brotosudarmo, H. Harutyunyan, A. Hartschuh, A. O. Govorov, H. Scheer and C. Brauchle, Nano Lett., 2008, 8, 558–564 CrossRef CAS.
- L. Zheng, F. S. Hong, S. P. Lu and C. Liu, Biol. Trace Elem. Res., 2005, 104, 83–91 CrossRef CAS.
- F. Yang, C. Liu, F. Q. Gao, M. Y. Su, X. Wu, L. Zheng, F. S. Hong and P. Yang, Biol. Trace Elem. Res., 2007, 119, 77–88 CrossRef CAS.
- M. Y. Su, X. Wu, C. Liu, C. X. Qu, X. Q. Liu, L. Chen, H. Huang and F. S. Hong, Biol. Trace Elem. Res., 2007, 119, 183–192 CrossRef CAS.
- A. C. S. Samia, X. B. Chen and C. Burda, J. Am. Chem. Soc., 2003, 125, 15736–15737 CrossRef CAS.
- J. M. Tsay, M. Trzoss, L. X. Shi, X. X. Kong, M. Selke, M. E. Jung and S. Weiss, J. Am. Chem. Soc., 2007, 129, 6865–6871 CrossRef CAS.
- E. R. Goldman, E. D. Balighian, H. Mattoussi, M. K. Kuno, J. M. Mauro, P. T. Tran and G. P. Anderson, J. Am. Chem. Soc., 2002, 124, 6378–6382 CrossRef CAS.
- Y. Sun, Z. L. Chen, X. X. Yang, P. Huang, X. P. Zhou and X. X. Du, Nanotechnology, 2009, 20, 135102 CrossRef.
- T. Sakakura, J. C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS.
- S. C. Roy, O. K. Varghese, M. Paulose and C. A. Grimes, ACS Nano, 2010, 4, 1259–1278 CrossRef CAS.
- T. W. Woolerton, S. Sheard, E. Reisner, E. Pierce, S. W. Ragsdale and F. A. Armstrong, J. Am. Chem. Soc., 2010, 132, 2132–2133 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |